Macrophage Autophagy and Silicosis: Current Perspective and Latest Insights


Abstract
:1. Silica Dust and Silicosis
1.1. Influencing Factors of Silica Dust to Lung Toxicity and Related Derivative Drugs
1.2. Silicosis Lesion Changes and Animal Model Establishments
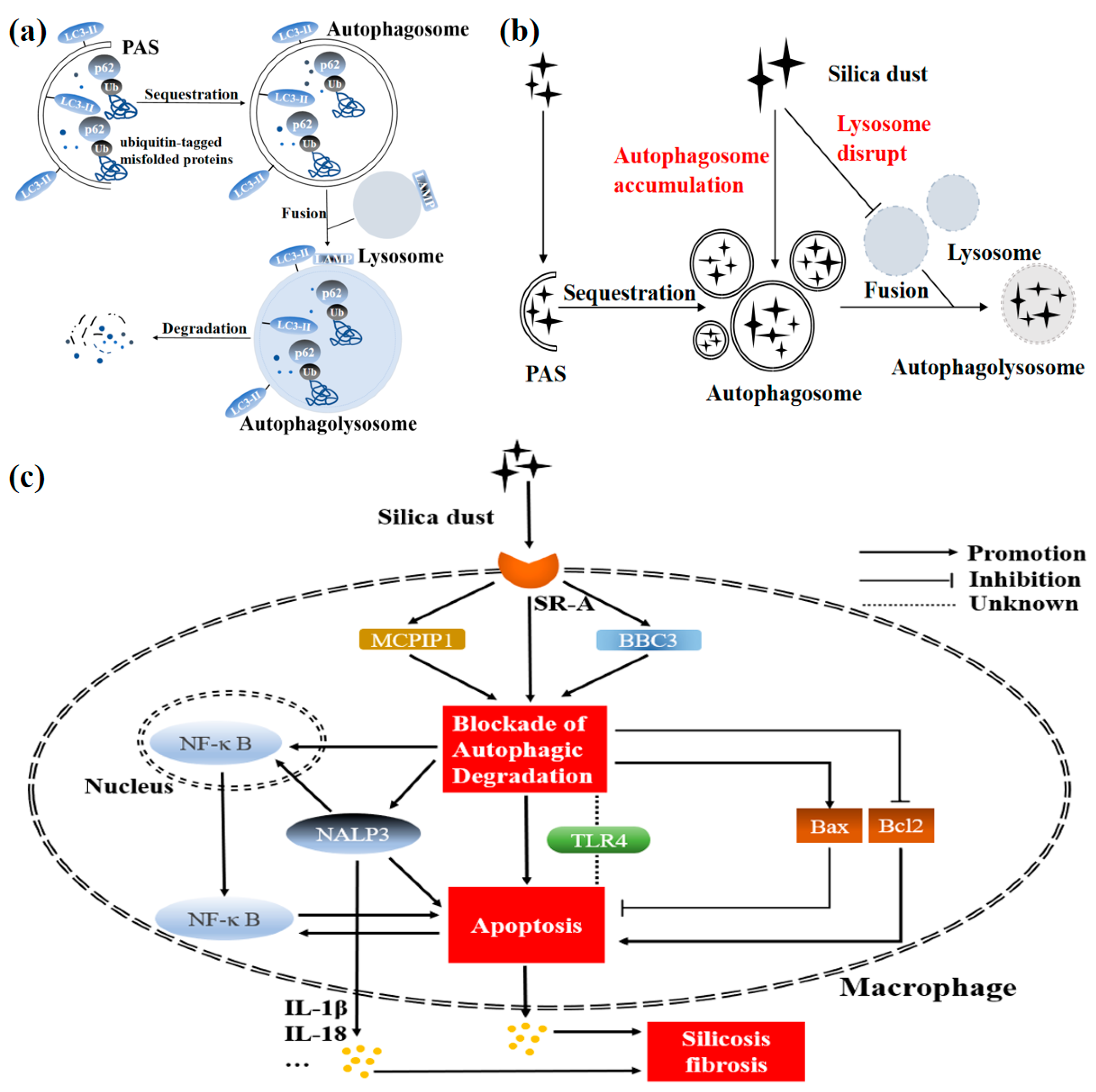
2. Alveolar Macrophage, a “Gatekeeper” for Defense against Silica Dust
3. Some Views about the Autophagy-Related Proteins and Silicosis Pathogenesis
3.1. Static Detection of LC3-II Alone Is Not Sufficient to Reflect Autophagy Activity
3.2. p62 May Perform a Complex Function in the Development of Silicosis
4. Macrophage Autophagy Plays an Important Role in the Silicosis Progression
4.1. Some Critical Proteins Targeting Autophagy Exist in Silicosis Pathogenesis
4.2. Exogenous Irritants-Mediated Autophagy Aggravates Silicosis Fibrosis
4.3. Autophagy Mediated by Natural Products Delays Silicosis Progression

{kind=link}
{kind=link}
| Stimulus | Cell Type | Autophagy-Related Protein Level | Significance |
|---|---|---|---|
| Silica [49] | AMs from silicosis patients | Increased ratio of LC3II/I; Increased p62 level; Decreased LAMP2 level; Decreased TLR4 level | Silica leads to accumulation of autophagosomes and blockade of autophagic degradation in AMs of human silicosis |
| Pro-fibrotic Stimulus | |||
| LPS [49] | AMs from silicosis patients | Increased ratio of LC3II/I; Increased p62 level; Increased MYD88 level; Increased TICAM1 level; Increased Beclin1 level | LPS aggravates silica-induced accumulation of autophagosomes and blockade of autophagic degradation in AMs of human silicosis |
| Smoking [67] | AMs from silicosis patients | Increased ratio of LC3II/I; Increased p62 level; Increased Beclin1 level | Smoking promotes silica-induced lack of autophagy function in AMs of human silicosis |
| Nicotine [68] | AMs from silicosis patients | Increased ratio of LC3II/I; Increased p62 level; Decreased LAMP2 level | Nicotine aggravates silica-induced lack of autophagy function in AMs of human silicosis |
| Potential anti-fibrotic targets | |||
| MCPIP1-siRNA [60] | The human monocytic cell line U937 cells | Decreased ratio of LC3II/I; Decreased Beclin1 level; Decreased p53 level | MCPIP1-siRNA induces autophagy which is mediated by the p53 signaling pathway in macrophages exposed to silica |
| BBC-siRNA [57] | The human monocytic cell line U937 cells | Decreased ratio of LC3II/I; Decreased p62 level | BBC3-siRNA accelerates the process of autophagic degradation in macrophages exposed to silica |
| the exogenous administration of BMSCs [70] | AMs from rats | Decreased ratio of LC3II/I; Decreased Beclin1 level | Inhibition of autophagy is regulated by the administration of BMSCs in AMs of the silicosis rat model |
| Protective natural medicine | |||
