Role of Macrophages and RhoA Pathway in Atherosclerosis




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
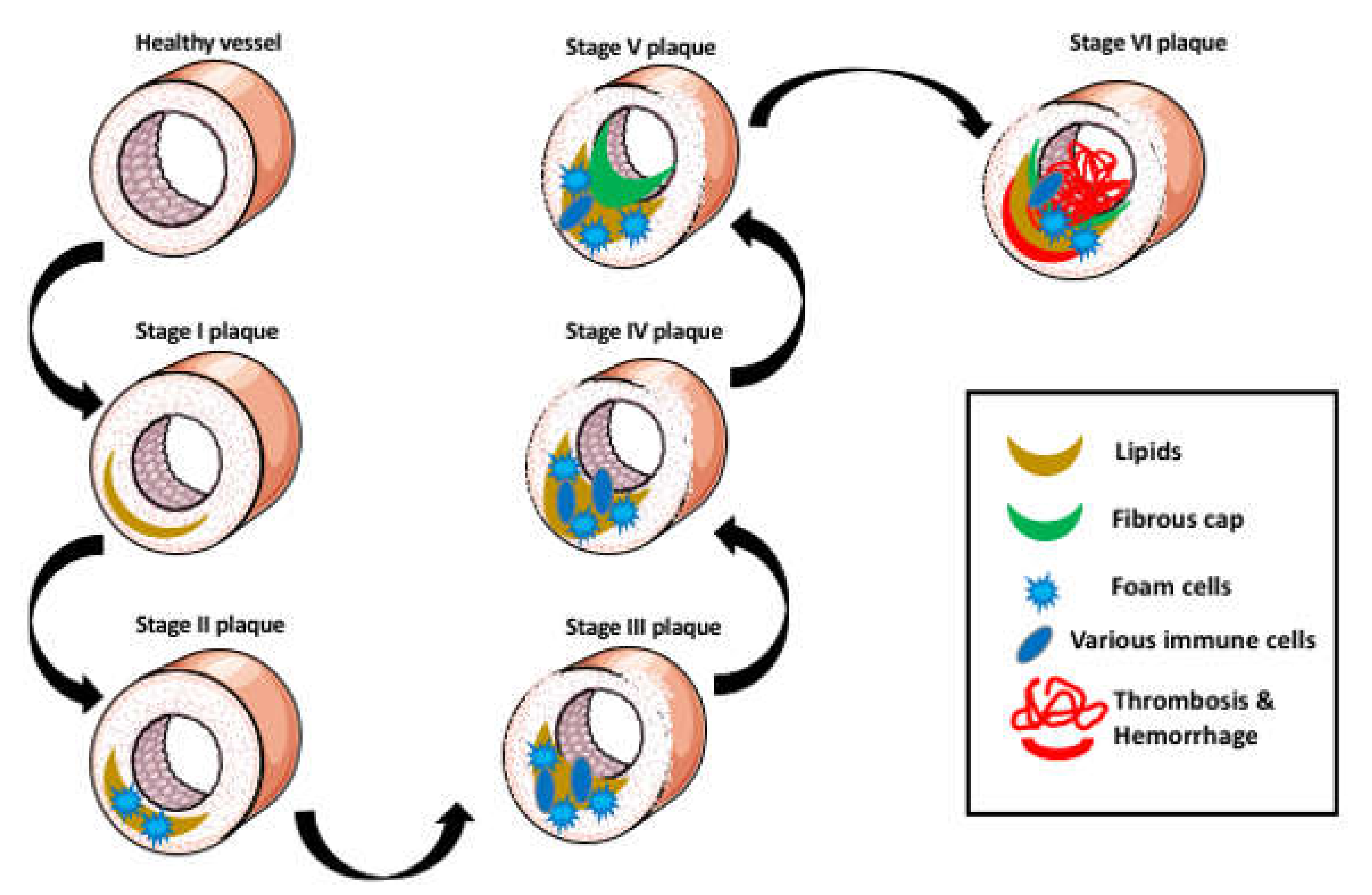
2. Aggregation of Lipids in the Arterial Wall
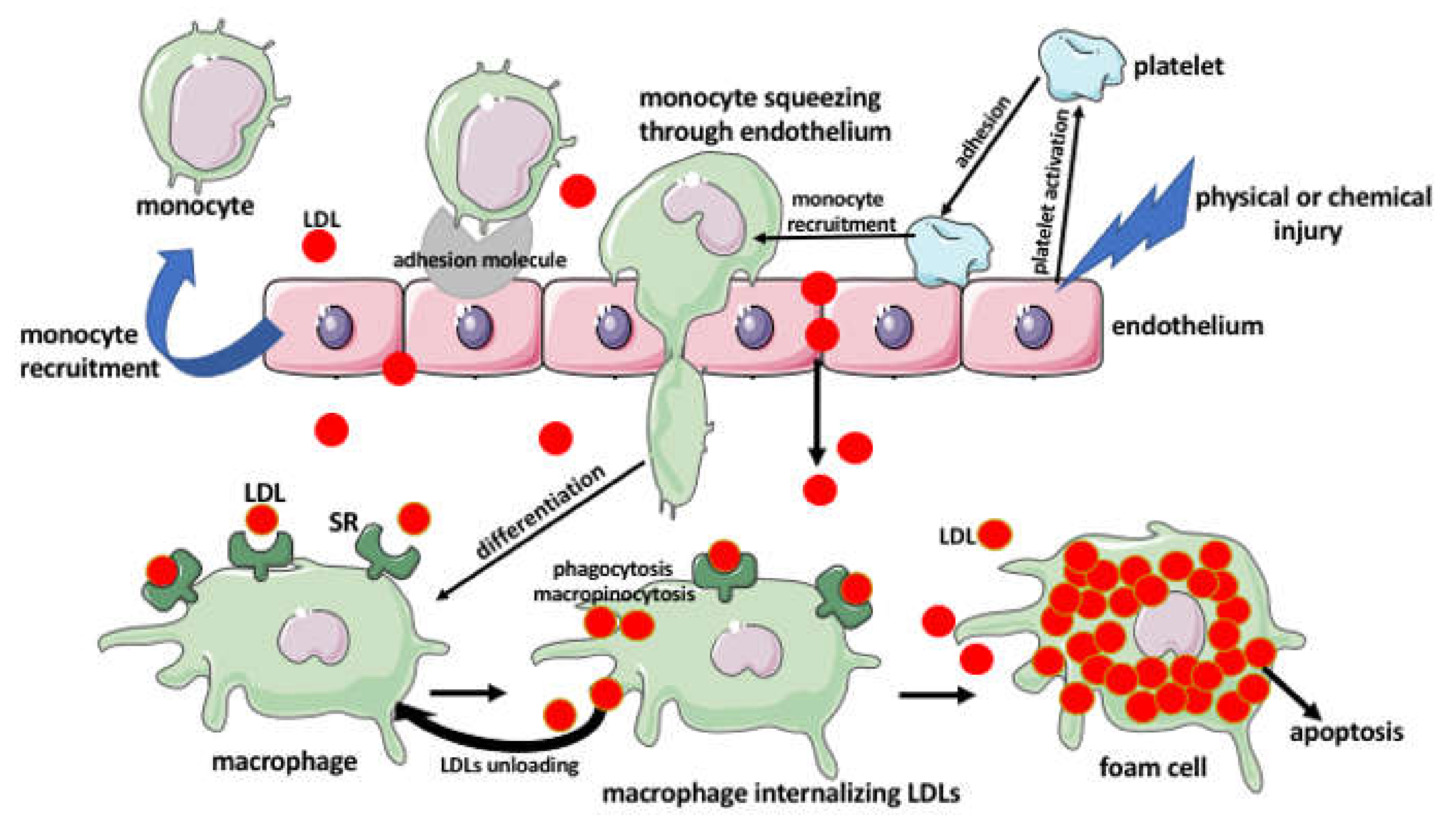
3. The Foam Cells
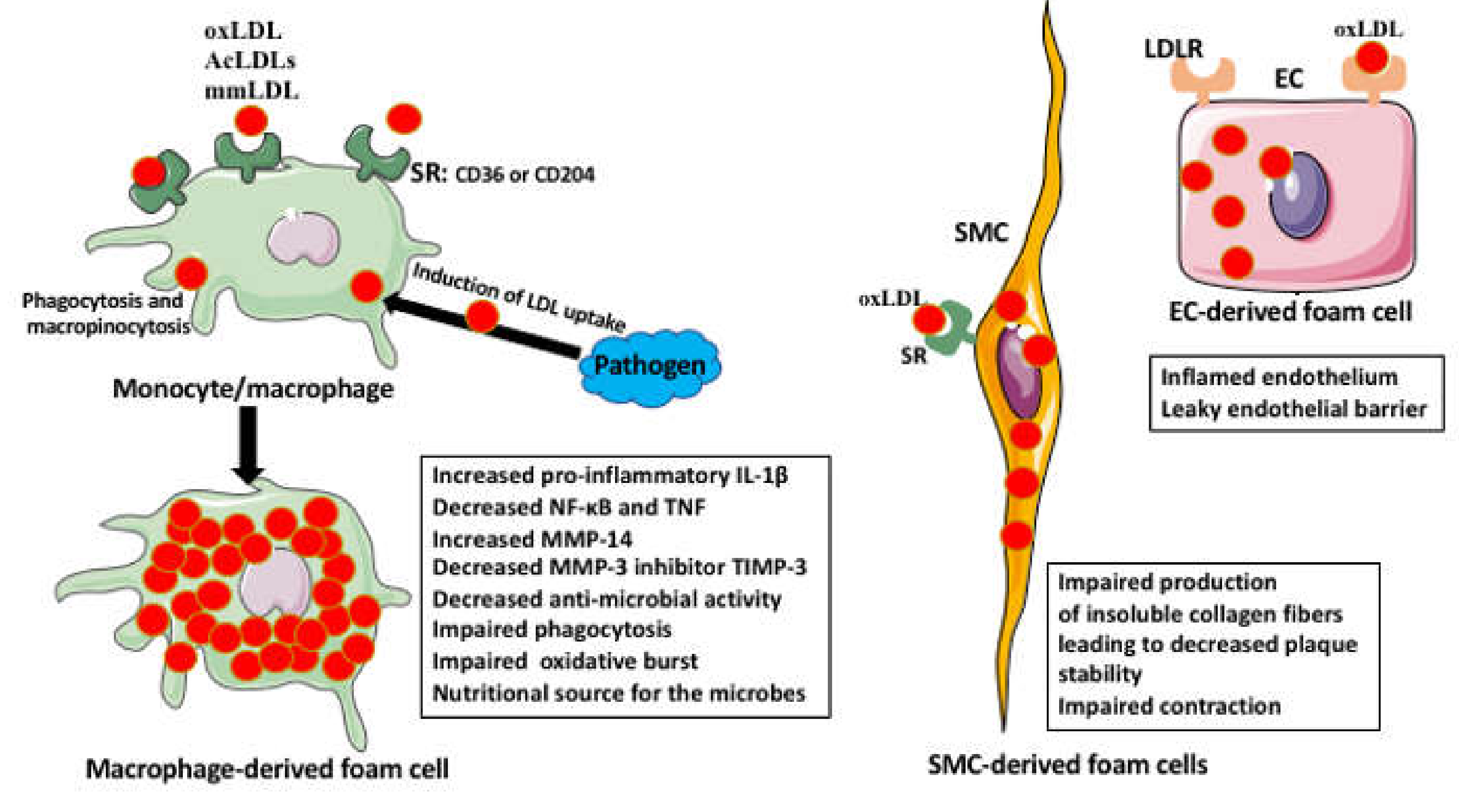
Foam Cell Activities
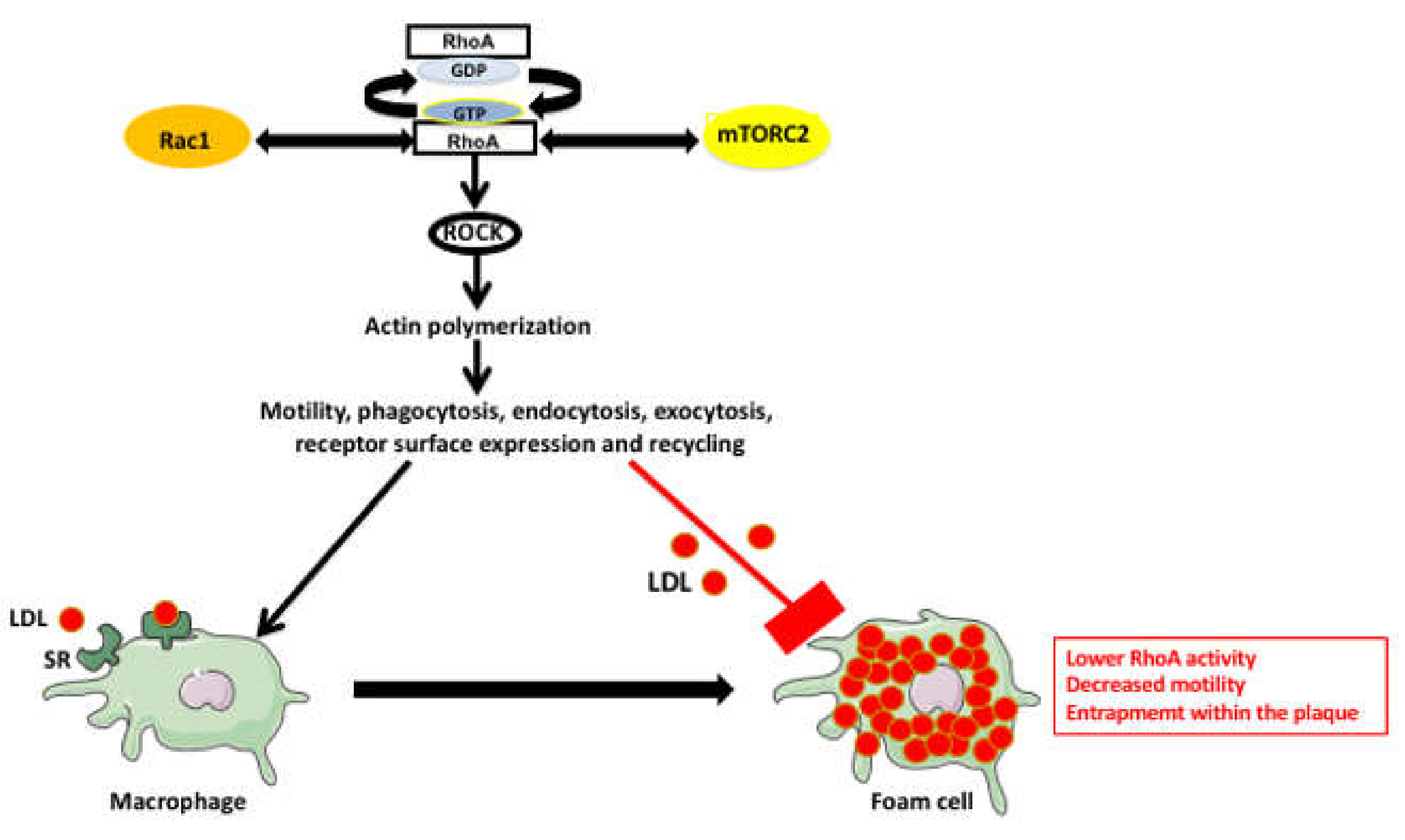
4. RhoA Pathway Involvement in Atherosclerosis
5. Summary and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kloc, M.; Ghobrial, R.M. Chronic