Exploring Large Domain Motions in Proteins Using Atomistic Molecular Dynamics with Enhanced Conformational Sampling


Abstract
:1. Introduction
2. Results
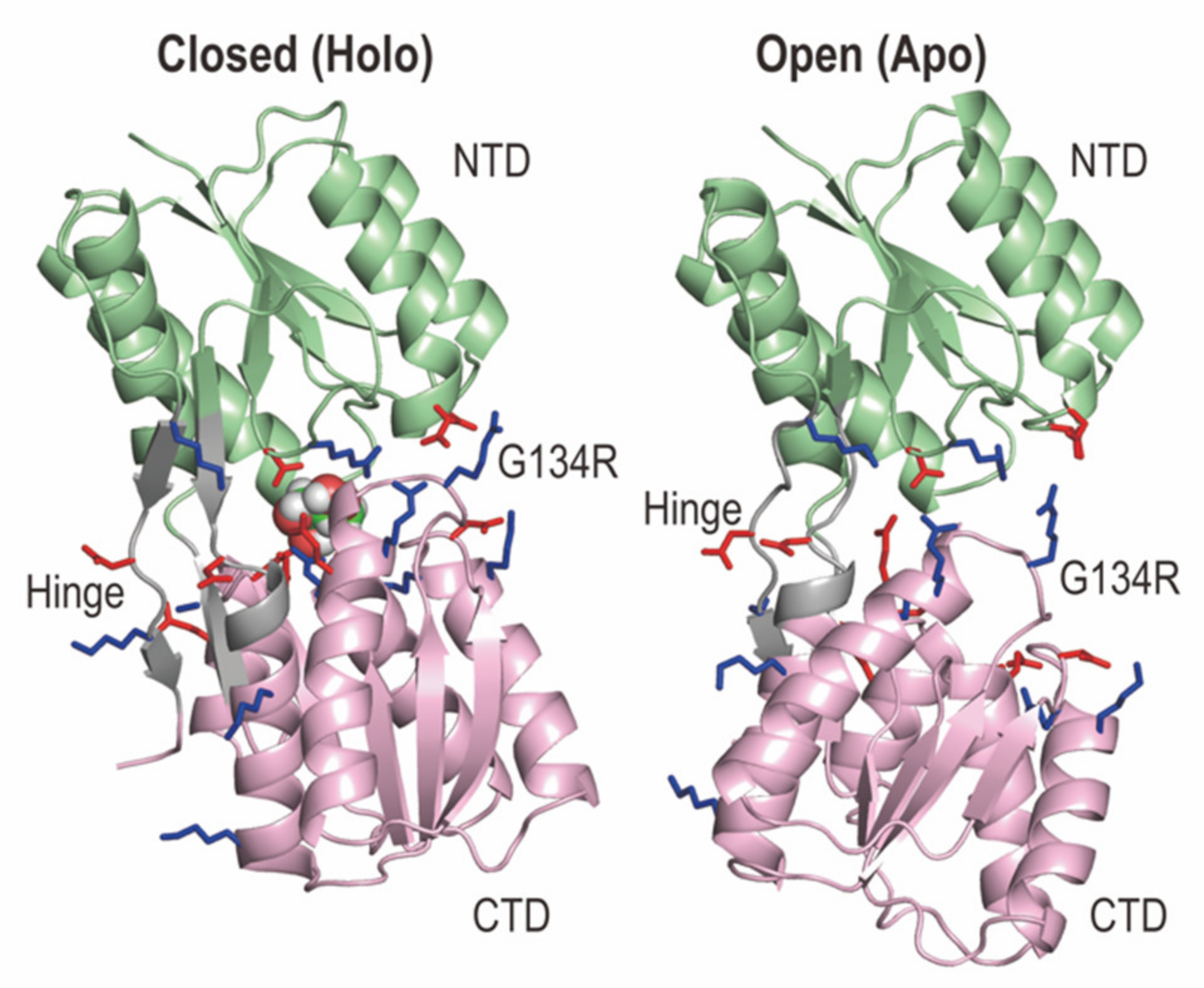
2.1. Structures of RBPG134R in the Apo and Holo States
2.2. gREST_SSCR Simulations of RBPG134R in the Apo and Holo States
2.2.1. How gREST_SSCR Works in RBPG134R Simulations
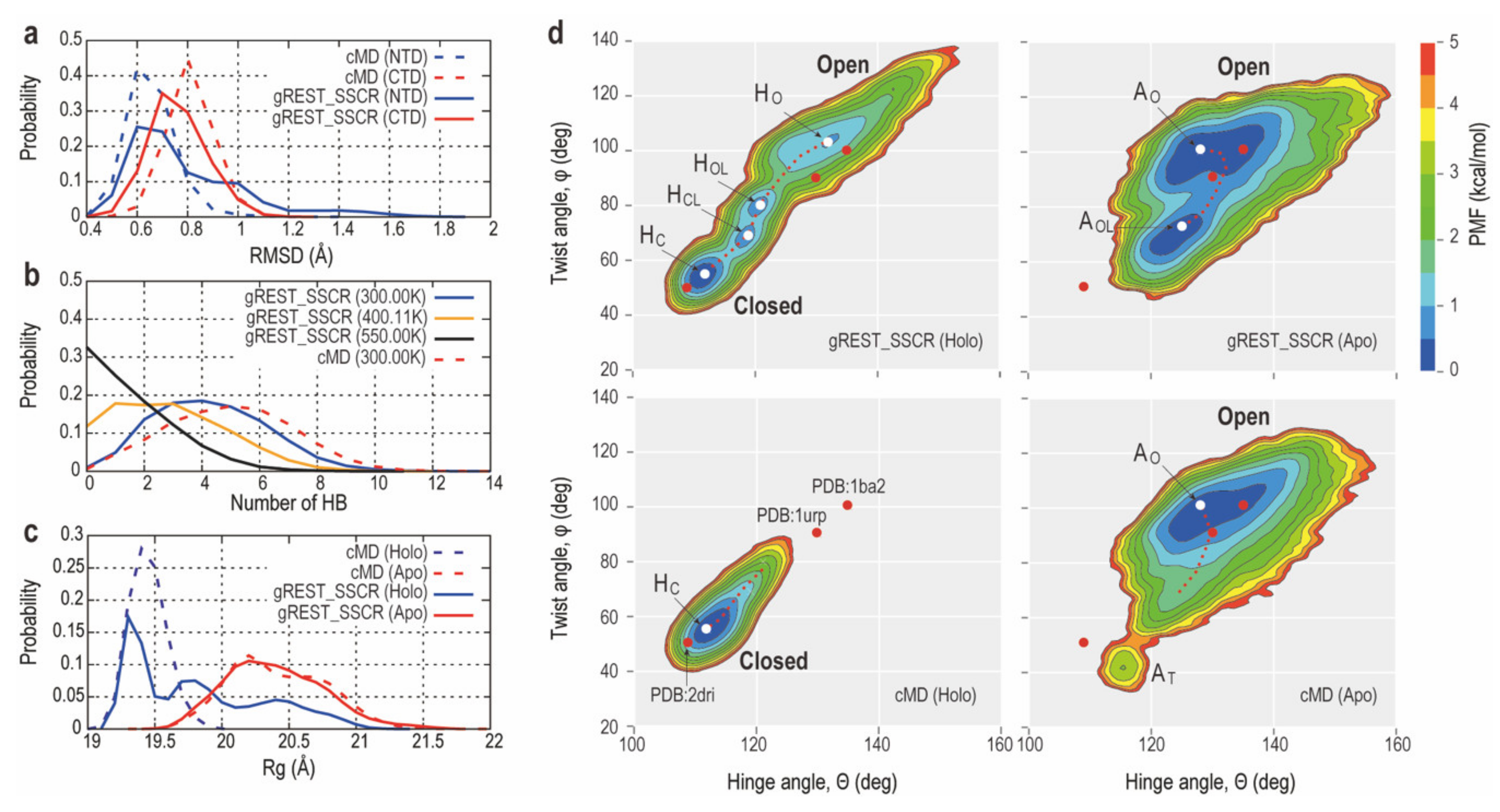
2.2.2. Comparison of Conformational Sampling Abilities between cMD and gREST_SSCR
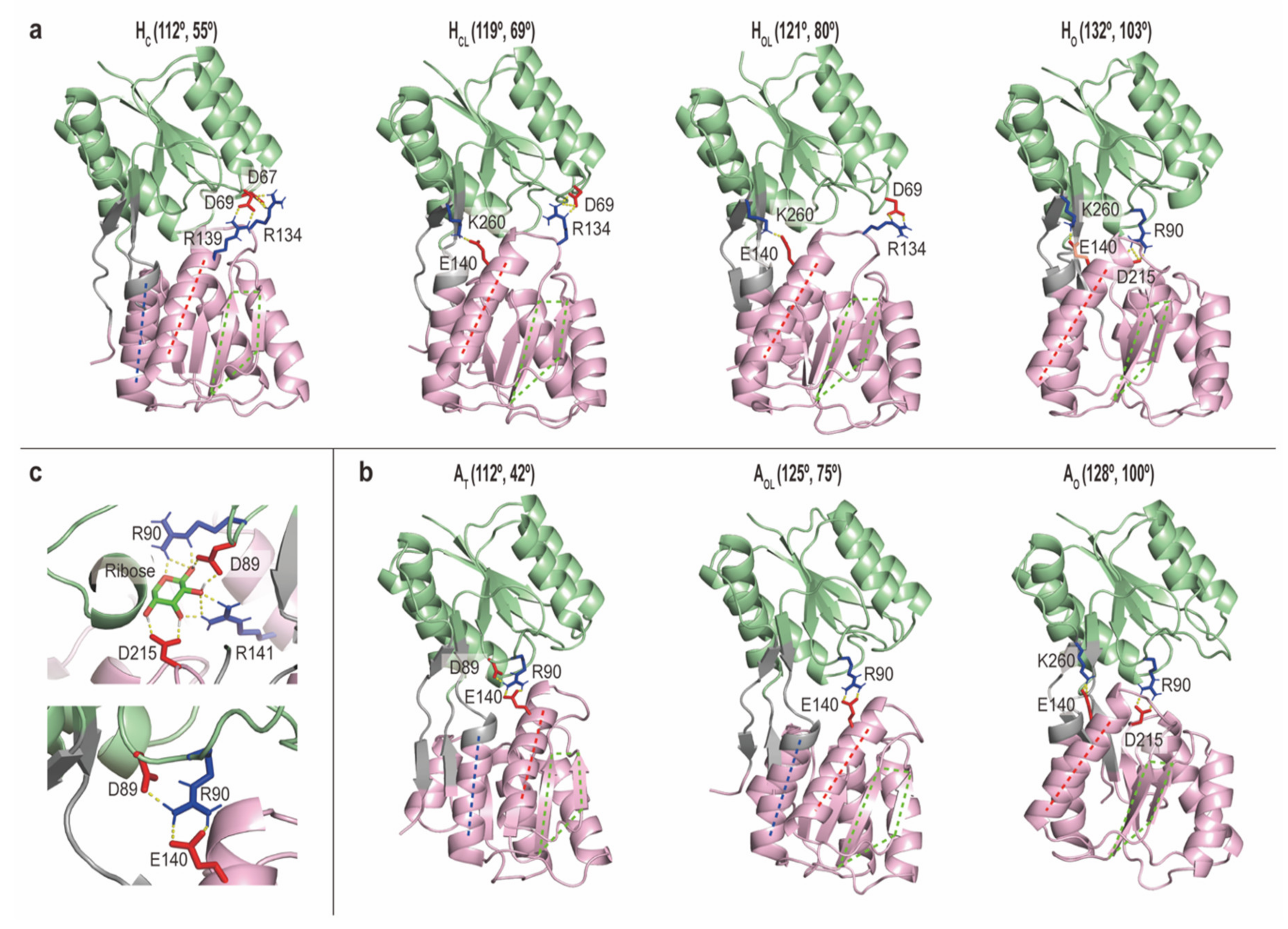
2.2.3. Intermediate Structures of RBPG134R Stabilized by the Inter-Domain Salt-Bridge Interactions
3. Discussion
3.1. How gREST_SSCR Can Enhance Conformational Sampling of Large-Scale Domain Motions of Proteins
3.2. Molecular Mechanisms Underlying Ligand-Induced Conformational Changes of RBP
3.3. General Applications of gREST and gREST_SSCR
4. Materials and Methods
4.1. Modeling of RBPG134R for MD Simulations
4.2. cMD Simulations
4.3. gREST_SSCR Simulations
4.4. Simulation Trajectory Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular dynamics |
| gREST | Generalized replica exchange with solute tempering |
| gREST_SSCR | gREST selected surface charged residues |
| RBP | Ribose binding protein |
| NTD | N-terminal domain |
| CTD | C-terminal domain |
| Rg | Radius of gyration |
| RMSD | Root mean square deviation |
References
- Gerstein, M.; Lesk, A.M.; Chothia, C. Structural Mechanisms for Domain Movements in Proteins. Biochemistry 1994, 33, 6739–6749. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.A.; Duderstadt, K.; Watkins, L.P.; Bhattacharyya, S.; Brokaw, J.; Chu, J.-W.; Yang, H. Illuminating the mechanistic roles of enzyme conformational dynamics. Proc. Natl. Acad. Sci. USA 2007, 104, 18055–18060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, D.; Zuiderweg, E.R. The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 2003, 13, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Bashton, M.; Kerrison, N.D.; Chothia, C.; Teichmann, S.A. Structure, function and evolution of multidomain proteins. Curr. Opin. Struct. Biol. 2004, 14, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, S.L.; Björkman, A. Conformational changes of ribose-binding protein and two related repressors are tailored to fit the functional need. J. Mol. Biol. 1999, 294, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Vishwanath, S.; De Brevern, A.G.; Srinivasan, N. Same but not alike: Structure, flexibility and energetics of domains in multi-domain proteins are influenced by the presence of other domains. PLoS Comput. Biol. 2018, 14, e1006008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michielssens, S.; De Groot, B.L.; Grubmüller, H. Binding Affinities Controlled by Shifting Conformational Equilibria: Opportunities and Limitations. Biophys. J. 2015, 108, 2585–2590. [Google Scholar] [CrossRef] [Green Version]
- Tiefenbrunn, T.; Forli, S.; Baksh, M.M.; Chang, M.W.; Happer, M.; Lin, Y.-C.; Perryman, A.L.; Rhee, J.-K.; Torbett, B.E.; Olson, A.J.; et al. Small Molecule Regulation of Protein Conformation by Binding in the Flap of HIV Protease. ACS Chem. Biol. 2013, 8, 1223–1231. [Google Scholar] [CrossRef]
- Millet, O.; Hudson, R.P.; Kay, L.E. The energetic cost of domain reorientation in maltose-binding protein as studied by NMR and fluorescence spectroscopy. Proc. Natl. Acad. Sci. USA 2003, 100, 12700–12705. [Google Scholar] [CrossRef] [Green Version]
- Kooshapur, H.; Ma, J.; Tjandra, N.; Bermejo, G.A. Nmr analysis of apo glutamine-binding protein exposes challenges in the study of interdomain dynamics. Angew. Chem. Int. Ed. Engl. 2019, 58, 16899–16902. [Google Scholar] [CrossRef]
- Tang, C.; Schwieters, C.D.; Clore, G.M. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature 2007, 449, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Han, W.; Liu, C.; Zang, Y.; Li, J.; Wang, F.; Wang, Y.; Cong, Y. An ensemble of cryo-EM structures of TRiC reveal its conformational landscape and subunit specificity. Proc. Natl. Acad. Sci. USA 2019, 116, 19513–19522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershenson, A.; Gosavi, S.; Faccioli, P.; Wintrode, P.L. Successes and challenges in simulating the folding of large proteins. J. Biol. Chem. 2020, 295, 15–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana, L. Large-Scale Conformational Changes and Protein Function: Breaking the in silico Barrier. Front. Mol. Biosci. 2019, 6, 117. [Google Scholar] [CrossRef] [PubMed]
- Barz, B.; Loschwitz, J.; Strodel, B. Large-scale, dynamin-like motions of the human guanylate binding protein 1 revealed by multi-resolution simulations. PLoS Comput. Biol. 2019, 15, e1007193. [Google Scholar] [CrossRef] [Green Version]