| Dioscin [72] | MH-S cell line | Increased ratio of LC3II/I; Decreased p62 level; Increased Beclin1 level; Increased PINK1 level; Increased PARKIN level | Dioscin-induced AM mitophagy protects against the mitochondria dysfunction by silica inhalation |
| Tre or TFEB Over-expression [11,77] | MH-S cell line or AMs from silicosis patients | Decreased ratio of LC3II/I; Decreased p62 level; Increased LAMP1 level | Nuclear transfer of TFEB activated by Tre improves disorder of autophagic substrates degradation in AM exposed to silica |
| ATL-III [78] | AMs from silicosis patients | Decreased ratio of LC3II/I; Decreased p62 level; Increased p-mTOR level | ATL-III protects the autophagy-lysosomal system by an mTOR-dependent pathway in AMs of human silicosis |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gaida, A.; Piasco, D. The Diffuse Interstitial Fibrosis Component of Pulmonary Silicosis. Minerva Med. 1963, 54, 1856–1861. [Google Scholar] [PubMed]
- Leung, C.C.; Yu, I.T.; Chen, W. Silicosis. Lancet 2012, 379, 2008–2018. [Google Scholar] [CrossRef]
- Pavan, C.; Santalucia, R.; Leinardi, R.; Fabbiani, M.; Yakoub, Y.; Uwambayinema, F.; Ugliengo, P.; Tomatis, M.; Martra, G.; Turci, F.; et al. Nearly Free Surface Silanols Are the Critical Molecular Moieties That Initiate the Toxicity of Silica Particles. Proc. Natl. Acad. Sci. USA 2020, 117, 27836–27846. [Google Scholar] [CrossRef] [PubMed]
- Pavan, C.; Delle Piane, M.; Gullo, M.; Filippi, F.; Fubini, B.; Hoet, P.; Horwell, C.J.; Huaux, F.; Lison, D.; Lo Giudice, C.; et al. The Puzzling Issue of Silica Toxicity: Are Silanols Bridging the Gaps between Surface States and Pathogenicity? Part. Fibre Toxicol. 2019, 16, 32. [Google Scholar] [CrossRef]
- Nagelschmidt, G.; Nelson, E.S.; King, E.J.; Attygalle, D.; Yoganathan, M. The Recovery of Quartz and Other Minerals from the Lungs of Rats; A Study in Experimental Silicosis. AMA Arch. Ind. Health 1957, 16, 188–202. [Google Scholar] [PubMed]
- Goldstein, S.; Czapski, G.; Heller, A. Mode of Action of Poly(vinylpyridine-N-oxide) in Preventing Silicosis: Effective Scavenging of Carbonate Anion Radical. Chem. Res. Toxicol. 2006, 19, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.; Rendall, R.E. The Prophylactic Use of Polyvinylpyridine-N-oxide (PVNO) in Baboons Exposed to Quartz Dust. Environ. Res. 1987, 42, 469–481. [Google Scholar] [CrossRef]
- Feng, L.F.; Jia, Z.Y.; Zhu, L.J.; Ju, L.; Chen, J.Q.; Jiang, Z.Q.; Chen, R.P.; Ma, Z.; Zhang, X. Pirfenidone Diminishes SiO2 Induced Lung Fibrosis in Rats. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2010, 28, 772–775. [Google Scholar]
- Guo, J.; Yang, Z.; Jia, Q.; Bo, C.; Shao, H.; Zhang, Z. Pirfenidone Inhibits Epithelial-Mesenchymal Transition and Pulmonary Fibrosis in the Rat Silicosis Model. Toxicol. Lett. 2019, 300, 59–66. [Google Scholar] [CrossRef]
