Regenerative Therapies to Restore Interneuron Disturbances in Experimental Models of Encephalopathy of Prematurity

 ,
, 
Abstract
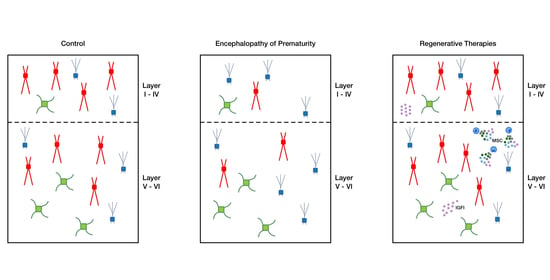
:
1. Introduction
2. Results
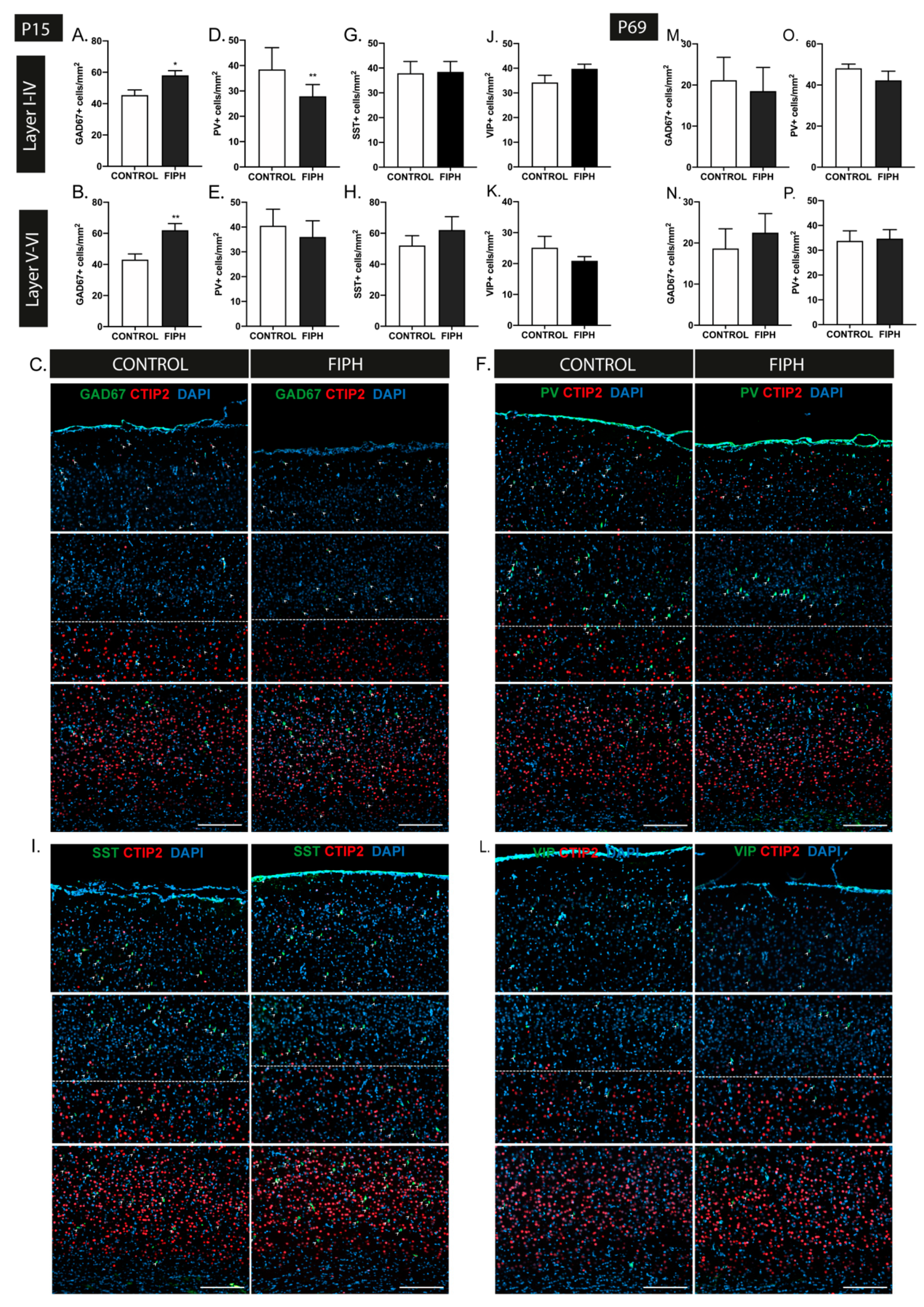
2.1. Fetal Inflammation and Postnatal Hypoxia in Rats Leads to Transient Disturbances in GABAergic Interneuron Distribution in the Cortex and Hippocampus
2.2. Intranasal MSC and IGF1 Therapy Restore Subtype-Specific Interneuron Deficits in the Cortex and Hippocampus after Postnatal Hypoxia-Ischemia and Systemic Inflammation in Mice
2.3. Treatment with MSCs or IGF1 Partly Restores Sociability in EoP Mice
3. Discussion
4. Materials and Methods
4.1. Animal Models of Encephalopathy of Prematurity
4.1.1. Rat Model
4.1.2. Mouse Model
4.2. Treatments in the Mouse EoP Model
4.2.1. Mesenchymal Stem Cells
4.2.2. Insulin-Like Growth Factor 1
4.3. Arx, Lhx6, and Sox6 Expression in EoP Mice
4.4. Three Chamber Social Test in EoP Mice
4.5. Immunohistochemistry
4.6. Microscopy and Image Analysis
4.7. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EoP | Encephalopathy of Prematurity |
| MSC | Mesenchymal Stem Cell |
| IGF1 | Insulin-like Growth Factor I |
| PV | Parvalbumin |
| SST | Somatostatin |
| VIP | Vasoactive Intestinal Polypeptide |
| GAD | Glutamate Decarboxylase |
| WMI | White Matter Injury |
| ADD | Attention Deficit Disorder |
| ASD | Autism Spectrum Disorder |
| FIPH | Fetal Inflammation and Postnatal Hypoxia |
| HI+LPS | Hypoxia/Ischemia and Lipopolysaccharide |
| DG | Dentate Gyrus |
| CA | Cornu Ammonis |
| P | Postnatal Day |
| i.p. | Intraperitoneal |
| LPS | Lipopolysaccharide |
| SEM | Standard Error of the Mean |
| HI | Hypoxia-ischemia |
| HGF | Hepatocyte Growth Factor |
| GDNF | Glial Cell Line-Derived Neurotrophic Factor |
| NGF | Nerve Growth Factor |
| MGE | Medial Ganglionic Eminence |
| CGE | Caudal Ganglionic Eminence |
References
- Deng, W. Neurobiology of injury to the developing brain. Nat. Rev. Neurol. 2010, 6, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Larroque, B.; Ancel, P.Y.; Marret, S.; Marchand, L.; Andre, M.; Arnaud, C.; Pierrat, V.; Roze, J.C.; Messer, J.; Thiriez, G.; et al. Neurodevelopmental disabilities and special care of 5-year-old children born before 33 weeks of gestation (the EPIPAGE study): A longitudinal cohort study. Lancet 2008, 371, 813–820. [Google Scholar] [CrossRef]
- Moster, D.; Lie, R.T.; Markestad, T. Long-term medical and social consequences of preterm birth. New Engl. J. Med. 2008, 359, 262–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsell, L.; Johnson, S.; Wolke, D.; O’Reilly, H.; Morris, J.K.; Kurinczuk, J.J.; Marlow, N. Cognitive trajectories from infancy to early adulthood following birth before 26 weeks of gestation: A prospective, population-based cohort study. Arch. Dis. Child. 2018, 103, 363–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, S.A.; Miller, S.P. Brain injury in premature neonates: A primary cerebral dysmaturation disorder? Ann. Neurol. 2014, 75, 469–486. [Google Scholar] [CrossRef]