allograft rejection: A significant hurdle to transplant success. Burn. Trauma 2014, 2, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Classification & Structure of Blood Vessels. Available online: https://training.seer.cancer.gov/anatomy/cardiovascular/blood/classification.html (accessed on 4 December 2020).
- Stary, H.C.; Chandler, A.B.; Glagov, S.; Guyton, J.R.; Insull, W., Jr.; Rosenfeld, M.E.; Schaffer, S.A.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1994, 89, 2462–2478. [Google Scholar] [CrossRef] [Green Version]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A Definition of Advanced Types of Atherosclerotic Lesions and a Histological Classification of Atherosclerosis. Circulation 1995, 92, 1355–1374. [Google Scholar] [CrossRef]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Finn, A.V.; Gold, H.K.; Tulenko, T.N.; Wrenn, S.P.; Narula, J. Atherosclerotic Plaque Progression and Vulnerability to Rupture. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2054–2061. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Gursky, O. Aggregation and fusion of low-density lipoproteins in vivo and in vitro. Biomol. Concepts 2013, 4, 501–518. [Google Scholar] [CrossRef] [Green Version]
- Frink, R.J. Chapter 1, The Beginnings. A Multicentric Disease. In Inflammatory Atherosclerosis: Characteristics of the Injurious Agent; Heart Research Foundation: Sacramento, CA, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK2029/ (accessed on 28 December 2020).