- Mossman, B.T.; Churg, A. Mechanisms in the Pathogenesis of Asbestosis and Silicosis. Am. J. Respir. Crit. Care Med. 1998, 157, 1666–1680. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Chen, S.; Li, C.; Ban, J.; Wei, Y.; He, Y.; Liu, F.; Chen, Y.; Chen, J. Trehalose Alleviates Crystalline Silica-Induced Pulmonary Fibrosis via Activation of the TFEB-Mediated Autophagy-Lysosomal System in Alveolar Macrophages. Cells 2020, 9, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessop, F.; Hamilton, R.F.; Rhoderick, J.F.; Shaw, P.K.; Holian, A. Autophagy Deficiency in Macrophages Enhances NLRP3 Inflammasome Activity and Chronic Lung Disease Following Silica Exposure. Toxicol. Appl. Pharmacol. 2016, 309, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yu, H.; Cao, Z.; Gu, J.; Pei, L.; Jia, M.; Su, M. Kaempferol Modulates Autophagy and Alleviates Silica-Induced Pulmonary Fibrosis. DNA Cell Biol. 2019, 38, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, E.; Rola-Pleszczynski, M. Pulmonary Inflammation and Fibrosis in a Murine Model of Asbestosis and Silicosis. Possible Role of Tumor Necrosis Factor. Inflammation 1989, 13, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Deb, U.; Lomash, V.; Raghuvanshi, S.; Pant, S.C.; Vijayaraghavan, R. Effects of 28 Days Silicon Dioxide Aerosol Exposure on Respiratory Parameters, Blood Biochemical Variables and Lung Histopathology in Rats. Environ. Toxicol. Pharmacol. 2012, 34, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Sager, T.M.; Umbright, C.M.; Mustafa, G.M.; Yanamala, N.; Leonard, H.D.; McKinney, W.G.; Kashon, M.L.; Joseph, P. Tobacco Smoke Exposure Exacerbated Crystalline Silica-Induced Lung Toxicity in Rats. Toxicol. Sci. 2020, 178, 375–390. [Google Scholar] [CrossRef]
- Rehn, B.; Bruch, J.; Zou, T.; Hobusch, G. Recovery of Rat Alveolar Macrophages by Bronchoalveolar Lavage under Normal and Activated Conditions. Environ. Health Perspect. 1992, 97, 11–16. [Google Scholar] [CrossRef]
- Dethloff, L.A.; Gladen, B.C.; Gilmore, L.B.; Hook, G.E. Quantitation of Cellular and Extracellular Constituents of the Pulmonary Lining in Rats by Using Bronchoalveolar Lavage. Effects of Silica-Induced Pulmonary Inflammation. Am. Rev. Respir. Dis. 1987, 136, 899–907. [Google Scholar] [CrossRef]
- Reynolds, H.Y. Bronchoalveolar Lavage. Am. Rev. Respir. Dis. 1987, 135, 250–263. [Google Scholar]
- Jonsson, S.; Musher, D.M.; Goree, A.; Lawrence, E.C. Human Alveolar Lining Material and Antibacterial Defenses. Am. Rev. Respir. Dis. 1986, 133, 136–140. [Google Scholar] [CrossRef]
- Johansson, A.; Lundborg, M.; Skold, C.M.; Lundahl, J.; Tornling, G.; Eklund, A.; Camner, P. Functional, Morphological, and Phenotypical Differences between Rat Alveolar and Interstitial Macrophages. Am. J. Respir. Cell Mol. Biol. 1997, 16, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Borensztajn, K.; Peppelenbosch, M.P.; Spek, C.A. Factor Xa: At the Crossroads between Coagulation and Signaling in Physiology and Disease. Trends Mol. Med. 2008, 14, 429–440. [Google Scholar] [CrossRef]