- Sacquin-Mora, S. Motions and mechanics: Investigating conformational transitions in multi-domain proteins with coarse-grain simulations. Mol. Simul. 2013, 40, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Hua, D.P.; Post, C.B. Analysis of Multidomain Protein Dynamics. J. Chem. Theory Comput. 2016, 12, 274–280. [Google Scholar] [CrossRef]
- Wriggers, W.; Schulten, K. Protein domain movements: Detection of rigid domains and visualization of hinges in comparisons of atomic coordinates. Proteins 1997, 29, 1–14. [Google Scholar] [CrossRef]
- Hayward, S.; Kitao, A.; Berendsen, H.J.C. Model-free methods of analyzing domain motions in proteins from simulation: A comparison of normal mode analysis and molecular dynamics simulation of lysozyme. Proteins Struct. Funct. Bioinform. 1997, 27, 425–437. [Google Scholar] [CrossRef]
- Poornam, G.P.; Matsumoto, A.; Ishida, H.; Hayward, S. A method for the analysis of domain movements in large biomolecular complexes. Proteins Struct. Funct. Bioinform. 2009, 76, 201–212. [Google Scholar] [CrossRef]
- Veevers, R.; Hayward, S. Methodological improvements for the analysis of domain movements in large biomolecular complexes. Biophys. Phys. 2018, 16, 328–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, G.; Lee, R.; Hayward, S. A comprehensive and non-redundant database of protein domain movements. Bioinformatics 2005, 21, 2832–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.; Cawley, G.C.; Hayward, S. Classification of Domain Movements in Proteins Using Dynamic Contact Graphs. PLoS ONE 2013, 8, e81224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, R.; Ota, M.; Kidera, A. Hierarchical description and extensive classification of protein structural changes by motion tree. J. Mol. Biol. 2014, 426, 752–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moritsugu, K.; Koike, R.; Yamada, K.; Kato, H.; Kidera, A. Motion Tree Delineates Hierarchical Structure of Protein Dynamics Observed in Molecular Dynamics Simulation. PLoS ONE 2015, 10, e0131583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinobu, A.; Kobayashi, C.; Matsunaga, Y.; Sugita, Y. Building a Macro-mixing Dual-basin Go Model using the Multistate Bennett Acceptance Ratio. Biophys. J. 2019, 16, 310–321. [Google Scholar] [CrossRef] [Green Version]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Sugita, Y.; Kitao, A.; Okamoto, Y. Multidimensional replica-exchange method for free-energy calculations. J. Chem. Phys. 2000, 113, 6042–6051. [Google Scholar] [CrossRef]