- Ross, R. The Pathogenesis of Atherosclerosis—An Update. N. Engl. J. Med. 1986, 314, 488–500. [Google Scholar] [CrossRef]
- Williams, K.J.; Tabas, I. The Response-to-Retention Hypothesis of Early Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Tedgui, A.; Mallat, Z. Cytokines in Atherosclerosis: Pathogenic and Regulatory Pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef] [Green Version]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Poole, J.C.F.; Florey, H.W. Changes in the endothelium of the aorta and the behaviour of macrophages in experimental atheroma of rabbits. J. Pathol. Bacteriol. 1958, 75, 245–251. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L. The Smad pathway. Cytokine Growth Factor Rev. 2000, 11, 5–13. [Google Scholar] [CrossRef]
- Mestas, J.; Ley, K. Monocyte-Endothelial Cell Interactions in the Development of Atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Huff, M.W.; Pickering, J.G. Can a Vascular Smooth Muscle–Derived Foam-Cell Really Change its Spots? Arterioscler. Thromb. Vasc. Biol. 2015, 35, 492–495. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. Cell biology of atherosclerosis. Annu. Rev. Physiol. 1995, 57, 791–804. [Google Scholar] [CrossRef]
- Pathogenesis of Atherosclerosis. Available online: https://sphweb.bumc.bu.edu/otlt/mphmodules/ph/ph709_heart/ph709_heart3.html (accessed on 4 December 2020).
- Ruuth, M.; Nguyen, S.D.; Vihervaara, T.; Hilvo, M.; Laajala, T.D.; Kondadi, P.K.; Gisterå, A.; Lähteenmäki, H.; Kittilä, T.; Huusko, J.; et al. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur. Heart J. 2018, 39, 2562–2573. [Google Scholar] [CrossRef] [Green Version]
- Tîrziu, D.; Dobrian, A.; Tasca, C.; Simionescu, M.; Simionescu, N. Intimal thickenings of human aorta contain modified reassembled lipoproteins. Atherosclerosis 1995, 112, 101–114. [Google Scholar] [CrossRef]
- Nievelstein, P.F.; Fogelman, A.M.; Mottino, G.; Frank, J.S. Lipid accumulation in rabbit aortic intima 2 hours after bolus infusion of low density lipoprotein. A deep-etch and immunolocalization study of ultrarapidly frozen tissue. Arterioscler. Thromb. J. Vasc. Biol. 1991, 11, 1795–1805. [Google Scholar] [CrossRef] [Green Version]
- Nievelstein-Post, P.; Mottino, G.; Fogelman, A.; Frank, J. An ultrastructural study of lipoprotein accumulation in cardiac valves of the rabbit. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 1151–1161. [Google Scholar] [CrossRef] [Green Version]
- Llorente-Cortes, V.; Badimon, L. LDL receptor-related protein and the vascular wall: Implications for atherothrombosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Siegel-Axel, D.; Daub, K.; Seizer, P.; Lindemann, S.; Gawaz, M. Platelet lipoprotein interplay: Trigger of foam cell formation and driver of atherosclerosis. Cardiovasc. Res. 2008, 78, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drechsler, M.; Duchêne, J.; Soehnlein, O. Chemokines Control Mobilization, Recruitment, and Fate of Monocytes in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1050–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eom, M.; Hudkins, K.L.; Alpers, C.E. Foam cells and the pathogenesis of kidney disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-H.; Fu, Y.-C.; Zhang, D.-W.; Yin, K.; Tang, C. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, P.N. Molecular Biology of Atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef]
- Chaabane, C.; Coen, M.; Bochaton-Piallat, M.-L. Smooth muscle cell phenotypic switch: Implications for foam cell formation. Curr. Opin. Lipidol. 2014, 25, 374–379. [Google Scholar] [CrossRef]