- Hamilton, R.F., Jr.; Thakur, S.A.; Mayfair, J.K.; Holian, A. MARCO Mediates Silica Uptake and Toxicity in Alveolar Macrophages from C57BL/6 Mice. J. Biol. Chem. 2006, 281, 34218–34226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlisle, E.M. Silicon. Nutr. Rev. 1975, 33, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Scabilloni, J.F.; Antonini, J.M.; Rojanasakul, Y.; Castranova, V.; Mercer, R.R. Induction of Secondary Apoptosis, Inflammation, and Lung Fibrosis after Intratracheal Instillation of Apoptotic Cells in Rats. Am. J. Physiol. Lung C. 2006, 290, L695–L702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehlet, S.N.; Willumsen, N.; Armbrecht, G.; Dietzel, R.; Brix, S.; Henriksen, K.; Karsdal, M.A. Age-Related Collagen Turnover of the Interstitial Matrix and Basement Membrane: Implications of Age- and Sex-Dependent Remodeling of the Extracellular Matrix. PLoS ONE 2018, 13, e0194458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, B.; Zhang, H.; Xue, R. miR-141 Alleviates LPS-Induced Inflammation Injury in WI-38 Fibroblasts by Up-regulation of NOX2. Life Sci. 2019, 216, 271–278. [Google Scholar] [CrossRef]
- Hu, S.; Zhao, H.; Al-Humadi, N.H.; Yin, X.J.; Ma, J.K. Silica-Induced Apoptosis in Alveolar Macrophages: Evidence of In Vivo Thiol Depletion and the Activation of Mitochondrial Pathway. J. Toxicol. Environ. Health A 2006, 69, 1261–1284. [Google Scholar] [CrossRef]
- McCabe, M.J., Jr. Mechanisms and Consequences of Silica-Induced Apoptosis. Toxicol. Sci. 2003, 76, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Thibodeau, M.; Giardina, C.; Hubbard, A.K. Silica-Induced Caspase Activation in Mouse Alveolar Macrophages is Dependent upon Mitochondrial Integrity and Aspartic Proteolysis. Toxicol. Sci. 2003, 76, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.Q.; He, Q.C.; Yuan, J.X.; Chen, J.; Chen, G.; Lu, Y.; Bai, Y.P.; Zhang, C.M.; Yuan, Y.; Xu, Y.J. Role of Fas/FasL Pathway-Mediated Alveolar Macrophages Releasing Inflammatory Cytokines in Human Silicosis. Biomed. Environ. Sci. 2013, 26, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.Q.; Rojanasakul, L.W.; Chen, Z.Y.; Xu, Y.J.; Bai, Y.P.; Chen, G.; Zhang, X.Y.; Zhang, C.M.; Yu, Y.Q.; Shen, F.H.; et al. Fas/FasL Pathway-Mediated Alveolar Macrophage Apoptosis Involved in Human Silicosis. Apoptosis 2011, 16, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Gozal, E.; Ortiz, L.A.; Zou, X.; Burow, M.E.; Lasky, J.A.; Friedman, M. Silica-Induced Apoptosis in Murine Macrophage: Involvement of Tumor Necrosis Factor-alpha and Nuclear Factor-kappaB Activation. Am. J. Respir. Cell Mol. Biol. 2002, 27, 91–98. [Google Scholar] [CrossRef]