- Fukunishi, H.; Watanabe, O.; Takada, S. On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: Application to protein structure prediction. J. Chem. Phys. 2002, 116, 9058–9067. [Google Scholar] [CrossRef]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; McCammon, J.A. Chapter six—Gaussian accelerated molecular dynamics: Theory, implementation, and applications. In Annual Reports in Computational Chemistry; Dixon, D.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 13, pp. 231–278. [Google Scholar]
- Camilloni, C.; Provasi, D.; Tiana, G.; Broglia, R.A. Exploring the protein G helix free-energy surface by solute tempering metadynamics. Proteins Struct. Funct. Bioinform. 2008, 71, 1647–1654. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Friesner, R.A.; Berne, B.J. Replica exchange with solute scaling: A more efficient version of replica exchange with solute tempering (rest2). J. Phys. Chem. B 2011, 115, 9431–9438. [Google Scholar] [CrossRef] [Green Version]
- Moors, S.L.C.; Michielssens, S.; Ceulemans, A. Improved Replica Exchange Method for Native-State Protein Sampling. J. Chem. Theory Comput. 2011, 7, 231–237. [Google Scholar] [CrossRef]
- Terakawa, T.; Kameda, T.; Takada, S. On easy implementation of a variant of the replica exchange with solute tempering in GROMACS. J. Comput. Chem. 2011, 32, 1228–1234. [Google Scholar] [CrossRef]
- Kamiya, M.; Sugita, Y. Flexible selection of the solute region in replica exchange with solute tempering: Application to protein-folding simulations. J. Chem. Phys. 2018, 149, 072304. [Google Scholar] [CrossRef]
- Re, S.; Oshima, H.; Kasahara, K.; Kamiya, M.; Sugita, Y. Encounter complexes and hidden poses of kinase-inhibitor binding on the free-energy landscape. Proc. Natl. Acad. Sci. USA 2019, 116, 18404–18409. [Google Scholar] [CrossRef] [Green Version]
- Niitsu, A.; Re, S.; Oshima, H.; Kamiya, M.; Sugita, Y. De Novo Prediction of Binders and Nonbinders for T4 Lysozyme by gREST Simulations. J. Chem. Inf. Model. 2019, 59, 3879–3888. [Google Scholar] [CrossRef]