- Van Tilborg, E.; Heijnen, C.J.; Benders, M.J.; van Bel, F.; Fleiss, B.; Gressens, P.; Nijboer, C.H. Impaired oligodendrocyte maturation in preterm infants: Potential therapeutic targets. Prog. Neurobiol. 2016, 136, 28–49. [Google Scholar] [CrossRef]
- Volpe, J.J. Brain injury in premature infants: A complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009, 8, 110–124. [Google Scholar] [CrossRef] [Green Version]
- Back, S.A. White matter injury in the preterm infant: Pathology and mechanisms. Acta Neuropathol. 2017, 134, 331–349. [Google Scholar] [CrossRef]
- Khwaja, O.; Volpe, J.J. Pathogenesis of cerebral white matter injury of prematurity. Arch. Dis. Child. Fetal Neonatal Ed. 2008, 93, F153–F161. [Google Scholar] [CrossRef]
- De Vries, L.S.; Benders, M.J.N.L.; Groenendaal, F. Imaging the premature brain: Ultrasound or MRI? Neuroradiology 2013, 55, 13–22. [Google Scholar] [CrossRef]
- Ment, L.R.; Hirtz, D.; Huppi, P.S. Imaging biomarkers of outcome in the developing preterm brain. Lancet Neurol. 2009, 8, 1042–1055. [Google Scholar] [CrossRef]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Levine, J.M.; Volpe, J.J.; Kinney, H.C. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 2001, 21, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Vinukonda, G.; Vose, L.R.; Diamond, D.; Bhimavarapu, B.B.R.; Hu, F.; Zia, M.T.; Hevner, R.; Zecevic, N.; Ballabh, P. Neurogenesis continues in the third trimester of pregnancy and is suppressed by premature birth. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 411–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleiss, B.; Gressens, P.; Stolp, H.B. Cortical Gray Matter Injury in Encephalopathy of Prematurity: Link to Neurodevelopmental Disorders. Front. Neurol. 2020, 11, 575. [Google Scholar] [CrossRef]
- Arshad, A.; Vose, L.R.; Vinukonda, G.; Hu, F.; Yoshikawa, K.; Csiszar, A.; Brumberg, J.C.; Ballabh, P. Extended Production of Cortical Interneurons into the Third Trimester of Human Gestation. Cereb. Cortex 2016, 26, 2242–2256. [Google Scholar] [CrossRef] [Green Version]
- Lim, L.; Mi, D.; Llorca, A.; Marín, O. Development and Functional Diversification of Cortical Interneurons. Neuron 2018, 100, 294–313. [Google Scholar] [CrossRef] [Green Version]
- Marín, O. Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 107–120. [Google Scholar] [CrossRef]
- Marín, O. Developmental timing and critical windows for the treatment of psychiatric disorders. Nat. Med. 2016, 22, 1229–1238. [Google Scholar] [CrossRef] [Green Version]
- Stolp, H.B.; Fleiss, B.; Arai, Y.; Supramaniam, V.; Vontell, R.; Birtles, S.; Yates, A.G.; Baburamani, A.A.; Thornton, C.; Rutherford, M.; et al. Interneuron Development Is Disrupted in Preterm Brains With Diffuse White Matter Injury: Observations in Mouse and Human. Front. Physiol. 2019, 10, 955. [Google Scholar] [CrossRef] [Green Version]