- Bandeali, S.; Farmer, J. High-Density Lipoprotein and Atherosclerosis: The Role of Antioxidant Activity. Curr. Atheroscler. Rep. 2012, 14, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Shashkin, P.; Dragulev, B.; Ley, K. Macrophage Differentiation to Foam Cells. Curr. Pharm. Des. 2005, 11, 3061–3072. [Google Scholar] [CrossRef] [Green Version]
- Baffy, G. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J. Hepatol. 2009, 51, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Dubland, J.A.; Allahverdian, S.; Asonye, E.; Sahin, B.; Jaw, J.E.; Sin, D.D.; Seidman, M.A.; Leeper, N.J.; Francis, G.A. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 876–887. [Google Scholar] [CrossRef]
- Owsiany, K.M.; Alencar, G.F.; Owens, G.K. Revealing the Origins of Foam Cells in Atherosclerotic Lesions. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 836–838. [Google Scholar] [CrossRef]
- Guerrini, V.; Gennaro, M.L. Foam Cells: One Size Doesn’t Fit All. Trends Immunol. 2019, 40, 1163–1179. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, V.; Prideaux, B.; Blanc, L.; Bruiners, N.; Arrigucci, R.; Singh, S.; Ho-Liang, H.P.; Salamon, H.; Chen, P.-Y.; Lakehal, K.; et al. Storage lipid studies in tuberculosis reveal that foam cell biogenesis is disease-specific. PLoS Pathog. 2018, 14, e1007223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grajchen, E.; Hendriks, J.J.A.; Bogie, J.F. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol. Commun. 2018, 6, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.W.; Saunders, B.T.; Kim, K.-W.; Epelman, S.; Lavine, K.; Zinselmeyer, B.H.; Randolph, G.J. Abstract 2: Aorta Intima-Resident Macrophages Contribute to Atherosclerotic Lesion Initiation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, A2. [Google Scholar]
- Michael, D.R.; Ashlin, T.G.; Davies, C.S.; Gallagher, H.; Stoneman, T.W.; Buckley, M.L.; Ramji, D.P. Differential regulation of macropinocytosis in macrophages by cytokines: Implications for foam cell formation and atherosclerosis. Cytokine 2013, 64, 357–361. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef] [Green Version]
- Kelley, J.L.; Ozment, T.R.; Li, C.; Schweitzer, J.B.; Williams, D.L. Scavenger Receptor-A (CD204): A Two-Edged Sword in Health and Disease. Crit. Rev. Immunol. 2014, 34, 241–261. [Google Scholar] [CrossRef]
- Shaik-Dasthagirisaheb, Y.B.; Mekasha, S.; He, X.; Gibson, F.C., III; Ingalls, R.R. Signaling events in pathogen-induced macrophage foam cell formation. Pathog. Dis. 2016, 74, ftw074. [Google Scholar] [CrossRef] [Green Version]
- Thorp, E.; Tabas, I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J. Leukoc. Biol. 2009, 86, 1089–1095. [Google Scholar] [CrossRef] [Green Version]
- Kojima, Y.; Weissman, I.L.; Leeper, N.J. The Role of Efferocytosis in Atherosclerosis. Circulation 2017, 135, 476–489. [Google Scholar] [CrossRef] [Green Version]
- Doran, A.C.; Ozcan, L.; Cai, B.; Zheng, Z.; Fredman, G.; Rymond, C.C.; Dorweiler, B.; Sluimer, J.C.; Hsieh, J.; Kuriakose, G.; et al. CAMKIIγ suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J. Clin. Investig. 2017, 127, 4075–4089. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy Links Inflammasomes to Atherosclerotic Progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robblee, M.M.; Kim, C.C.; Abate, J.P.; Valdearcos, M.; Sandlund, K.L.M.; Shenoy, M.K.; Volmer, R.; Iwawaki, T.; Koliwad, S.K. Saturated Fatty Acids Engage an IRE1α-Dependent Pathway to Activate the NLRP3 Inflammasome in Myeloid Cells. Cell Rep. 2016, 14, 2611–2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Riek, A.E.; Weng, S.; Petty, M.; Kim, D.; Colonna, M.; Cella, M.; Bernal-Mizrachi, C. Endoplasmic Reticulum Stress Controls M2 Macrophage Differentiation and Foam Cell Formation. J. Biol. Chem. 2012, 287, 11629–11641. [Google Scholar] [CrossRef] [Green Version]