- Matsuoka, D.; Kamiya, M.; Sato, T.; Sugita, Y. Role of the N-Terminal Transmembrane Helix Contacts in the Activation of FGFR3. J. Comput. Chem. 2020, 41, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Tam, R.; Saier, M.H. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 1993, 57, 320–346. [Google Scholar] [CrossRef] [PubMed]
- Björkman, A.J.; Binnie, R.A.; Zhang, H.; Cole, L.B.; Hermodson, M.A.; Mowbray, S.L. Probing protein-protein interactions. The ribose-binding protein in bacterial transport and chemotaxis. J. Biol. Chem. 1994, 269, 30206–30211. [Google Scholar] [PubMed]
- Unione, L.; Ortega, G.; Mallagaray, A.; Corzana, F.; Pérez-Castells, J.; Canales, A.; Jimenez-Barbero, J.; Millet, O. Unraveling the Conformational Landscape of Ligand Binding to Glucose/Galactose-Binding Protein by Paramagnetic NMR and MD Simulations. ACS Chem. Biol. 2016, 11, 2149–2157. [Google Scholar] [CrossRef]
- Loeffler, H.H.; Kitao, A. Collective Dynamics of Periplasmic Glutamine Binding Protein upon Domain Closure. Biophys. J. 2009, 97, 2541–2549. [Google Scholar] [CrossRef] [Green Version]
- Björkman, A.; Mowbray, S.L. Multiple open forms of ribose-binding protein trace the path of its conformational change. J. Mol. Biol. 1998, 279, 651–664. [Google Scholar] [CrossRef]
- Cuneo, M.J.; Beese, L.S.; Hellinga, H.W. Ligand-induced conformational changes in a thermophilic ribose-binding protein. BMC Struct. Biol. 2008, 8, 50. [Google Scholar] [CrossRef] [Green Version]
- Ravindranathan, K.P.; Gallicchio, E.; Levy, R.M. Conformational Equilibria and Free Energy Profiles for the Allosteric Transition of the Ribose-binding Protein. J. Mol. Biol. 2005, 353, 196–210. [Google Scholar] [CrossRef]
- Borrok, M.J.; Kiessling, L.L.; Forest, K.T. Conformational changes of glucose/galactose-binding protein illuminated by open, unliganded, and ultra-high-resolution ligand-bound structures. Protein Sci. 2007, 16, 1032–1041. [Google Scholar] [CrossRef]
- Sun, Y.-J.; Rose, J.; Wang, B.-C.; Hsiao, C.-D. The structure of glutamine-binding protein complexed with glutamine at 1.94 Å resolution: Comparisons with other amino acid binding proteins. J. Mol. Biol. 1998, 278, 219–229. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, L.; Wu, S.; Liu, Z.; Gao, X.; Zhang, X.; Liu, M.; Liu, J.; Huang, X.; Wang, W. Conformational dynamics of apo-glnbp revealed by experimental and computational analysis. Angew. Chem. Int. Ed. Engl. 2016, 55, 13990–13994. [Google Scholar] [CrossRef] [PubMed]