- Panda, S.; Dohare, P.; Jain, S.; Parikh, N.; Singla, P.; Mehdizadeh, R.; Klebe, D.W.; Kleinman, G.M.; Cheng, B.; Ballabh, P. Estrogen Treatment Reverses Prematurity-Induced Disruption in Cortical Interneuron Population. J. Neurosci. 2018, 38, 7378–7391. [Google Scholar] [CrossRef] [Green Version]
- Lacaille, H.; Vacher, C.M.; Bakalar, D.; O’Reilly, J.J.; Salzbank, J.; Penn, A.A. Impaired Interneuron Development in a Novel Model of Neonatal Brain Injury. eNeuro 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, S.; Li, Q.; Dechant, A.; Cohen, M.L. Neonatal loss of gamma-aminobutyric acid pathway expression after human perinatal brain injury. J. Neurosurg. 2006, 104, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Ardalan, M.; Svedin, P.; Baburamani, A.A.; Supramaniam, V.G.; Ek, J.; Hagberg, H.; Mallard, C. Dysmaturation of Somatostatin Interneurons Following Umbilical Cord Occlusion in Preterm Fetal Sheep. Front. Physiol. 2019, 10, 563. [Google Scholar] [CrossRef] [Green Version]
- Tibrewal, M.; Cheng, B.; Dohare, P.; Hu, F.; Mehdizadeh, R.; Wang, P.; Zheng, D.; Ungvari, Z.; Ballabh, P. Disruption of Interneuron Neurogenesis in Premature Newborns and Reversal with Estrogen Treatment. J. Neurosci. 2018, 38, 1100–1113. [Google Scholar] [CrossRef] [PubMed]
- Duchatel, R.J.; Harms, L.R.; Meehan, C.L.; Michie, P.T.; Bigland, M.J.; Smith, D.W.; Jobling, P.; Hodgson, D.M.; Tooney, P.A. Reduced cortical somatostatin gene expression in a rat model of maternal immune activation. Psychiatry Res. 2019, 282, 112621. [Google Scholar] [CrossRef]
- Canetta, S.; Bolkan, S.; Padilla-Coreano, N.; Song, L.J.; Sahn, R.; Harrison, N.L.; Gordon, J.A.; Brown, A.; Kellendonk, C. Maternal immune activation leads to selective functional deficits in offspring parvalbumin interneurons. Mol. Psychiatry 2016, 21, 956–968. [Google Scholar] [CrossRef]
- Komitova, M.; Xenos, D.; Salmaso, N.; Tran, K.M.; Brand, T.; Schwartz, M.L.; Ment, L.; Vaccarino, F.M. Hypoxia-induced developmental delays of inhibitory interneurons are reversed by environmental enrichment in the postnatal mouse forebrain. J. Neurosci. 2013, 33, 13375–13387. [Google Scholar] [CrossRef]
- Vaes, J.E.G.; Vink, M.A.; de Theije, C.G.M.; Hoebeek, F.E.; Benders, M.; Nijboer, C.H.A. The Potential of Stem Cell Therapy to Repair White Matter Injury in Preterm Infants: Lessons Learned From Experimental Models. Front. Physiol. 2019, 10, 540. [Google Scholar] [CrossRef]
- van Velthoven, C.T.; Kavelaars, A.; Heijnen, C.J. Mesenchymal stem cells as a treatment for neonatal ischemic brain damage. Pediatr. Res. 2012, 71, 474–481. [Google Scholar] [CrossRef]
- Ophelders, D.; Gussenhoven, R.; Klein, L.; Jellema, R.K.; Westerlaken, R.J.J.; Hütten, M.C.; Vermeulen, J.; Wassink, G.; Gunn, A.J.; Wolfs, T. Preterm Brain Injury, Antenatal Triggers, and Therapeutics: Timing Is Key. Cells 2020, 9, 1871. [Google Scholar] [CrossRef]