- Wang, L.; Bowman, L.; Lu, Y.; Rojanasakul, Y.; Mercer, R.R.; Castranova, V.; Ding, M. Essential Role of p53 in Silica-Induced Apoptosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L488–L496. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-Mediated Autophagy and Endosomal Microautophagy: Joint by a Chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germic, N.; Frangez, Z.; Yousefi, S.; Simon, H.U. Regulation of the Innate Immune System by Autophagy: Monocytes, Macrophages, Dendritic Cells and Antigen Presentation. Cell Death Differ. 2019, 26, 715–727. [Google Scholar] [CrossRef]
- Pyo, J.O.; Nah, J.; Jung, Y.K. Molecules and Their Functions in Autophagy. Exp. Mol. Med. 2012, 44, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Fujita, N.; Noda, T.; Yoshimori, T. Monitoring Autophagy in Mammalian Cultured Cells Through the Dynamics of Lc3. Methods Enzymol. 2009, 452, 1–12. [Google Scholar] [CrossRef]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal Turnover, but Not a Cellular Level, of Endogenous LC3 Is a Marker for Autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Streeter, A.; Menzies, F.M.; Rubinsztein, D.C. LC3-II Tagging and Western Blotting for Monitoring Autophagic Activity in Mammalian Cells. Methods Mol. Biol. 2016, 1303, 161–170. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 Links the Autophagy Pathway and the Ubiqutin-Proteasome System upon Ubiquitinated Protein Degradation. Cell Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Wu, R.; Zheng, J.; Li, P.; Yu, L. Polyubiquitin Chain-Induced p62 Phase Separation Drives Autophagic Cargo Segregation. Cell Res. 2018, 28, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Min, Y.; Im, J.S.; Son, J.; Lee, J.S.; Lee, K.Y. p62 Is Negatively Implicated in the TRAF6-BECN1 Signaling Axis for Autophagy Activation and Cancer Progression by Toll-Like Receptor 4 (TLR4). Cells 2020, 9, 1142. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Ezaki, J.; Komatsu, M.; Takahashi, K.; Mineki, R.; Taka, H.; Kikkawa, M.; Fujimura, T.; Takeda-Ezaki, M.; Ueno, T.; et al. Comprehensive Proteomics Analysis of Autophagy-Deficient Mouse Liver. Biochem. Biophys. Res. Commun. 2008, 368, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 Degradation by Autophagy for the Maintenance of Redox Homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Harder, B.; Rojo, D.L.V.M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 Links Autophagy and Nrf2 Signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Jin, Y.; Chen, S.; Yao, S.; Zhu, L.; Duan, J.; Yuan, J. The Study of Autophagy in Alveolar Macrophages of Patients with Coal Workers’ Pneumoconiosis. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2015, 33, 41–44. [Google Scholar]
- Chen, S.; Jin, Y.L.; Yao, S.Q.; Bai, Y.P.; Fan, X.Y.; Xu, Y.J.; Yuan, J.X. Autophagy in Lung Tissue of Rats Exposed to Silica Dust. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2013, 31, 607–610. [Google Scholar]
