Astroglial Connexin43 as a Potential Target for a Mood Stabiliser


Abstract
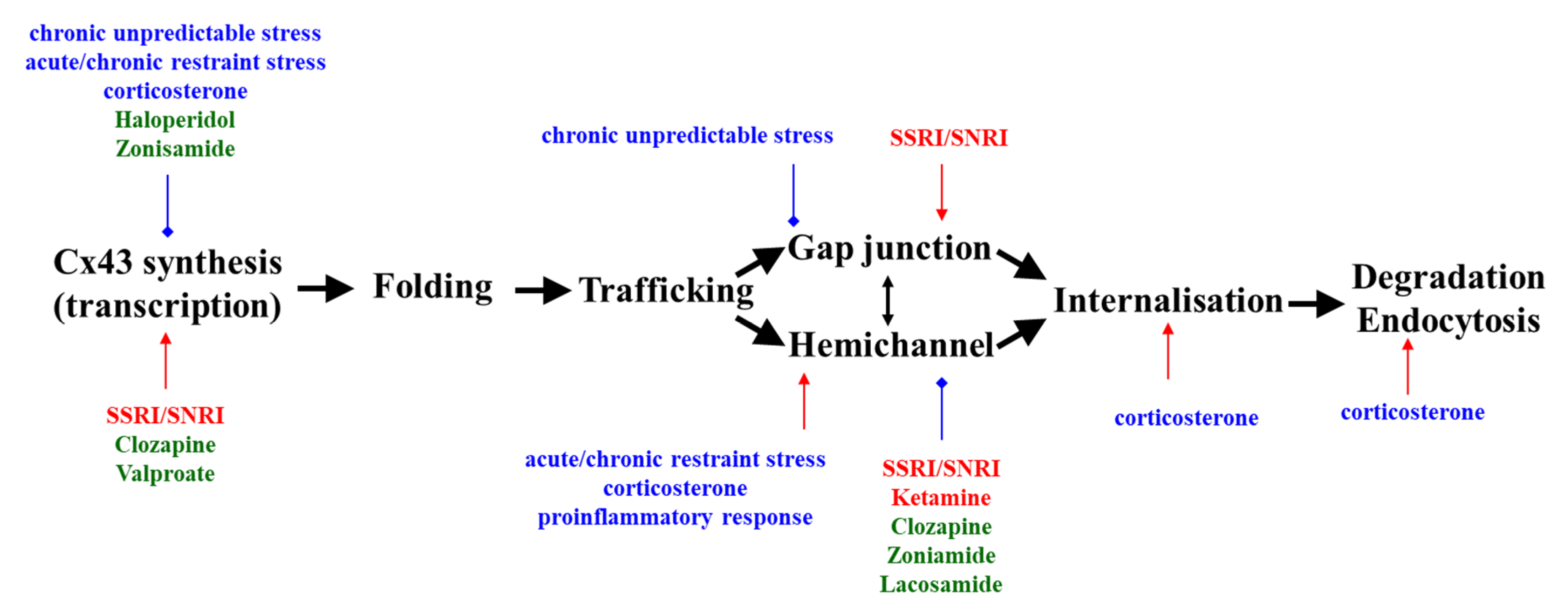
:1. Introduction
2. Abnormalities of Cx43 in Depression
3. Cx43 and Behaviour
4. Effects of Monoamine Transporter-Inhibiting Antidepressants on Cx43
5. Effects of Antipsychotics and Ketamine on Cx43
6. Effects of Anticonvulsants on Cx43

{kind=link}
| Agent | Model (Region) | Treatment (Dose, Duration) | Effect (Hemichannel) | Reference |
|---|---|---|---|---|
| Carbamazepine | rat cortical astrocyte | in vitro (40–400 µM for 24 h) | no effect (protein) | [110] |
| rat cortical astrocyte | in vitro (100 µM for 7 days) | no effect (protein) (no effect) | [2] | |
| Lacosamide | rat cortical astrocyte | in vitro (30–100 µM for 7 days) | no effect (protein) (inhibition) | [2] |
| Zonisamide | rat cortical astrocyte | in vitro (30 µM for 7 days) | decrease (protein) (inhibition) | [2] |
| Valproate | Rat (frontal) | in vivo (300 mg/kg for 21 days) | no effect (protein) | [76] |
| rat cortical astrocyte | in vitro (350–1400 µM for 24 h) | no effect (protein) | [110] | |
| rat cortical astrocyte | in vitro (1000–3000 µM for 7 days) | increase (protein) (activation) | [3] | |
| Gabapentin | rat cortical astrocyte | in vitro (60–600 µM for 24 h) | no effect (protein) | [110] |
| Phenytoin | rat cortical astrocyte | in vitro (40–400 µM for 24 h) | no effect (protein) | [110] |
| Diazepam | rat cortical astrocyte | in vitro (25 µM for 48 h) | no effect (protein) | [77] |
7. Candidate Pathophysiology of Mood Disorders associated with Cx43
7.1. Candidate Pathophysiology of Major Depression Associated with Cx43
7.2. Candidate Pathophysiology of Other Mood Disorders Associated with Cx43
7.3. Potential of Cx43 as a Target for Mood Stabilisers
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CBX | Carbenoxolone |
| Cx43 | Connexin43 |
| IL 1β | Interleukin 1β |
| NRI | Norepinephrine reuptake inhibitor |
| PKB | Protein kinase B |
| SNRI | Serotonin norepinephrine reuptake inhibitor |
| SSRI | Selective serotonin reuptake inhibitor |
| TNF α | Tumour necrosis factor α |
| VDSC | Voltage-dependent sodium channel |
References
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Murata, M. A Working Hypothesis Regarding Identical Pathomechanisms between Clinical Efficacy and Adverse Reaction of Clozapine via the Activation of Connexin43. Int. J. Mol. Sci. 2020, 21, 7019. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Ueda, Y.; Okada, M. Effects of Carbamazepine, Lacosamide and Zonisamide on Gliotransmitter Release Associated with Activated Astroglial Hemichannels. Pharmaceuticals 2020, 13, 117. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Okubo, R.; Murata, M.; Shiroyama, T.; Okada, M. Activation of Astroglial Connexin is Involved in Concentration-Dependent Double-Edged Sword Clinical Action of Clozapine. Cells 2020, 9, 414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuyama, K.; Fukuzawa, M.; Ruri, O.; Okada, M. Upregulated Connexin 43 Induced by Loss-of-Functional S284L-Mutant alpha4 Subunit of Nicotinic ACh Receptor Contributes to Pathomechanisms of Autosomal Dominant Sleep-Related Hypermotor Epilepsy. Pharmaceuticals 2020, 13, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuyama, K.; Fukuzawa, M.; Okada, M. Upregulated and hyperactivated thalamic connexin 43 plays important roles in pathomechanisms of cognitive impairment and seizure of autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant α4 subunit of nicotinic ACh receptor. Pharmaceuticals 2020, 13, 99. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Carbamazepine Attenuates Astroglial L-Glutamate Release Induced by Pro-Inflammatory Cytokines via Chronically Activation of Adenosine A2A Receptor. Int. J. Mol. Sci. 2019, 20, 3727. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Ueda, Y. Memantine protects thalamocortical hyper-glutamatergic transmission induced by NMDA receptor antagonism via activation of system xc−. Pharm. Res. Perspect. 2019, 7, e00457. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Kato, R.; Murata, M.; Shiroyama, T.; Okada, M. Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules 2019, 9, 234. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Okada, M. Effects of levetiracetam on astroglial release of kynurenine-pathway metabolites. Br. J. Pharm. 2018, 175, 4253–4265. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Tanahashi, S.; Hoshikawa, M.; Shinagawa, R.; Okada, M. Zonisamide regulates basal ganglia transmission via astroglial kynurenine pathway. Neuropharmacology 2014, 76 Pt A, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Hoshikawa, M.; Dai, K.; Saito, H.; Suzuki, N.; Niwa, O.; Okada, M. ONO-2506 inhibits spike-wave discharges in a genetic animal model without affecting traditional convulsive tests via gliotransmission regulation. Br. J. Pharm. 2013, 168, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial D-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharm. 2012, 165, 1543–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesseveur, G.; Gardier, A.M.; Guiard, B.P. The monoaminergic tripartite synapse: A putative target for currently available antidepressant drugs. Current Drug Targets 2013, 14, 1277–1294. [Google Scholar] [CrossRef] [PubMed]
