Human Nitric Oxide Synthase—Its Functions, Polymorphisms, and Inhibitors in the Context of Inflammation, Diabetes and Cardiovascular Diseases


Abstract
:1. Introduction
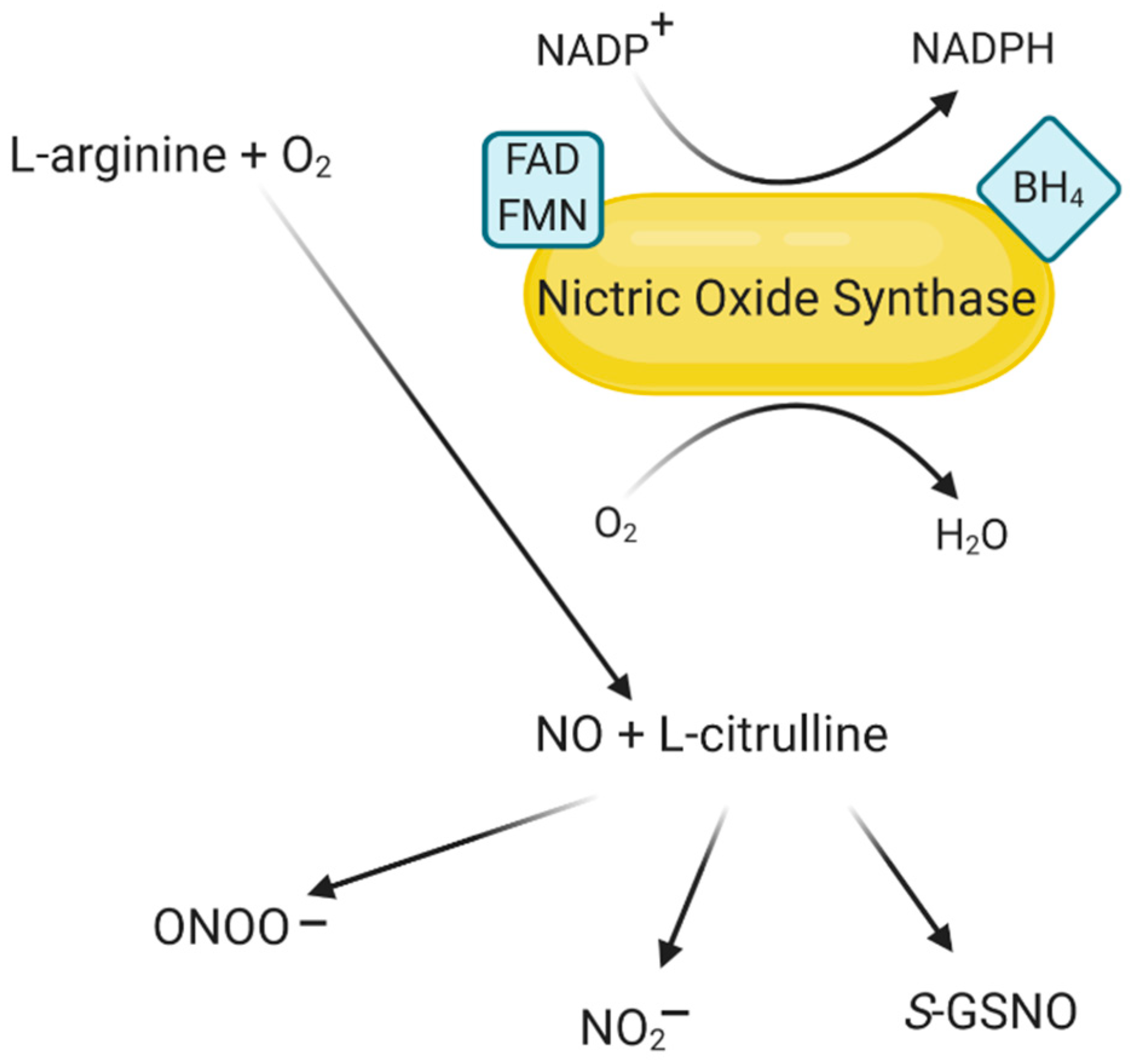
2. Characteristics of NO and Its Derivatives
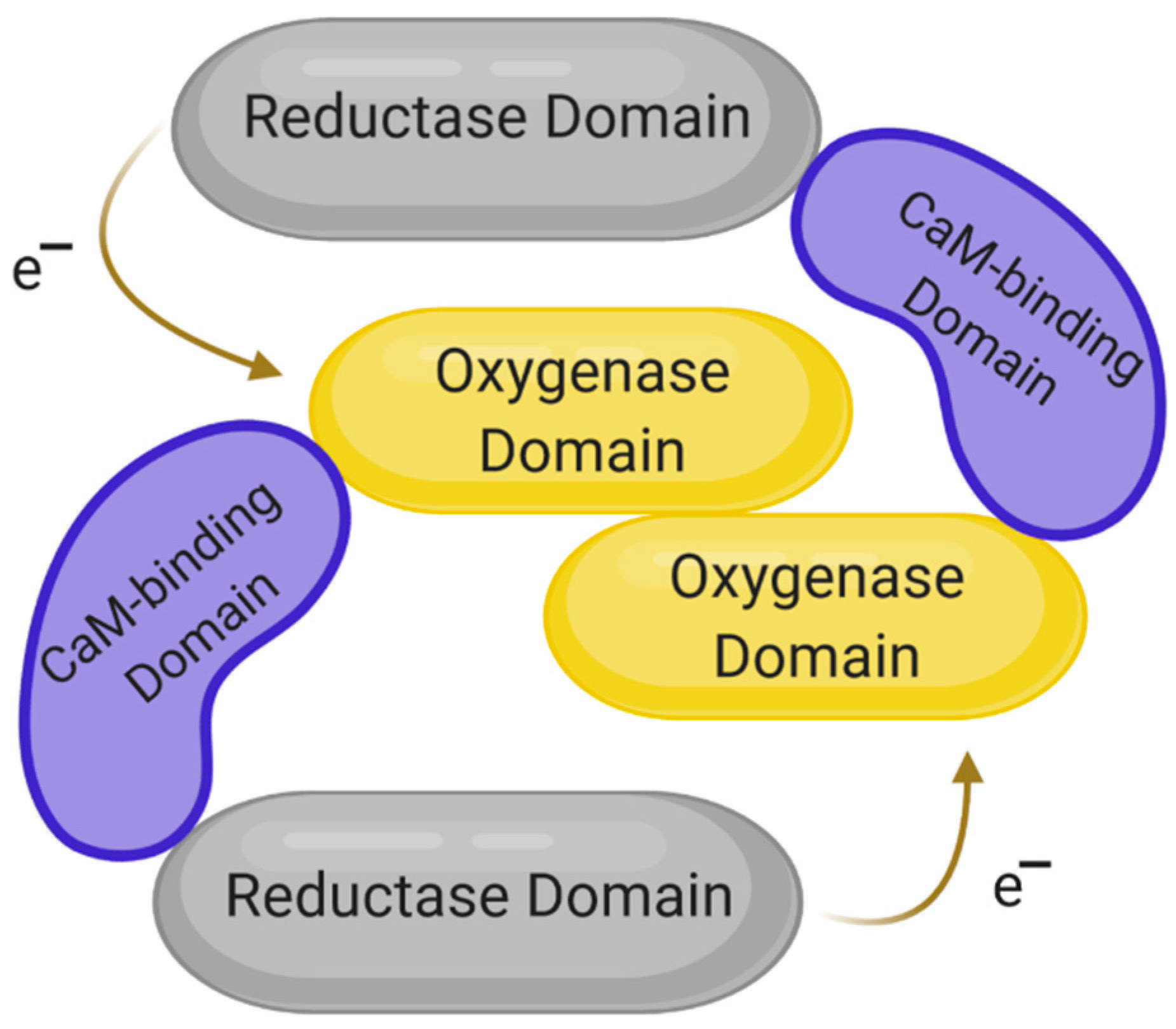
3. The Structure of NOS and Its Isoforms
4. The Role of NO and NOS in the Context of Inflammation, Diabetes and Cardiovascular Diseases
4.1. The Role of NO and NOS in Inflammation
4.2. The Role of NOS in Obesity
4.3. The Role of NOS in Insulin Resistance and Diabetes
4.4. The Role of NOS in Heart Disease
5. Selected NOS Inhibitors and Their Role in Therapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Togliatto, G.; Lombardo, G.; Brizzi, M.F. The future challenge of reactive oxygen species (ros) in hypertension: From bench to bed side. Int. J. Mol. Sci. 2017, 18, 1988. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M. Unraveling the truth about antioxidants: Mitohormesis explains ros-induced health benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef]
- Ferreira, C.A.; Ni, D.; Rosenkrans, Z.T.; Cai, W. Scavenging of reactive oxygen and nitrogen species with nanomaterials. Nano Res. 2018, 10, 4955–4984. [Google Scholar] [CrossRef]