- Chen, S.; Yuan, J.; Yao, S.; Jin, Y.; Chen, G.; Tian, W.; Xi, J.; Xu, Z.; Weng, D.; Chen, J. Lipopolysaccharides May Aggravate Apoptosis Through Accumulation of Autophagosomes in Alveolar Macrophages of Human Silicosis. Autophagy 2015, 11, 2346–2357. [Google Scholar] [CrossRef] [Green Version]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-Like Receptors Control Autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Jagannath, C.; Liu, X.D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-Like Receptor 4 Is a Sensor for Autophagy Associated with Innate Immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.Y.W.; Tsui, J.C.C.; Law, P.T.W.; So, W.K.W.; Leung, D.Y.P.; Sham, M.M.K.; Tsui, S.K.W.; Chan, C.W.H. Regulation of TLR4 in Silica-Induced Inflammation: An Underlying Mechanism of Silicosis. Int. J. Med. Sci. 2018, 15, 986–991. [Google Scholar] [CrossRef] [Green Version]
- Li, X.X.; Jiang, D.Y.; Huang, X.X.; Guo, S.L.; Yuan, W.; Dai, H.P. Toll-Like Receptor 4 Promotes Fibrosis in Bleomycin-Induced Lung Injury in Mice. Genet. Mol. Res. 2015, 14, 17391–17398. [Google Scholar] [CrossRef]
- He, Z.; Zhu, Y.; Jiang, H. Inhibiting Toll-Like Receptor 4 Signaling Ameliorates Pulmonary Fibrosis during Acute Lung Injury Induced by Lipopolysaccharide: An Experimental Study. Respir. Res. 2009, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.Z.; Wang, J.P.; Mi, S.; Liu, H.Z.; Cui, B.; Yan, H.M.; Yan, J.; Li, Z.; Liu, H.; Hua, F.; et al. TLR4 Activity Is Required in the Resolution of Pulmonary Inflammation and Fibrosis after Acute and Chronic Lung Injury. Am. J. Pathol. 2012, 180, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a Novel Proapoptotic Gene, Is Induced by p53. Mol. Cell. 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Liu, H.; Cheng, Y.; Yang, J.; Wang, W.; Fang, S.; Zhang, W.; Han, B.; Zhou, Z.; Yao, H.; Chao, J.; et al. BBC3 in Macrophages Promoted Pulmonary Fibrosis Development Through Inducing Autophagy during Silicosis. Cell Death Dis. 2017, 8, e2657. [Google Scholar] [CrossRef]
- Zhou, L.; Azfer, A.; Niu, J.; Graham, S.; Choudhury, M.; Adamski, F.M.; Younce, C.; Binkley, P.F.; Kolattukudy, P.E. Monocyte Chemoattractant Protein-1 Induces a Novel Transcription Factor that Causes Cardiac Myocyte Apoptosis and Ventricular Dysfunction. Circ. Res. 2006, 98, 1177–1185. [Google Scholar] [CrossRef]
- Qu, B.; Cao, J.C.; Zhang, F.F.; Cui, H.J.; Teng, J.L.; Li, J.; Liu, Z.; Morehouse, C.; Jallal, B.; Tang, Y.J.; et al. Type I Interferon Inhibition of MicroRNA-146a Maturation Through Up-regulation of Monocyte Chemotactic Protein-Induced Protein 1 in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2015, 67, 3209–3218. [Google Scholar] [CrossRef]