- Czeh, B.; Fuchs, E.; Wiborg, O.; Simon, M. Animal models of major depression and their clinical implications. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 64, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G.; Miguel-Hidalgo, J.J.; Wei, J.; Dilley, G.; Pittman, S.D.; Meltzer, H.Y.; Overholser, J.C.; Roth, B.L.; Stockmeier, C.A. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 1999, 45, 1085–1098. [Google Scholar] [CrossRef]
- Ongur, D.; Drevets, W.C.; Price, J.L. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. USA 1998, 95, 13290–13295. [Google Scholar] [CrossRef] [Green Version]
- Cotter, D.; Mackay, D.; Chana, G.; Beasley, C.; Landau, S.; Everall, I.P. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb. Cortex 2002, 12, 386–394. [Google Scholar] [CrossRef]
- Bowley, M.P.; Drevets, W.C.; Ongur, D.; Price, J.L. Low glial numbers in the amygdala in major depressive disorder. Biol. Psychiatry 2002, 52, 404–412. [Google Scholar] [CrossRef]
- Chana, G.; Landau, S.; Beasley, C.; Everall, I.P.; Cotter, D. Two-dimensional assessment of cytoarchitecture in the anterior cingulate cortex in major depressive disorder, bipolar disorder, and schizophrenia: Evidence for decreased neuronal somal size and increased neuronal density. Biol. Psychiatry 2003, 53, 1086–1098. [Google Scholar] [CrossRef]
- Maes, M.; Yirmyia, R.; Noraberg, J.; Brene, S.; Hibbeln, J.; Perini, G.; Kubera, M.; Bob, P.; Lerer, B.; Maj, M. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: Leads for future research and new drug developments in depression. Metab. Brain Dis. 2009, 24, 27–53. [Google Scholar] [PubMed]
- Willner, P.; Scheel-Kruger, J.; Belzung, C. The neurobiology of depression and antidepressant action. Neurosci. Biobehav. Rev. 2013, 37, 2331–2371. [Google Scholar] [CrossRef] [PubMed]
- Mulders, P.C.; van Eijndhoven, P.F.; Schene, A.H.; Beckmann, C.F.; Tendolkar, I. Resting-state functional connectivity in major depressive disorder: A review. Neurosci. Biobehav. Rev. 2015, 56, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G.; Selemon, L.D.; Goldman-Rakic, P.S. Neuronal and glial somal size in the prefrontal cortex: A postmortem morphometric study of schizophrenia and Huntington disease. Arch. Gen. Psychiatry 1998, 55, 215–224. [Google Scholar] [CrossRef]
- Selemon, L.D.; Rajkowska, G.; Goldman-Rakic, P.S. Elevated neuronal density in prefrontal area 46 in brains from schizophrenic patients: Application of a three-dimensional, stereologic counting method. J. Comp. Neurol. 1998, 392, 402–412. [Google Scholar] [CrossRef]
- Czeh, B.; Simon, M.; Schmelting, B.; Hiemke, C.; Fuchs, E. Astroglial plasticity in the hippocampus is affected by chronic psychosocial stress and concomitant fluoxetine treatment. Neuropsychopharmacology 2006, 31, 1616–1626. [Google Scholar] [CrossRef] [Green Version]
- Banasr, M.; Chowdhury, G.M.; Terwilliger, R.; Newton, S.S.; Duman, R.S.; Behar, K.L.; Sanacora, G. Glial pathology in an animal model of depression: Reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol. Psychiatry 2010, 15, 501–511. [Google Scholar] [CrossRef]
- Araya-Callis, C.; Hiemke, C.; Abumaria, N.; Flugge, G. Chronic psychosocial stress and citalopram modulate the expression of the glial proteins GFAP and NDRG2 in the hippocampus. Psychopharmacology 2012, 224, 209–222. [Google Scholar] [CrossRef] [Green Version]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Caceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef] [Green Version]
- Hertz, L.; Xu, J.; Song, D.; Du, T.; Li, B.; Yan, E.; Peng, L. Astrocytic glycogenolysis: Mechanisms and functions. Metab. Brain Dis. 2015, 30, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G. Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol. Psychiatry 2000, 48, 766–777. [Google Scholar] [CrossRef]
- Rajkowska, G.; Halaris, A.; Selemon, L.D. Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol. Psychiatry 2001, 49, 741–752. [Google Scholar] [CrossRef]
- Uranova, N.A.; Vostrikov, V.M.; Orlovskaya, D.D.; Rachmanova, V.I. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: A study from the Stanley Neuropathology Consortium. Schizophr. Res. 2004, 67, 269–275. [Google Scholar] [CrossRef]
- Brauch, R.A.; Adnan El-Masri, M.; Parker, J.C., Jr.; El-Mallakh, R.S. Glial cell number and neuron/glial cell ratios in postmortem brains of bipolar individuals. J. Affect. Disord. 2006, 91, 87–90. [Google Scholar] [CrossRef]
- Butt, A.M.; Kalsi, A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: A special role for Kir4.1 in glial functions. J. Cell Mol. Med. 2006, 10, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Kofuji, P.; Newman, E.A. Potassium buffering in the central nervous system. Neuroscience 2004, 129, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Suzumura, A. Gap junctions and hemichannels composed of connexins: Potential therapeutic targets for neurodegenerative diseases. Front. Cell. Neurosci. 2014, 8, 189. [Google Scholar] [CrossRef] [Green Version]