- Bucher, D.; Grant, B.J.; McCammon, J.A. Induced Fit or Conformational Selection? The Role of the Semi-closed State in the Maltose Binding Protein. Biochemistry 2011, 50, 10530–10539. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Palotai, R.; Nussinov, R. Induced fit, conformational selection and independent dynamic segments: An extended view of binding events. Trends Biochem. Sci. 2010, 35, 539–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. Charmm-gui input generator for namd, gromacs, amber, openmm, and charmm/openmm simulations using the charmm36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Mori, T.; Kobayashi, C.; Matsunaga, Y.; Yoda, T.; Feig, M.; Sugita, Y. GENESIS: A hybrid-parallel and multi-scale molecular dynamics simulator with enhanced sampling algorithms for biomolecular and cellular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, C.; Jung, J.; Matsunaga, Y.; Mori, T.; Ando, T.; Tamura, K.; Kamiya, M.; Sugita, Y. GENESIS 1.1: A hybrid-parallel molecular dynamics simulator with enhanced sampling algorithms on multiple computational platforms. J. Comput. Chem. 2017, 38, 2193–2206. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, S.R.B.L.D.G.H.; MacKerell, J.A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Guvench, O.; Mallajosyula, S.S.; Raman, E.P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T.J.; Jamison, F.W., 2nd; Mackerell, A.D., Jr. Charmm additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Berendsen, H.J.C. A Leap-frog Algorithm for Stochastic Dynamics. Mol. Simul. 2007, 1, 173–185. [Google Scholar] [CrossRef]
- Quigley, D.; Probert, M.I.J. Langevin dynamics in constant pressure extended systems. J. Chem. Phys. 2004, 120, 11432–11441. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Zykova-Timan, T.; Parrinello, M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys. 2009, 130, 074101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.; Kobayashi, C.; Sugita, Y. Kinetic energy definition in velocity Verlet integration for accurate pressure evaluation. J. Chem. Phys. 2018, 148, 164109. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 1.4; Schrödinger, LLC: New York, NY, USA, 2011.