- Liu, H.; Fang, S.; Wang, W.; Cheng, Y.; Zhang, Y.; Liao, H.; Yao, H.; Chao, J. Macrophage-Derived MCPIP1 Mediates Silica-Induced Pulmonary Fibrosis via Autophagy. Part. Fibre Toxicol. 2016, 13, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, Y.; Zhang, W.; Liu, H.; Zhou, Z.; Dai, X.; Cheng, Y.; Fang, S.; Zhang, Y.; Yao, H.; et al. MCPIP1 Regulates Alveolar Macrophage Apoptosis and Pulmonary Fibroblast Activation after In Vitro Exposure to Silica. Toxicol. Sci. 2016, 151, 126–138. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Peeters, P.M.; Eurlings, I.M.; Perkins, T.N.; Wouters, E.F.; Schins, R.P.; Borm, P.J.; Drommer, W.; Reynaert, N.L.; Albrecht, C. Silica-Induced NLRP3 Inflammasome Activation In Vitro and in Rat Lungs. Part. Fibre Toxicol. 2014, 11, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.H.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica Crystals and Aluminum Salts Activate the NALP3 Inflammasome Through Phagosomal Destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Abdel, F.E.; Bhattacharya, A.; Herron, A.; Safdar, Z.; Eissa, N.T. Critical Role for IL-18 in Spontaneous Lung Inflammation Caused by Autophagy Deficiency. J. Immunol. 2015, 194, 5407–5416. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.K.; Tan, S.Y.; Wang, Y.R.; Li, S.H.; Chen, G.; Chen, S. The Study of Smoking Impact on Autophagy in Alveolar Macrophages of Human Silicosis. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2020, 38, 738–741. [Google Scholar] [CrossRef]
- Chen, S.; Tan, S.; Yang, S.; Chen, G.; Zhu, L.; Sun, Z.; Li, H.; Yao, S. Nicotine Induces Apoptosis Through Exacerbation of Blocked Alveolar Macrophage Autophagic Degradation in Silicosis. Toxicol. Lett. 2020, 334, 94–101. [Google Scholar] [CrossRef]
- Luan, Y.; Zhang, X.; Kong, F.; Cheng, G.H.; Qi, T.G.; Zhang, Z.H. Mesenchymal Stem Cell Prevention of Vascular Remodeling in High Flow-Induced Pulmonary Hypertension Through a Paracrine Mechanism. Int. Immunopharmacol. 2012, 14, 432–437. [Google Scholar] [CrossRef]
- Aslam, M.; Baveja, R.; Liang, O.D.; Fernandez-Gonzalez, A.; Lee, C.; Mitsialis, S.A.; Kourembanas, S. Bone Marrow Stromal Cells Attenuate Lung Injury in a Murine Model of Neonatal Chronic Lung Disease. Am. J. Respir. Crit. Care Med. 2009, 180, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.X.; Gao, J.L.; Zhao, M.M.; Li, R.; Tian, Y.X.; Wang, X.; Zhang, J.; Yuan, J.X.; Cui, J.Z. Effects of Bone Marrow-Derived Mesenchymal Stem Cells on the Autophagic Activity of Alveolar Macrophages in a Rat Model of Silicosis. Exp. Ther. Med. 2016, 11, 2577–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, S.T.; Li, C.; Lu, Y.; Lei, X.; Zhang, Y.; Li, S.; Liu, F.; Chen, Y.; Weng, D.; Chen, J. Dioscin Alleviates Crystalline Silica-Induced Pulmonary Inflammation and Fibrosis Through Promoting Alveolar Macrophage Autophagy. Theranostics 2019, 9, 1878–1892. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lu, Y.; Du, S.; Li, S.; Zhang, Y.; Liu, F.; Chen, Y.; Weng, D.; Chen, J. Dioscin Exerts Protective Effects against Crystalline Silica-Induced Pulmonary Fibrosis in Mice. Theranostics 2017, 7, 4255–4275. [Google Scholar] [CrossRef]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and Artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestrini, M.J.; Johnson, J.R.; Kumar, A.V.; Thakurta, T.G.; Blais, K.; Neill, Z.A.; Marion, S.W.; St Amand, V.; Reenan, R.A.; Lapierre, L.R. Nuclear Export Inhibition Enhances HLH-30/TFEB Activity, Autophagy, and Lifespan. Cell Rep. 2018, 23, 1915–1921. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Yang, S.; Chen, G.; Zhu, L.; Sun, Z.; Chen, S. Trehalose Alleviates Apoptosis by Protecting the Autophagy-Lysosomal System in Alveolar Macrophages during Human Silicosis. Life Sci. 2020, 257, 118043. [Google Scholar] [CrossRef]
- Chen, S.; Tang, K.; Hu, P.; Tan, S.; Yang, S.; Yang, C.; Chen, G.; Luo, Y.; Zou, H. Atractylenolide III Alleviates the Apoptosis Through Inhibition of Autophagy by the mTOR-Dependent Pathway in Alveolar Macrophages of Human Silicosis. Mol. Cell Biochem. 2020. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Saqcena, M.; Chatterjee, A.; Garcia, A.; Frias, M.A.; Foster, D.A. Reciprocal Regulation of AMP-Activated Protein Kinase and Phospholipase D. J. Biol. Chem. 2015, 290, 6986–6993. [Google Scholar] [CrossRef] [Green Version]


| Methods | Route of Administration | Advantages | Disadvantages |
|---|---|---|---|
| Exposed or non-exposed intratracheal instillation [9,14] | Instillation of SiO2 suspension into the trachea | One-time instillation; Simple operation; Low cost | Cannot reflect the mechanism of actual human silicosis; The exposed way may increase animal infection |
| Dynamic or static inhalation [15,16] | Inhalation of SiO2 aerosol | Better for simulating the pathologic process of actual human silicosis | Long modeling time; Expensive equipment |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, S.; Chen, S. Macrophage Autophagy and Silicosis: Current Perspective and Latest Insights. Int. J. Mol. Sci. 2021, 22, 453. https://doi.org/10.3390/ijms22010453
Tan S, Chen S. Macrophage Autophagy and Silicosis: Current Perspective and Latest Insights. International Journal of Molecular Sciences. 2021; 22(1):453. https://doi.org/10.3390/ijms22010453
Chicago/Turabian StyleTan, Shiyi, and Shi Chen. 2021. "Macrophage Autophagy and Silicosis: Current Perspective and Latest Insights" International Journal of Molecular Sciences 22, no. 1: 453. https://doi.org/10.3390/ijms22010453
APA StyleTan, S., & Chen, S. (2021). Macrophage Autophagy and Silicosis: Current Perspective and Latest Insights. International Journal of Molecular Sciences, 22(1), 453. https://doi.org/10.3390/ijms22010453