- Lapato, A.S.; Tiwari-Woodruff, S.K. Connexins and pannexins: At the junction of neuro-glial homeostasis & disease. J. Neurosci. Res. 2018, 96, 31–44. [Google Scholar]
- Li, Q.; Li, Q.Q.; Jia, J.N.; Liu, Z.Q.; Zhou, H.H.; Mao, X.Y. Targeting gap junction in epilepsy: Perspectives and challenges. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 109, 57–65. [Google Scholar] [CrossRef]
- Loewenstein, W.R. Junctional intercellular communication: The cell-to-cell membrane channel. Physiol. Rev. 1981, 61, 829–913. [Google Scholar] [CrossRef]
- Chever, O.; Lee, C.Y.; Rouach, N. Astroglial connexin43 hemichannels tune basal excitatory synaptic transmission. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 11228–11232. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T.; Morel, S.; Kwak, B.R.; Girao, H. Role of connexin 43 in different forms of intercellular communication—Gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630. [Google Scholar] [CrossRef] [Green Version]
- Naus, C.C.; Laird, D.W. Implications and challenges of connexin connections to cancer. Nat. Rev. Cancer 2010, 10, 435–441. [Google Scholar] [CrossRef]
- Wang, F.; Qi, X.; Zhang, J.; Huang, J.H. Astrocytic modulation of potassium under seizures. Neural Regen Res. 2020, 15, 980–987. [Google Scholar]
- Fukuyama, K.; Okada, M. Age-dependent and sleep/seizure-induced pathomechanisms of autosomal dominant sleep-related hypermotor epilepsy. Int. J. Mol. Sci. 2020, 21, 8142. [Google Scholar] [CrossRef]
- Murphy, S.; Pearce, B. Eicosanoids in the CNS: Sources and effects. Prostaglandins Leukot Essent Fatty Acids 1988, 31, 165–170. [Google Scholar]
- Portal, B.; Delcourte, S.; Rovera, R.; Lejards, C.; Bullich, S.; Malnou, C.E.; Haddjeri, N.; Deglon, N.; Guiard, B.P. Genetic and pharmacological inactivation of astroglial connexin 43 differentially influences the acute response of antidepressant and anxiolytic drugs. Acta Physiol. 2020, 229, e13440. [Google Scholar] [CrossRef]
- Bernard, R.; Kerman, I.A.; Thompson, R.C.; Jones, E.G.; Bunney, W.E.; Barchas, J.D.; Schatzberg, A.F.; Myers, R.M.; Akil, H.; Watson, S.J. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol. Psychiatry 2011, 16, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Ernst, C.; Nagy, C.; Kim, S.; Yang, J.P.; Deng, X.; Hellstrom, I.C.; Choi, K.H.; Gershenfeld, H.; Meaney, M.J.; Turecki, G. Dysfunction of astrocyte connexins 30 and 43 in dorsal lateral prefrontal cortex of suicide completers. Biol. Psychiatry 2011, 70, 312–319. [Google Scholar] [CrossRef]
- Nagy, C.; Torres-Platas, S.G.; Mechawar, N.; Turecki, G. Repression of Astrocytic Connexins in Cortical and Subcortical Brain Regions and Prefrontal Enrichment of H3K9me3 in Depression and Suicide. Int. J. Neuropsychopharmacol. 2017, 20, 50–57. [Google Scholar] [CrossRef] [Green Version]
- Miguel-Hidalgo, J.J.; Wilson, B.A.; Hussain, S.; Meshram, A.; Rajkowska, G.; Stockmeier, C.A. Reduced connexin 43 immunolabeling in the orbitofrontal cortex in alcohol dependence and depression. J. Psychiatr. Res. 2014, 55, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Kawano, Y.; Fukuyama, K.; Motomura, E.; Shiroyama, T. Candidate Strategies for Development of a Rapid-Acting Antidepressant Class That Does Not Result in Neuropsychiatric Adverse Effects: Prevention of Ketamine-Induced Neuropsychiatric Adverse Reactions. Int. J. Mol. Sci. 2020, 21, 7951. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathogenesis and pathophysiology of autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant alpha4 subunit of nicotinic ACh receptor. Br. J. Pharm. 2020, 177, 2143–2162. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Lurasidone inhibits NMDA antagonist-induced functional abnormality of thalamocortical glutamatergic transmission via 5-HT7 receptor blockade. Br. J. Pharm. 2019, 176, 4002–4018. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Okubo, R.; Shiroyama, T.; Ueda, Y. Lurasidone Sub-Chronically Activates Serotonergic Transmission via Desensitization of 5-HT1A and 5-HT7 Receptors in Dorsal Raphe Nucleus. Pharmaceuticals 2019, 12, 149. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Suzuki, D.; Ueda, Y. Effects of acute and sub-chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology 2019, 156, 107547. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Nakano, T.; Ueda, Y. Pharmacological Discrimination of Effects of MK801 on Thalamocortical, Mesothalamic, and Mesocortical Transmissions. Biomolecules 2019, 9, 746. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.D.; Liu, Y.; Yuan, Y.H.; Li, J.; Chen, N.H. Gap junction dysfunction in the prefrontal cortex induces depressive-like behaviors in rats. Neuropsychopharmacology 2012, 37, 1305–1320. [Google Scholar] [CrossRef]
- Miguel-Hidalgo, J.J.; Moulana, M.; Deloach, P.H.; Rajkowska, G. Chronic Unpredictable Stress Reduces Immunostaining for Connexins 43 and 30 and Myelin Basic Protein in the Rat Prelimbic and Orbitofrontal Cortices. Chronic Stress 2018, 2, 2470547018814186. [Google Scholar] [CrossRef]