- Wagenaar, N.; Nijboer, C.H.; van Bel, F. Repair of neonatal brain injury: Bringing stem cell-based therapy into clinical practice. Dev. Med. Child. Neurol. 2017, 59, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaes, J.E.G.; van Kammen, C.M.; Trayford, C.; van der Toorn, A.; Ruhwedel, T.; Benders, M.J.N.L.; Dijkhuizen, R.M.; Möbius, W.; van Rijt, S.H.; Nijboer, C.H. Intranasal mesenchymal stem cell therapy to boost myelination after encephalopathy of prematurity. Glia 2020. [Google Scholar] [CrossRef] [PubMed]
- van Velthoven, C.T.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Mesenchymal stem cell treatment after neonatal hypoxic-ischemic brain injury improves behavioral outcome and induces neuronal and oligodendrocyte regeneration. Brain Behav. Immunity 2010, 24, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Oppliger, B.; Joerger-Messerli, M.; Reinhart, U.; Barnea, E.; Paidas, M.; Kramer, B.W.; Surbek, D.V.; Schoeberlein, A. Wharton’s Jelly Mesenchymal Stem Cells Protect the Immature Brain in Rats and Modulate Cell Fate. Stem Cells Dev. 2017, 26, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Paton, M.C.B.; Allison, B.J.; Fahey, M.C.; Li, J.; Sutherland, A.E.; Pham, Y.; Nitsos, I.; Bischof, R.J.; Moss, T.J.; Polglase, G.R.; et al. Umbilical cord blood versus mesenchymal stem cells for inflammation-induced preterm brain injury in fetal sheep. Pediatr. Res. 2019, 86, 165–173. [Google Scholar] [CrossRef]
- Hellstrom, A.; Ley, D.; Hansen-Pupp, I.; Hallberg, B.; Lofqvist, C.; van Marter, L.; van Weissenbruch, M.; Ramenghi, L.A.; Beardsall, K.; Dunger, D.; et al. Insulin-like growth factor 1 has multisystem effects on foetal and preterm infant development. Acta Paediatr. 2016, 105, 576–586. [Google Scholar] [CrossRef] [Green Version]
- Hansen-Pupp, I.; Hovel, H.; Lofqvist, C.; Hellstrom-Westas, L.; Fellman, V.; Huppi, P.S.; Hellstrom, A.; Ley, D. Circulatory insulin-like growth factor-I and brain volumes in relation to neurodevelopmental outcome in very preterm infants. Pediatr. Res. 2013, 74, 564–569. [Google Scholar] [CrossRef]
- Beck, K.D.; Powell-Braxton, L.; Widmer, H.R.; Valverde, J.; Hefti, F. Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron 1995, 14, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Hansen-Pupp, I.; Hovel, H.; Hellstrom, A.; Hellstrom-Westas, L.; Lofqvist, C.; Larsson, E.M.; Lazeyras, F.; Fellman, V.; Huppi, P.S.; Ley, D. Postnatal decrease in circulating insulin-like growth factor-I and low brain volumes in very preterm infants. J. Clin. Endocrinol. Metab. 2011, 96, 1129–1135. [Google Scholar] [CrossRef]
- Nieto-Estévez, V.; Defterali, Ç.; Vicario-Abejón, C. IGF-I: A Key Growth Factor that Regulates Neurogenesis and Synaptogenesis from Embryonic to Adult Stages of the Brain. Front. Neurosci. 2016, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Hellström, A.; Ley, D.; Hansen-Pupp, I.; Hallberg, B.; Ramenghi, L.A.; Löfqvist, C.; Smith, L.E.; Hård, A.L. Role of Insulinlike Growth Factor 1 in Fetal Development and in the Early Postnatal Life of Premature Infants. Am. J. Perinatol. 