- Daniel, J.; Maamar, H.; Deb, C.; Sirakova, T.D.; Kolattukudy, P.E. Mycobacterium tuberculosis Uses Host Triacylglycerol to Accumulate Lipid Droplets and Acquires a Dormancy-Like Phenotype in Lipid-Loaded Macrophages. PLoS Pathog. 2011, 7, e1002093. [Google Scholar] [CrossRef] [Green Version]
- Hung, J.; Scanlon, J.P.; Mahmoud, A.D.; Rodor, J.; Ballantyne, M.; Fontaine, M.A.; Temmerman, L.; Kaczynski, J.; Connor, K.L.; Bhushan, R.; et al. Novel Plaque Enriched Long Noncoding RNA in Atherosclerotic Macrophage Regulation (PELATON). Arterioscler. Thromb. Vasc. Biol. 2020, 40, 697–713. [Google Scholar] [CrossRef]
- Yao, R.-W.; Wang, Y.; Chen, L.-L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Kloc, M.; Li, X.C.; Ghobrial, R.M. RhoA cytoskeletal pathway to transplantation. J. Immunol. Clin. Res. 2014, 2, 1012. [Google Scholar]
- Liu, Y.; Chen, W.; Wu, C.; Minze, L.J.; Kubiak, J.Z.; Li, X.C.; Kloc, M.; Ghobrial, R.M. Macrophage/monocyte-specific deletion of Ras homolog gene family member A (RhoA) downregulates fractalkine receptor and inhibits chronic rejection of mouse cardiac allografts. J. Heart Lung Transplant. 2017, 36, 340–354. [Google Scholar] [CrossRef] [Green Version]
- Mooren, O.L.; Galletta, B.J.; Cooper, J.A. Roles for Actin Assembly in Endocytosis. Annu. Rev. Biochem. 2012, 81, 661–686. [Google Scholar] [CrossRef]
- Porat-Shliom, N.; Milberg, O.; Masedunskas, A.; Weigert, R. Multiple roles for the actin cytoskeleton during regulated exocytosis. Cell. Mol. Life Sci. 2013, 70, 2099–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxfield, F.R.; McGraw, T.E. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 2004, 5, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanley, J.G. Actin-dependent mechanisms in AMPA receptor trafficking. Front. Cell. Neurosci. 2014, 8, 381. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Finnemann, S.C. Regulation of phagocytosis by Rho GTPases. Small GTPases 2015, 6, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Southwick, F.S.; Purich, D.L. Actin-based phagosome motility. Cell Motil. Cytoskelet. 2002, 53, 81–88. [Google Scholar] [CrossRef]
- Merrifield, C.J.; Moss, S.E.; Ballestrem, C.; Imhof, B.A.; Giese, G.; Wunderlich, I.; Almers, W. Endocytic vesicles move at the tips of actin tails in cultured mast cells. Nat. Cell Biol. 1999, 1, 72–74. [Google Scholar] [CrossRef]
- Cai, A.; Zhou, Y.; Li, L. Rho-GTPase and Atherosclerosis: Pleiotropic Effects of Statins. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Nagao, T.; Qin, C.; Grosheva, I.; Maxfield, F.R.; Pierini, L.M. Elevated Cholesterol Levels in the Plasma Membranes of Macrophages Inhibit Migration by Disrupting RhoA Regulation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1596–1602. [Google Scholar] [CrossRef] [Green Version]
- Khan, O.M.; Akula, M.K.; Skålen, K.; Karlsson, C.; Ståhlman, M.; Young, S.G.; Borén, J.; Bergo, M.O. Targeting GGTase-I Activates RHOA, Increases Macrophage Reverse Cholesterol Transport, and Reduces Atherosclerosis in Mice. Circulation 2013, 127, 782–790. [Google Scholar] [CrossRef] [Green Version]