- Jin, C.; Wang, Z.Z.; Zhou, H.; Lou, Y.X.; Chen, J.; Zuo, W.; Tian, M.T.; Wang, Z.Q.; Du, G.H.; Kawahata, I.; et al. Ginsenoside Rg1-induced antidepressant effects involve the protection of astrocyte gap junctions within the prefrontal cortex. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 75, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.X.; Wang, Z.Z.; Xia, C.Y.; Mou, Z.; Ren, Q.; Liu, D.D.; Zhang, X.; Chen, N.H. The protective effect of ginsenoside Rg1 on depression may benefit from the gap junction function in hippocampal astrocytes. Eur. J. Pharmacol. 2020, 882, 173309. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Moraga-Amaro, R.; Diaz-Galarce, R.; Rojas, S.; Maturana, C.J.; Stehberg, J.; Saez, J.C. Restraint stress increases hemichannel activity in hippocampal glial cells and neurons. Front. Cell. Neurosci. 2015, 9, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesseveur, G.; Portal, B.; Basile, J.A.; Ezan, P.; Mathou, A.; Halley, H.; Leloup, C.; Fioramonti, X.; Deglon, N.; Giaume, C.; et al. Attenuated Levels of Hippocampal Connexin 43 and its Phosphorylation Correlate with Antidepressant- and Anxiolytic-Like Activities in Mice. Front. Cell. Neurosci. 2015, 9, 490. [Google Scholar] [CrossRef] [Green Version]
- Miguel-Hidalgo, J.J.; Carter, K.; Deloach, P.H.; Sanders, L.; Pang, Y. Glucocorticoid-Induced Reductions of Myelination and Connexin 43 in Mixed Central Nervous System Cell Cultures Are Prevented by Mifepristone. Neuroscience 2019, 411, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Zhang, X.; Zhang, Z.; Mitsui, T.; Kamiyama, M.; Takeda, M.; Yao, J. Connexin43 hemichannels contributes to the disassembly of cell junctions through modulation of intracellular oxidative status. Redox Biol. 2016, 9, 198–209. [Google Scholar] [CrossRef] [Green Version]
- Hurtubise, J.L.; Howland, J.G. Effects of stress on behavioral flexibility in rodents. Neuroscience 2017, 345, 176–192. [Google Scholar] [CrossRef]
- Xia, C.Y.; Wang, Z.Z.; Zhang, Z.; Chen, J.; Wang, Y.Y.; Lou, Y.X.; Gao, Y.; Luo, P.; Ren, Q.; Du, G.H.; et al. Corticosterone impairs gap junctions in the prefrontal cortical and hippocampal astrocytes via different mechanisms. Neuropharmacology 2018, 131, 20–30. [Google Scholar] [CrossRef]
- Xia, C.Y.; Chu, S.F.; Zhang, S.; Gao, Y.; Ren, Q.; Lou, Y.X.; Luo, P.; Tian, M.T.; Wang, Z.Q.; Du, G.H.; et al. Ginsenoside Rg1 alleviates corticosterone-induced dysfunction of gap junctions in astrocytes. J. Ethnopharmacol. 2017, 208, 207–213. [Google Scholar] [CrossRef]
- Lampe, P.D.; TenBroek, E.M.; Burt, J.M.; Kurata, W.E.; Johnson, R.G.; Lau, A.F. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 2000, 149, 1503–1512. [Google Scholar] [CrossRef]
- Cone, A.C.; Cavin, G.; Ambrosi, C.; Hakozaki, H.; Wu-Zhang, A.X.; Kunkel, M.T.; Newton, A.C.; Sosinsky, G.E. Protein kinase Cdelta-mediated phosphorylation of Connexin43 gap junction channels causes movement within gap junctions followed by vesicle internalization and protein degradation. J. Biol. Chem. 2014, 289, 8781–8798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, C.; Suderman, M.; Yang, J.; Szyf, M.; Mechawar, N.; Ernst, C.; Turecki, G. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol. Psychiatry 2015, 20, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, T.J.; Kloth, A.D.; Hsueh, B.; Runkle, M.B.; Kane, G.A.; Wang, S.S.; Gould, E. Gap junctions in the ventral hippocampal-medial prefrontal pathway are involved in anxiety regulation. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 15679–15688. [Google Scholar] [CrossRef] [Green Version]
- Eiland, L.; Romeo, R.D. Stress and the developing adolescent brain. Neuroscience 2013, 249, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Jeanson, T.; Pondaven, A.; Ezan, P.; Mouthon, F.; Charveriat, M.; Giaume, C. Antidepressants Impact Connexin 43 Channel Functions in Astrocytes. Front. Cell. Neurosci. 2015, 9, 495. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, S.H.; Folsom, T.D.; Reutiman, T.J.; Pandian, T.; Braun, N.N.; Haug, K. Chronic psychotropic drug treatment causes differential expression of connexin 43 and GFAP in frontal cortex of rats. Schizophr. Res. 2008, 104, 127–134. [Google Scholar] [CrossRef]
- Morioka, N.; Suekama, K.; Zhang, F.F.; Kajitani, N.; Hisaoka-Nakashima, K.; Takebayashi, M.; Nakata, Y. Amitriptyline up-regulates connexin43-gap junction in rat cultured cortical astrocytes via activation of the p38 and c-Fos/AP-1 signalling pathway. Br. J. Pharmacol. 2014, 171, 2854–2867. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.V.; Contreras, J.E.; Bukauskas, F.F.; Saez, J.C. New roles for astrocytes: Gap junction hemichannels have something to communicate. Trends Neurosci. 2003, 26, 610–617. [Google Scholar] [CrossRef] [Green Version]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Saez, P.J.; Saez, J.C.; Giaume, C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef]