2016, 33, 1067–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Fan, L.W.; Lin, S.; Pang, Y.; Rhodes, P.G. Intranasal administration of insulin-like growth factor-1 protects against lipopolysaccharide-induced injury in the developing rat brain. Neuroscience 2011, 194, 195–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Fan, L.W.; Rhodes, P.G.; Cai, Z. Intranasal administration of IGF-1 attenuates hypoxic-ischemic brain injury in neonatal rats. Exp. Neurol. 2009, 217, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Bennet, L.; George, S.; Wu, D.; Waldvogel, H.J.; Gluckman, P.D.; Faull, R.L.; Crosier, P.S.; Gunn, A.J. Insulin-like growth factor-1 reduces postischemic white matter injury in fetal sheep. J. Cereb. Blood Flow Metab. 2001, 21, 493–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Gunn, A.J.; Bennet, L.; Wu, D.; George, S.; Gluckman, P.D.; Shao, X.M.; Guan, J. Insulin-like growth factor (IGF)-1 suppresses oligodendrocyte caspase-3 activation and increases glial proliferation after ischemia in near-term fetal sheep. J. Cereb. Blood Flow Metab. 2003, 23, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Hevner, R.F. Layer-specific markers as probes for neuron type identity in human neocortex and malformations of cortical development. J. Neuropathol. Exp. Neurol. 2007, 66, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Roby, K.D.; Callaway, E.M. Immunochemical characterization of inhibitory mouse cortical neurons: Three chemically distinct classes of inhibitory cells. J. Comp. Neurol. 2010, 518, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 2016, 91, 260–292. [Google Scholar] [CrossRef] [Green Version]
- Rudy, B.; Fishell, G.; Lee, S.; Hjerling-Leffler, J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 2011, 71, 45–61. [Google Scholar] [CrossRef] [Green Version]
- van Tilborg, E.; Achterberg, E.J.M.; van Kammen, C.M.; van der Toorn, A.; Groenendaal, F.; Dijkhuizen, R.M.; Heijnen, C.J.; Vanderschuren, L.; Benders, M.; Nijboer, C.H.A. Combined fetal inflammation and postnatal hypoxia causes myelin deficits and autism-like behavior in a rat model of diffuse white matter injury. Glia 2018, 66, 78–93. [Google Scholar] [CrossRef]
- Pelkey, K.A.; Chittajallu, R.; Craig, M.T.; Tricoire, L.; Wester, J.C.; McBain, C.J. Hippocampal GABAergic Inhibitory Interneurons. Physiol. Rev. 2017, 97, 1619–1747. [Google Scholar] [CrossRef] [PubMed]
- Moy, S.S.; Nadler, J.J.; Perez, A.; Barbaro, R.P.; Johns, J.M.; Magnuson, T.R.; Piven, J.; Crawley, J.N. Sociability and preference for social novelty in five inbred strains: An approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004, 3, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashemi, E.; Ariza, J.; Rogers, H.; Noctor, S.C.; Martínez-Cerdeño, V. The Number of Parvalbumin-Expressing Interneurons Is Decreased in the Prefrontal Cortex in Autism. Cereb. Cortex 2017, 27, 1931–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunden, J.W.; Durens, M.; Phillips, A.W.; Nestor, M.W. Cortical interneuron function in autism spectrum condition. Pediatr. Res. 2019, 85, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiebe, S.; Nagpal, A.; Truong, V.T.; Park, J.; Skalecka, A.; He, A.J.; Gamache, K.; Khoutorsky, A.; Gantois, I.; Sonenberg, N. Inhibitory interneurons mediate autism-associated behaviors via 4E-BP2. Proc. Natl. Acad. Sci. USA 2019, 116, 18060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Lee, J.; Kim, E. Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders. Biol. Psychiatry 2017, 81, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.; Marlow, N. Preterm Birth and Childhood Psychiatric Disorders. Pediatr. Res. 2011, 69, 11–18. [Google Scholar] [CrossRef]
- Hu, J.S.; Vogt, D.; Sandberg, M.; Rubenstein, J.L. Cortical interneuron development: A tale of time and space. Development 2017, 144, 3867–3878. [Google Scholar] [CrossRef] [Green Version]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef]
- Kepecs, A.; Fishell, G. Interneuron cell types are fit to function. Nature 2014, 505, 318–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasistha, N.A.; Pardo-Navarro, M.; Gasthaus, J.; Weijers, D.; Müller, M.K.; García-González, D.; Malwade, S.; Korshunova, I.; Pfisterer, U.; von Engelhardt, J.; et al. Maternal inflammation has a profound effect on cortical interneuron development in a stage and subtype-specific manner. Mol. Psychiatry 2020, 25, 2313–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpe, J.J. Dysmaturation of Premature Brain: Importance, Cellular Mechanisms, and Potential Interventions. Pediatr. Neurol. 2019, 95, 42–66. [Google Scholar] [CrossRef] [PubMed]
- Laclef, C.; Métin, C. Conserved rules in embryonic development of cortical interneurons. Semin. Cell Dev. Biol. 2018, 76, 86–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wamsley, B.; Fishell, G. Genetic and activity-dependent mechanisms underlying interneuron diversity. Nat. Rev. Neurosci. 2017, 18, 299–309. [Google Scholar] [CrossRef]
- Miyoshi, G.; Fishell, G. GABAergic interneuron lineages selectively sort into specific cortical layers during early postnatal development. Cereb. Cortex 2011, 21, 845–852. [Google Scholar] [CrossRef] [Green Version]
- Denaxa, M.; Neves, G.; Rabinowitz, A.; Kemlo, S.; Liodis, P.; Burrone, J.; Pachnis, V. Modulation of Apoptosis Controls Inhibitory Interneuron Number in the Cortex. Cell Rep. 2018, 22, 1710–1721. [Google Scholar] [CrossRef] [Green Version]
- Liodis, P.; Denaxa, M.; Grigoriou, M.; Akufo-Addo, C.; Yanagawa, Y.; Pachnis, V. Lhx6 Activity Is Required for the Normal Migration and Specification of Cortical Interneuron Subtypes. J. Neurosci. 2007, 27, 3078. [Google Scholar] [CrossRef]