- Khan, O.M.; Ibrahim, M.X.; Jonsson, I.-M.; Karlsson, C.; Liu, M.; Sjogren, A.-K.M.; Olofsson, F.J.; Brisslert, M.; Andersson, S.; Ohlsson, C.; et al. Geranylgeranyltransferase type I (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. J. Clin. Investig. 2011, 121, 628–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkawara, H.; Ishibashi, T.; Shiomi, M.; Sugimoto, K.; Uekita, H.; Kamioka, M.; Takuwa, Y.; Teramoto, T.; Maruyama, Y.; Takeishi, Y. RhoA and Rac1 Changes in the Atherosclerotic Lesions of WHHLMI Rabbits. J. Atheroscler. Thromb. 2009, 16, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liao, J.K. Rho Kinase: An Important Mediator of Atherosclerosis and Vascular Disease. Curr. Pharm. Des. 2009, 15, 3108–3115. [Google Scholar] [CrossRef] [Green Version]
- Shimokawa, H.; Morishige, K.; Miyata, K.; Kandabashi, T.; Eto, Y.; Ikegaki, I.; Asano, T.; Kaibuchi, K.; Takeshita, A. Long-term inhibition of Rho-kinase induces a regression of arteriosclerotic coronary lesions in a porcine model in vivo. Cardiovasc. Res. 2001, 51, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Mallat, Z.; Gojova, A.; Sauzeau, V.; Brun, V.; Silvestre, J.-S.; Esposito, B.; Merval, R.; Groux, H.; Loirand, G.; Tedgui, A. Rho-Associated Protein Kinase Contributes to Early Atherosclerotic Lesion Formation in Mice. Circ. Res. 2003, 93, 884–888. [Google Scholar] [CrossRef] [Green Version]
- Woodside, D.G.; Wooten, D.K.; McIntyre, B.W. Adenosine Diphosphate (ADP)-Ribosylation of the Guanosine Triphosphatase (GTPase) Rho in Resting Peripheral Blood Human T Lymphocytes Results in Pseudopodial Extension and the Inhibition of T Cell Activation. J. Exp. Med. 1998, 188, 1211–1221. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.L.; Chadeuf, G.; Heurtebise-Chrétien, S.; Prieur, X.; Quillard, T.; Goueffic, Y.; Vaillant, N.; Rio, M.; Castan, L.; Durand, M.; et al. Leukocyte RhoA exchange factor Arhgef1 mediates vascular inflammation and atherosclerosis. J. Clin. Investig. 2017, 127, 4516–4526. [Google Scholar] [CrossRef] [Green Version]
- Kloc, M.; Ghobrial, R.M. The multiple sclerosis (MS) drugs as a potential treatment of ARDS in COVID-19 patients. Mult. Scler. Relat. Disord. 2020, 45, 102437. [Google Scholar] [CrossRef]
- Uosef, A.; Vaughn, N.; Chu, X.; Elshawwaf, M.; Abdelshafy, A.A.A.; Elsaid, K.M.K.; Ghobrial, R.; Kloc, M. Siponimod (Mayzent) Downregulates RhoA and Cell Surface Expression of the S1P1 and CX3CR1 Receptors in Mouse RAW 264.7 Macrophages. Arch. Immunol. Ther. Exp. 2020, 68, 19. [Google Scholar] [CrossRef]
- Chen, W.; Ghobrial, R.M.; Li, X.C.; Kloc, M. Inhibition of RhoA and mTORC2/Rictor by Fingolimod (FTY720) induces p21-activated kinase 1, PAK-1 and amplifies podosomes in mouse peritoneal macrophages. Immunobiology 2018, 223, 634–647. [Google Scholar] [CrossRef]
- Chen, W.; Chen, W.; Chen, S.; Uosef, A.; Ghobrial, R.M.; Kloc, M. Fingolimod (FTY720) prevents chronic rejection of rodent cardiac allografts through inhibition of the RhoA pathway. Transpl. Immunol. 2020, 101347. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kloc, M.; Uosef, A.; Kubiak, J.Z.; Ghobrial, R.M. Role of Macrophages and RhoA Pathway in Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 216. https://doi.org/10.3390/ijms22010216
Kloc M, Uosef A, Kubiak JZ, Ghobrial RM. Role of Macrophages and RhoA Pathway in Atherosclerosis. International Journal of Molecular Sciences. 2021; 22(1):216. https://doi.org/10.3390/ijms22010216
Chicago/Turabian StyleKloc, Malgorzata, Ahmed Uosef, Jacek Z. Kubiak, and Rafik Mark Ghobrial. 2021. "Role of Macrophages and RhoA Pathway in Atherosclerosis" International Journal of Molecular Sciences 22, no. 1: 216. https://doi.org/10.3390/ijms22010216
APA StyleKloc, M., Uosef, A., Kubiak, J. Z., & Ghobrial, R. M. (2021). Role of Macrophages and RhoA Pathway in Atherosclerosis. International Journal of Molecular Sciences, 22(1), 216. https://doi.org/10.3390/ijms22010216