- Mostafavi, H.; Khaksarian, M.; Joghataei, M.T.; Hassanzadeh, G.; Soleimani, M.; Eftekhari, S.; Soleimani, M.; Mousavizadeh, K.; Hadjighassem, M.R. Fluoxetin upregulates connexin 43 expression in astrocyte. Basic Clin. Neurosci. 2014, 5, 74–79. [Google Scholar]
- Singh, J.B.; Fedgchin, M.; Daly, E.J.; De Boer, P.; Cooper, K.; Lim, P.; Pinter, C.; Murrough, J.W.; Sanacora, G.; Shelton, R.C.; et al. A Double-Blind, Randomized, Placebo-Controlled, Dose-Frequency Study of Intravenous Ketamine in Patients With Treatment-Resistant Depression. Am. J. Psychiatry 2016, 173, 816–826. [Google Scholar] [CrossRef] [PubMed]
- DiazGranados, N.; Ibrahim, L.A.; Brutsche, N.E.; Ameli, R.; Henter, I.D.; Luckenbaugh, D.A.; Machado-Vieira, R.; Zarate, C.A., Jr. Rapid resolution of suicidal ideation after a single infusion of an N-methyl-D-aspartate antagonist in patients with treatment-resistant major depressive disorder. J. Clin. Psychiatry 2010, 71, 1605–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Liu, X.; Gangoso, E.; Yi, C.; Jeanson, T.; Kandelman, S.; Mantz, J.; Giaume, C. General anesthetics have differential inhibitory effects on gap junction channels and hemichannels in astrocytes and neurons. Glia 2016, 64, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.L.; Chan, S.L.; Way, W.L.; Trevor, A.J. Distribution in the brain and metabolism of ketamine in the rat after intravenous administration. Anesthesiology 1973, 39, 370–376. [Google Scholar] [CrossRef]
- Idvall, J.; Ahlgren, I.; Aronsen, K.R.; Stenberg, P. Ketamine infusions: Pharmacokinetics and clinical effects. Br. J. Anaesth. 1979, 51, 1167–1173. [Google Scholar] [CrossRef]
- Verdolini, N.; Hidalgo-Mazzei, D.; Murru, A.; Pacchiarotti, I.; Samalin, L.; Young, A.H.; Vieta, E.; Carvalho, A.F. Mixed states in bipolar and major depressive disorders: Systematic review and quality appraisal of guidelines. Acta Psychiatr. Scand. 2018, 138, 196–222. [Google Scholar] [CrossRef]
- Delgado, A.; Velosa, J.; Zhang, J.; Dursun, S.M.; Kapczinski, F.; de Azevedo Cardoso, T. Clozapine in bipolar disorder: A systematic review and meta-analysis. J. Psychiatr. Res. 2020, 125, 21–27. [Google Scholar] [CrossRef]
- Fornaro, M.; Carvalho, A.F.; Fusco, A.; Anastasia, A.; Solmi, M.; Berk, M.; Sim, K.; Vieta, E.; de Bartolomeis, A. The concept and management of acute episodes of treatment-resistant bipolar disorder: A systematic review and exploratory meta-analysis of randomized controlled trials. J. Affect. Disord. 2020, 276, 970–983. [Google Scholar] [CrossRef]
- Rey Souto, D.; Pinzon Espinosa, J.; Vieta, E.; Benabarre Hernandez, A. Clozapine in patients with schizoaffective disorder: A systematic review. Rev. Psiquiatr. Salud. Ment 2020, in press. [Google Scholar] [CrossRef]
- Yatham, L.N.; Kennedy, S.H.; Parikh, S.V.; Schaffer, A.; Bond, D.J.; Frey, B.N.; Sharma, V.; Goldstein, B.I.; Rej, S.; Beaulieu, S.; et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) 2018 guidelines for the management of patients with bipolar disorder. Bipolar Disord. 2018, 20, 97–170. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, G.M.; Haddad, P.M.; Ferrier, I.N.; Aronson, J.K.; Barnes, T.; Cipriani, A.; Coghill, D.R.; Fazel, S.; Geddes, J.R.; Grunze, H.; et al. Evidence-based guidelines for treating bipolar disorder: Revised third edition recommendations from the British Association for Psychopharmacology. J. Psychopharmacol. 2016, 30, 495–553. [Google Scholar] [CrossRef] [PubMed]
- Escudero, M.A.G.; Gutierrez-Rojas, L.; Lahera, G. Second Generation Antipsychotics Monotherapy as Maintenance Treatment for Bipolar Disorder: A Systematic Review of Long-Term Studies. Psychiatr. Q. 2020, 91, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Muhiudeen-Russell, I.A.; Miller-Hance, W.C.; Silverman, N.H. Unrecognized esophageal perforation in a neonate during transesophageal echocardiography. J. Am. Soc. Echocardiogr. 2001, 14, 747–749. [Google Scholar] [CrossRef]
- Kalinin, V.V. Suicidality and antiepileptic drugs: Is there a link? Drug Saf 2007, 30, 123–142. [Google Scholar] [CrossRef]
- Piedad, J.; Rickards, H.; Besag, F.M.; Cavanna, A.E. Beneficial and adverse psychotropic effects of antiepileptic drugs in patients with epilepsy: A summary of prevalence, underlying mechanisms and data limitations. CNS Drugs 2012, 26, 319–335. [Google Scholar] [CrossRef]
- Murakami, T.; Okada, M.; Kawata, Y.; Zhu, G.; Kamata, A.; Kaneko, S. Determination of effects of antiepileptic drugs on SNAREs-mediated hippocampal monoamine release using in vivo microdialysis. Br. J. Pharm. 2001, 134, 507–520. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Kaneko, S.; Hirano, T.; Ishida, M.; Kondo, T.; Otani, K.; Fukushima, Y. Effects of zonisamide on extracellular levels of monoamine and its metabolite, and on Ca2+ dependent dopamine release. Epilepsy Res. 1992, 13, 113–119. [Google Scholar] [CrossRef]
- Okada, M.; Hirano, T.; Kawata, Y.; Murakami, T.; Wada, K.; Mizuno, K.; Kondo, T.; Kaneko, S. Biphasic effects of zonisamide on serotonergic system in rat hippocampus. Epilepsy Res. 1999, 34, 187–197. [Google Scholar] [CrossRef]