- Kessaris, N.; Magno, L.; Rubin, A.N.; Oliveira, M.G. Genetic programs controlling cortical interneuron fate. Curr. Opin. Neurobiol. 2014, 26, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Gonchar, Y.; Wang, Q.; Burkhalter, A. Multiple distinct subtypes of GABAergic neurons in mouse visual cortex identified by triple immunostaining. Front. Neuroanat. 2007, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Thion, M.S.; Mosser, C.A.; Férézou, I.; Grisel, P.; Baptista, S.; Low, D.; Ginhoux, F.; Garel, S.; Audinat, E. Biphasic Impact of Prenatal Inflammation and Macrophage Depletion on the Wiring of Neocortical Inhibitory Circuits. Cell Rep. 2019, 28, 1119–1126.e1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, H.M.; Mishra, P.; Kohn, J. The overwhelming use of rat models in nerve regeneration research may compromise designs of nerve guidance conduits for humans. J. Mater. Sci. Mater. Med. 2015, 26, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butt, S.J.; Stacey, J.A.; Teramoto, Y.; Vagnoni, C. A role for GABAergic interneuron diversity in circuit development and plasticity of the neonatal cerebral cortex. Curr. Opin. Neurobiol. 2017, 43, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Sato, W.; Kochiyama, T.; Uono, S.; Yoshimura, S.; Kubota, Y.; Sawada, R.; Sakihama, M.; Toichi, M. Reduced Gray Matter Volume in the Social Brain Network in Adults with Autism Spectrum Disorder. Front. Hum. Neurosci. 2017, 11, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, M.H.; Bissonette, G.B.; Mars, W.M.; Michalopoulos, G.K.; Achim, C.L.; Depireux, D.A.; Powell, E.M. Hepatocyte growth factor (HGF) modulates GABAergic inhibition and seizure susceptibility. Exp. Neurol. 2010, 221, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Powell, E.M.; Mars, W.M.; Levitt, P. Hepatocyte Growth Factor/Scatter Factor Is a Motogen for Interneurons Migrating from the Ventral to Dorsal Telencephalon. Neuron 2001, 30, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Pozas, E.; Ibáñez, C.F. GDNF and GFRα1 Promote Differentiation and Tangential Migration of Cortical GABAergic Neurons. Neuron 2005, 45, 701–713. [Google Scholar] [CrossRef] [Green Version]
- Canty, A.J.; Dietze, J.; Harvey, M.; Enomoto, H.; Milbrandt, J.; Ibáñez, C.F. Regionalized Loss of Parvalbumin Interneurons in the Cerebral Cortex of Mice with Deficits in GFRα1 Signaling. J. Neurosci. 2009, 29, 10695. [Google Scholar] [CrossRef] [Green Version]
- Mei, L.; Nave, K.-A. Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 2014, 83, 27–49. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-Chong, A.; Yusta-Boyo, M.J.; Vergaño-Vera, E.; Bulfone, A.; de Pablo, F.; Vicario-Abejón, C. IGF-I promotes neuronal migration and positioning in the olfactory bulb and the exit of neuroblasts from the subventricular zone. Eur. J. Neurosci. 2009, 30, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; McGuire, T.; Peng, C.Y.; Kessler, J.A. Differential effects of BMP signaling on parvalbumin and somatostatin interneuron differentiation. Development 2009, 136, 2633–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benamer, N.; Vidal, M.; Angulo, M.C. The cerebral cortex is a substrate of multiple interactions between GABAergic interneurons and oligodendrocyte lineage cells. Neurosci. Lett. 2020, 715, 134615. [Google Scholar] [CrossRef] [PubMed]