{kind=link}
{kind=link}
{kind=link}
| Method (State) | gREST_SSCR (Holo) | gREST_SSCR (Apo) | cMD (Holo) | cMD (Apo) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Residue (domain) | Residue (domain) | HC | HCL | HOL | HO | AOL | AO | All | HC | All | AT |
| Asp67 * (NTD) | Arg134 * (CTD) | 82.8 ± 1.4 | 5.0 ± 0.6 | 0 | 0 | 0 | 0 | 47.7 ± 3.8 | 79.8 ± 1.1 | <0.1 | 0 |
| Asp69 * (NTD) | Arg134 * (CTD) | 19.4 ± 1.3 | 78.0 ± 2.4 | 30.2 ± 4.5 | <0.4 | <0.1 | <0.1 | 19.0 ± 1.8 | 16.0 ± 0.8 | <0.1 | 0 |
| Asp69 (NTD) | Arg139 (CTD) | 23.5 ± 1.0 | <0.5 | 0 | 0 | 0.1 | 0 | 18.0 ± 2.5 | 39.7 ± 2.4 | 0 | <0.1 |
| Arg90 (NTD) | Glu140 (CTD) | 0 | 0 | 0 | 0 | 86.8 ± 0.9 | <1.1 | 11.8 ± 3.0 | 0 | 1.8 ± 0.3 | 74.5 |
| Arg90 (NTD) | Asp215 (CTD) | 0 | 0 | <0.5 | 34.8 ± 4.2 | 0 | 37.2 ± 3.9 | 0 | 0 | 30.9 ± 2.3 | 0 |
| Glu140 (CTD) | Lys260 (Hinge) | 3.9 ± 0.3 | 44.5 ± 3.8 | 80.4 ± 1.2 | 79.2 ± 1.4 | <0.7 | 59.0 ± 3.5 | 6.7 ± 1.3 | 3.6 ± 0.1 | 62.4 ± 1.1 | 0 |
| Glu221 (CTD) | Lys266 (Hinge) | 24.0 ± 1.2 | 26.9 ± 1.1 | <0.2 | 0 | <0.6 | <0.3 | 19.4 ± 1.8 | 31.2 ± 0.8 | <0.3 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dokainish, H.M.; Sugita, Y. Exploring Large Domain Motions in Proteins Using Atomistic Molecular Dynamics with Enhanced Conformational Sampling. Int. J. Mol. Sci. 2021, 22, 270. https://doi.org/10.3390/ijms22010270
Dokainish HM, Sugita Y. Exploring Large Domain Motions in Proteins Using Atomistic Molecular Dynamics with Enhanced Conformational Sampling. International Journal of Molecular Sciences. 2021; 22(1):270. https://doi.org/10.3390/ijms22010270
Chicago/Turabian StyleDokainish, Hisham M., and Yuji Sugita. 2021. "Exploring Large Domain Motions in Proteins Using Atomistic Molecular Dynamics with Enhanced Conformational Sampling" International Journal of Molecular Sciences 22, no. 1: 270. https://doi.org/10.3390/ijms22010270
APA StyleDokainish, H. M., & Sugita, Y. (2021). Exploring Large Domain Motions in Proteins Using Atomistic Molecular Dynamics with Enhanced Conformational Sampling. International Journal of Molecular Sciences, 22(1), 270. https://doi.org/10.3390/ijms22010270