- Kawata, Y.; Okada, M.; Murakami, T.; Mizuno, K.; Wada, K.; Kondo, T.; Kaneko, S. Effects of zonisamide on K+ and Ca2+ evoked release of monoamine as well as K+ evoked intracellular Ca2+ mobilization in rat hippocampus. Epilepsy Res. 1999, 35, 173–182. [Google Scholar] [CrossRef]
- Kaneko, S.; Okada, M.; Hirano, T.; Kondo, T.; Otani, K.; Fukushima, Y. Carbamazepine and zonisamide increase extracellular dopamine and serotonin levels in vivo, and carbamazepine does not antagonize adenosine effect in vitro: Mechanisms of blockade of seizure spread. Jpn. J. Psychiatry Neurol. 1993, 47, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Hirano, T.; Mizuno, K.; Kawata, Y.; Wada, K.; Murakami, T.; Tasaki, H.; Kaneko, S. Effects of carbamazepine on hippocampal serotonergic system. Epilepsy Res. 1998, 31, 187–198. [Google Scholar] [CrossRef]
- Kawata, Y.; Okada, M.; Murakami, T.; Kamata, A.; Zhu, G.; Kaneko, S. Pharmacological discrimination between effects of carbamazepine on hippocampal basal, Ca(2+)- and K(+)-evoked serotonin release. Br. J. Pharm. 2001, 133, 557–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamura, S.; Hamaguchi, T.; Ohoyama, K.; Sugiura, Y.; Suzuki, D.; Kanehara, S.; Nakagawa, M.; Motomura, E.; Matsumoto, T.; Tanii, H.; et al. Topiramate and zonisamide prevent paradoxical intoxication induced by carbamazepine and phenytoin. Epilepsy Res. 2009, 84, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Effect of lamotrigine and carbamazepine on corticotropin-releasing factor-associated serotonergic transmission in rat dorsal raphe nucleus. Psychopharmacology 2012, 220, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Zhu, G.; Yoshida, S.; Kanai, K.; Hirose, S.; Kaneko, S. Exocytosis mechanism as a new targeting site for mechanisms of action of antiepileptic drugs. Life Sci. 2002, 72, 465–473. [Google Scholar] [CrossRef]
- Garbelli, R.; Frassoni, C.; Condorelli, D.F.; Trovato Salinaro, A.; Musso, N.; Medici, V.; Tassi, L.; Bentivoglio, M.; Spreafico, R. Expression of connexin 43 in the human epileptic and drug-resistant cerebral cortex. Neurology 2011, 76, 895–902. [Google Scholar] [CrossRef]
- Das, A.; Wallace, G.C.t.; Holmes, C.; McDowell, M.L.; Smith, J.A.; Marshall, J.D.; Bonilha, L.; Edwards, J.C.; Glazier, S.S.; Ray, S.K.; et al. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 2012, 220, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Hussein, A.M.; Ghalwash, M.; Magdy, K.; Abulseoud, O.A. Beta Lactams Antibiotic Ceftriaxone Modulates Seizures, Oxidative Stress and Connexin 43 Expression in Hippocampus of Pentylenetetrazole Kindled Rats. J. Epilepsy Res. 2016, 6, 8–15. [Google Scholar] [CrossRef]
- Dambach, H.; Hinkerohe, D.; Prochnow, N.; Stienen, M.N.; Moinfar, Z.; Haase, C.G.; Hufnagel, A.; Faustmann, P.M. Glia and epilepsy: Experimental investigation of antiepileptic drugs in an astroglia/microglia co-culture model of inflammation. Epilepsia 2014, 55, 184–192. [Google Scholar] [CrossRef]
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, T.; Kumada, T.; Saito, K.; Fujii, T. Autism in siblings with autosomal dominant nocturnal frontal lobe epilepsy. Brain Dev. 2013, 35, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Asioli, G.M.; Rossi, S.; Bisulli, F.; Licchetta, L.; Tinuper, P.; Provini, F. Therapy in Sleep-Related Hypermotor Epilepsy (SHE). Curr Treat. Options Neurol. 2020, 22, 1. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Kobayashi, K.; Fujii, T.; Okuno, T.; Hirose, S.; Iwata, H.; Mitsudome, A.; Kaneko, S. Electroclinical picture of autosomal dominant nocturnal frontal lobe epilepsy in a Japanese family. Epilepsia 2000, 41, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Zhu, G.; Yoshida, S.; Kaneko, S. Validation criteria for genetic animal models of epilepsy. Epilepsy Seizure 2010, 3, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Arif, H.; Buchsbaum, R.; Weintraub, D.; Pierro, J.; Resor, S.R., Jr.; Hirsch, L.J. Patient-reported cognitive side effects of antiepileptic drugs: Predictors and comparison of all commonly used antiepileptic drugs. Epilepsy Behav. E&B 2009, 14, 202–209. [Google Scholar]
- Meador, K.J.; Loring, D.W.; Boyd, A.; Echauz, J.; LaRoche, S.; Velez-Ruiz, N.; Korb, P.; Byrnes, W.; Dilley, D.; Borghs, S.; et al. Randomized double-blind comparison of cognitive and EEG effects of lacosamide and carbamazepine. Epilepsy Behav. E&B 2016, 62, 267–275. [Google Scholar]
- Toniolo, S.; Di Lorenzo, F.; Bozzali, M.; Yogarajah, M. The impact of lacosamide on mood disorders in adult patients with epilepsy: A systematic review. Epilepsy Behav. E&B 2020, 111, 107179. [Google Scholar]
- Kanba, S.; Yagi, G.; Kamijima, K.; Suzuki, T.; Tajima, O.; Otaki, J.; Arata, E.; Koshikawa, H.; Nibuya, M.; Kinoshita, N.; et al. The first open study of zonisamide, a novel anticonvulsant, shows efficacy in mania. Prog. Neuro Psychopharmacol. Biol. Psychiatry 1994, 18, 707–715. [Google Scholar]