- Zonouzi, M.; Scafidi, J.; Li, P.; McEllin, B.; Edwards, J.; Dupree, J.L.; Harvey, L.; Sun, D.; Hübner, C.A.; Cull-Candy, S.G.; et al. GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nat. Neurosci. 2015, 18, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Plane, J.M.; Deng, W. Mouse Models of Periventricular Leukomalacia. J. Vis. Exp. 2010, 39, 1951. [Google Scholar] [CrossRef] [PubMed]
- Danielyan, L.; Schafer, R.; von Ameln-Mayerhofer, A.; Buadze, M.; Geisler, J.; Klopfer, T.; Burkhardt, U.; Proksch, B.; Verleysdonk, S.; Ayturan, M.; et al. Intranasal delivery of cells to the brain. Eur. J. Cell Biol. 2009, 88, 315–324. [Google Scholar] [CrossRef]
- Nadler, J.J.; Moy, S.S.; Dold, G.; Simmons, N.; Perez, A.; Young, N.B.; Barbaro, R.P.; Piven, J.; Magnuson, T.R.; Crawley, J.N. Automated apparatus for quantitation of social approach behaviors in mice. Genes Brain Behav. 2004, 3, 303–314. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|
| Arx | GCACCACGTTCACCAGTTAC | TCTGTCAGGTCCAGCCTCAT |
| Lhx6 | CGTTGAGGAGAAGGTGCTTTGC | GCTTGGGCTGACTGTCCTGTTC |
| Sox6 | ATCTCTCATCCCGGCCTAAGAC | TCCCCAGGCTTCCTCCAATG |
| GAPDH | TGAAGCAGGCATCTGAGGG | CGAAGGTGGAAGAGTGGGAG |
| β-actin | AGAGGGAAATCGTGCGTGAC | CAATAGTGATGACCTGGCCGT |
| Antigen | Species (Host) | Company, Product Code | Dilution |
|---|---|---|---|
| Anti-GAD67 | Mouse | Sigma, MAB5406 | 1:200 |
| Anti-PV | Mouse Rabbit | Sigma, P3088 Novusbio, NB120-11427 | 1:1000 1:200 |
| Anti-SST | Mouse | Santa Cruz, SC-74556 | 1:50 |
| Anti-VIP | Rabbit | ImmunoStar, 20077 | 1:1000 |
| Anti-CTIP2 | Rat | Abcam, ab18465 | 1:1000 |
| Anti-NeuN | Mouse Rabbit | Merck Millipore, mab377 Abcam, ab177487 | 1:200 1:500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaes, J.E.G.; Kosmeijer, C.M.; Kaal, M.; van Vliet, R.; Brandt, M.J.V.; Benders, M.J.N.L.; Nijboer, C.H. Regenerative Therapies to Restore Interneuron Disturbances in Experimental Models of Encephalopathy of Prematurity. Int. J. Mol. Sci. 2021, 22, 211. https://doi.org/10.3390/ijms22010211
Vaes JEG, Kosmeijer CM, Kaal M, van Vliet R, Brandt MJV, Benders MJNL, Nijboer CH. Regenerative Therapies to Restore Interneuron Disturbances in Experimental Models of Encephalopathy of Prematurity. International Journal of Molecular Sciences. 2021; 22(1):211. https://doi.org/10.3390/ijms22010211
Chicago/Turabian StyleVaes, Josine E. G., Chantal M. Kosmeijer, Marthe Kaal, Rik van Vliet, Myrna J. V. Brandt, Manon J. N. L. Benders, and Cora H. Nijboer. 2021. "Regenerative Therapies to Restore Interneuron Disturbances in Experimental Models of Encephalopathy of Prematurity" International Journal of Molecular Sciences 22, no. 1: 211. https://doi.org/10.3390/ijms22010211
APA StyleVaes, J. E. G., Kosmeijer, C. M., Kaal, M., van Vliet, R., Brandt, M. J. V., Benders, M. J. N. L., & Nijboer, C. H. (2021). Regenerative Therapies to Restore Interneuron Disturbances in Experimental Models of Encephalopathy of Prematurity. International Journal of Molecular Sciences, 22(1), 211. https://doi.org/10.3390/ijms22010211