- Oyamada, M.; Takebe, K.; Oyamada, Y. Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim. Biophys. Acta 2013, 1828, 118–133. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Yamamura, S.; Ohoyama, K.; Nakagawa, M.; Motomura, E.; Kaneko, S.; Okada, M. Effects of valproate on neurotransmission associated with ryanodine receptors. Neurosci. Res. 2010, 68, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.B.; Chibane, F.L.; Wang, Z.; Chuang, D.M. Potential roles of HDAC inhibitors in mitigating ischemia-induced brain damage and facilitating endogenous regeneration and recovery. Curr. Pharm. Des. 2013, 19, 5105–5120. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.; Shao, Q.; Yang, X.J.; Luh, S.P.; Kandouz, M.; Batist, G.; Laird, D.W.; Alaoui-Jamali, M.A. A histone deacetylation-dependent mechanism for transcriptional repression of the gap junction gene cx43 in prostate cancer cells. Prostate 2006, 66, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Hayashi, T.; Tokunou, M.; Nakachi, K.; Trosko, J.E.; Chang, C.C.; Yorioka, N. Suberoylanilide hydroxamic acid enhances gap junctional intercellular communication via acetylation of histone containing connexin 43 gene locus. Cancer Res. 2005, 65, 9771–9778. [Google Scholar] [CrossRef] [Green Version]
- Khan, Z.; Akhtar, M.; Asklund, T.; Juliusson, B.; Almqvist, P.M.; Ekstrom, T.J. HDAC inhibition amplifies gap junction communication in neural progenitors: Potential for cell-mediated enzyme prodrug therapy. Exp. Cell Res. 2007, 313, 2958–2967. [Google Scholar] [CrossRef]
- Chen, T.Y.; Kamali, M.; Chu, C.S.; Yeh, C.B.; Huang, S.Y.; Mao, W.C.; Lin, P.Y.; Chen, Y.W.; Tseng, P.T.; Hsu, C.Y. Divalproex and its effect on suicide risk in bipolar disorder: A systematic review and meta-analysis of multinational observational studies. J. Affect. Disord. 2019, 245, 812–818. [Google Scholar] [CrossRef]
- Taylor, D.M.; Cornelius, V.; Smith, L.; Young, A.H. Comparative efficacy and acceptability of drug treatments for bipolar depression: A multiple-treatments meta-analysis. Acta Psychiatr. Scand. 2014, 130, 452–469. [Google Scholar] [CrossRef]
- Lindstrom, L.; Lindstrom, E.; Nilsson, M.; Hoistad, M. Maintenance therapy with second generation antipsychotics for bipolar disorder—A systematic review and meta-analysis. J. Affect. Disord. 2017, 213, 138–150. [Google Scholar] [CrossRef]
- Einoch, R.; Weinreb, O.; Mandiuk, N.; Youdim, M.B.H.; Bilker, W.; Silver, H. The involvement of BDNF-CREB signaling pathways in the pharmacological mechanism of combined SSRI- antipsychotic treatment in schizophrenia. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2017, 27, 470–483. [Google Scholar] [CrossRef]
- Aringhieri, S.; Kolachalam, S.; Gerace, C.; Carli, M.; Verdesca, V.; Brunacci, M.G.; Rossi, C.; Ippolito, C.; Solini, A.; Corsini, G.U.; et al. Clozapine as the most efficacious antipsychotic for activating ERK 1/2 kinases: Role of 5-HT2A receptor agonism. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2017, 27, 383–398. [Google Scholar] [CrossRef]
- Jochim, J.; Rifkin-Zybutz, R.P.; Geddes, J.; Cipriani, A. Valproate for acute mania. Cochrane Database Syst Rev. 2019, 10, CD004052. [Google Scholar] [CrossRef] [PubMed]
- Bahji, A.; Ermacora, D.; Stephenson, C.; Hawken, E.R.; Vazquez, G. Comparative efficacy and tolerability of pharmacological treatments for the treatment of acute bipolar depression: A systematic review and network meta-analysis. J. Affect. Disord. 2020, 269, 154–184. [Google Scholar] [CrossRef] [PubMed]
- Reus, G.Z.; Abelaira, H.M.; Agostinho, F.R.; Ribeiro, K.F.; Vitto, M.F.; Luciano, T.F.; Souza, C.T.; Quevedo, J. The administration of olanzapine and fluoxetine has synergistic effects on intracellular survival pathways in the rat brain. J. Psychiatr. Res. 2012, 46, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Walrave, L.; Vinken, M.; Leybaert, L.; Smolders, I. Astrocytic Connexin43 Channels as Candidate Targets in Epilepsy Treatment. Biomolecules 2020, 10, 1578. [Google Scholar] [CrossRef] [PubMed]

| Subject | Region (Cell) | Effect | Reference |
|---|---|---|---|
| Suicide | dorsal lateral prefrontal cortex (astrocyte) | decrease (mRNA) | [50] |
| Major depression | locus coeruleus | decrease (mRNA) | [49] |
| Major depression | orbitofrontal cortex | decrease (protein) | [52] |
| Major depression | prefrontal cortex | decrease (mRNA) | [72] |
| Major depression (suicide) | Neocortex, mediodorsal thalamus, caudate nucleus, cerebellum | decrease (mRNA) | [51] |
| Model | Region (Cell) | Effect | Reference |
|---|---|---|---|
| (In Vivo) | |||
| chronic unpredictable stress | prefrontal cortex (rat, in vivo) | decrease (mRNA and protein) suppresses gap junction permeability | [59,60,61] |
| Hippocampus (rat, in vivo) | decrease (protein) suppresses gap junction permeability | [62] | |
| acute restraint stress (2 h) | Hippocampus (mouse, in vivo) | No effect (protein) enhances hemichannel permeability | [63] |
| chronic restraint stress (2 h × 10 times) | Hippocampus (mouse, in vivo) | No effect (protein) enhances hemichannel permeability | [63] |
| Mouse corticosterone (5 mg/kg/day for 28 days) | Hippocampus (mouse, in vivo) | No effect (protein) increase (phosphorylated protein) | [64] |
| (In Vitro) | |||
| corticosterone (50 µM for 24 h) | cortical astrocyte (rat, in vitro) | decrease (protein in total lysate and plasma membrane) increase phosphorylated Cx43 in plasma membrane supresses gap junction permeability | [68] |
| Corticosterone (50 µM for 24 h) | hippocampal astrocyte (rat, in vitro) | decrease (protein in total lysate and plasma membrane) increase phosphorylated Cx43 in plasma membrane supresses gap junction permeability | [68] |
| corticosterone (5‒50 µM for 16 days) | cortical astrocyte (rat, in vitro) | decrease (protein) | [65] |
| Mouse lipopolysaccharide (1 µg/mL for 24 h) | cortical astrocyte (mouse, in vitro) | augmentation of hemichannel permeability | [75] |
| Agent (Class) | Model (Region) | Treatment (Dose, Duration) | Effect (Function) | Reference |
|---|---|---|---|---|
| Fluoxetine (SSRI) | Rat (frontal) | in vivo (20 mg/kg for 21 days) | Increase (protein) | [76] |
| rat (frontal) | in vivo (10 mg/kg for 21 days) | increase (mRNA/protein) (gap junction: no effect) | [59] | |
| Rat (frontal) chronic unpredictable stress | in vivo (10 mg/kg for 21 days) | increase (mRNA/protein) (gap junction: augmentation) | [59] | |
| astrocytoma cells (1321N1/U87MG) | in vitro (30‒60 µM for 24 h) | increase (mRNA/protein) | [80] | |
| Mouse (cortical astrocyte) | in vitro (10 µM for 24 h) | no effect (protein) (gap junction: inhibition) | [75] | |
| Mouse lipopolysaccharide (cortical astrocyte) | in vitro (10 µM for 24 h) | (hemichannel: inhibition) | [75] | |
| mouse exogenous corticosterone (hippocampus) | in vivo (18 mg/kg for 28 days) | Decreased (phosphorylated protein) | [64] | |
| Fluvoxamine (SSRI) | rat (cortical astrocyte) | in vitro (25 µM for 48 h) | increase (protein) | [77] |
| Paroxetine (SSRI) | Mouse (cortical astrocyte) | in vitro (5 µM for 24 h) | no effect (protein) (gap junction: augmentation) | [75] |
| Mouse lipopolysaccharide (cortical astrocyte) | in vitro (5 µM for 24 h) | (hemichannel: inhibition) | [75] | |
| Reboxetine (NRI) | Mouse (cortical astrocyte) | in vitro (10 µM for 24 h) | no effect (protein) (gap junction: no effect) | [75] |
| mouse lipopolysaccharide (cortical astrocyte) | in vitro (10 µM for 24 h) | (hemichannel: inhibition) | [75] | |
| Duloxetine (SNRI) | Rat (frontal) | in vivo (10 mg/kg for 21 days) | increase (mRNA/protein) (gap junction: no effect) | [59] |
| Rat (frontal) chronic unpredictable stress | in vivo (10 mg/kg for 21 days) | increase (mRNA/protein) (gap junction: augmentation) | [59] | |
| mouse (cortical astrocyte) | in vitro (5 µM for 24 h) | no effect (protein) (gap junction: no effect) | [75] | |
| mouse lipopolysaccharide (cortical astrocyte) | in vitro (5 µM for 24 h) | (hemichannel: inhibition) | [75] | |
| Venlafaxine (SNRI) | mouse (cortical astrocyte) | in vitro (5 µM for 24 h) | no effect (protein) (gap junction: inhibition) | [75] |
| mouse lipopolysaccharide (cortical astrocyte) | in vitro (5 µM for 24 h) | (hemichannel: inhibition) | [75] | |
| Milnacipran (SNRI) | rat cortical astrocyte | in vitro (25 µM for 48 h) | no effect (protein) | [77] |
| Cocaine | rat cortical astrocyte | in vitro (100 µM for 48 h) | no effect (protein) | [77] |
| (nonselective monoamine transporter inhibitor) | ||||
| Agent | Model (Region) | Treatment (Dose, Duration) | Cx43 Expression (Function) | Reference |
|---|---|---|---|---|
| Haloperidol | Rat (frontal) | in vivo (1.5 mg/kg for 21 days) | decrease (protein) | [76] |
| rat cortical astrocyte | in vitro (25 µM for 48 h) | no effect (protein) | [77] | |
| Clozapine | Rat (frontal) | in vivo (20 mg/kg for 21 days) | increase (protein) | [76] |
| rat cortical astrocyte | in vitro (30 µM for 7 days) | increase (protein) (activation) | [3,8] | |
| Olanzapine | Rat (frontal) | in vivo (2 mg/kg for 21 days) | no effect (protein) | [76] |
| Ketamine | mouse cortical astrocyte | in vitro (300 µM for 30 min) | Inhibition (gap junction) | [84] |
| mouse cortical astrocyte lipopolysaccharide (200 ng/mL) | in vitro (20 µM for 30 min) | Inhibition (hemichannel) | [84] | |
| mouse cortical astrocyte TNFα + IL1β (20 ng/mL) | in vitro (50 µM for 30 min) | Inhibition (hemichannel) | [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okada, M.; Oka, T.; Nakamoto, M.; Fukuyama, K.; Shiroyama, T. Astroglial Connexin43 as a Potential Target for a Mood Stabiliser. Int. J. Mol. Sci. 2021, 22, 339. https://doi.org/10.3390/ijms22010339
Okada M, Oka T, Nakamoto M, Fukuyama K, Shiroyama T. Astroglial Connexin43 as a Potential Target for a Mood Stabiliser. International Journal of Molecular Sciences. 2021; 22(1):339. https://doi.org/10.3390/ijms22010339
Chicago/Turabian StyleOkada, Motohiro, Tomoka Oka, Misaki Nakamoto, Kouji Fukuyama, and Takashi Shiroyama. 2021. "Astroglial Connexin43 as a Potential Target for a Mood Stabiliser" International Journal of Molecular Sciences 22, no. 1: 339. https://doi.org/10.3390/ijms22010339
APA StyleOkada, M., Oka, T., Nakamoto, M., Fukuyama, K., & Shiroyama, T. (2021). Astroglial Connexin43 as a Potential Target for a Mood Stabiliser. International Journal of Molecular Sciences, 22(1), 339. https://doi.org/10.3390/ijms22010339