- Ganten, D.; Ruckpaul, K. Nitrosative Stress. In Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar] [CrossRef]
- Pérez-Torres, I.; Manzano-Pech, L.; Rubio-Ruíz, M.E.; Soto, M.E.; Guarner-Lans, V. Nitrosative stress and its association with cardiometabolic disorders. Molecules 2020, 25, 2555. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Hedner, T. Endothelium-Derived relaxing factor (EDRF) and nitric oxide (NO). II. Physiology, pharmacology and pathophysiological implications. Clin. Physiol. 1990, 10, 503–526. [Google Scholar] [CrossRef] [PubMed]
- Naseem, K.M. The role of nitric oxide in cardiovascular diseases. Mol. Asp. Med. 2005, 26, 33–65. [Google Scholar] [CrossRef]
- Thomas, D.D.; Liu, X.; Kantrow, S.P.; Lancaster, J.R., Jr. The biological lifetime of nitric oxide: Implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad. Sci. USA 2001, 98, 355–360. [Google Scholar] [CrossRef]
- Ziche, M.; Morbidelli, L. Nitric oxide and angiogenesis. J. Neurooncol. 2000, 50, 139–148. [Google Scholar] [CrossRef]
- Ji, H.; Tan, S.; Igarashi, J.; Li, H.; Derrick, M.; Martásek, P.; Roman, L.J.; Vásquez-Vivar, J.; Poulos, T.L.; Silverman, R.B. Selective neuronal nitric oxide synthase inhibitors and the prevention of cerebral palsy. Ann. Neurol. 2009, 65, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Ługowski, M.; Saczko, J.; Kulbacka, J.; Banaś, T. Reaktywne formy tlenu i azotu. Pol. Merk. Lek. 2011, 185, 313–317. [Google Scholar] [PubMed]
- Bailey, S.M.; Landar, A.; Darley-Usmar, V. Mitochondrial proteomics in free radical research. Free Radic. Biol. Med. 2005, 38, 175–188. [Google Scholar] [CrossRef] [PubMed]
- PubChem Substance and Compound Databases—NCBI—NIH. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Nitrilooxonium (accessed on 18 December 2020).
- Hayton, T.W.; Legzdins, P.; Sharp, W.B. Coordination and organometallic chemistry of metal-NO complexes. Chem. Rev. 2002, 102, 935–992. [Google Scholar] [CrossRef] [PubMed]
- PubChem Substance and Compound Databases—NCBI—NIH. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Nitrogen-dioxide (accessed on 18 December 2020).
- PubChem Substance and Compound Databases—NCBI—NIH. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Dinitrogen-trioxide (accessed on 18 December 2020).
- Goldstein, S.; Merényi, G. The chemistry of peroxynitrite: Implications for biological activity. Methods Enzymol. 2008, 436, 49–61. [Google Scholar] [CrossRef] [PubMed]
- PubChem Substance and Compound Databases—NCBI—NIH. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Nitrite (accessed on 18 December 2020).
- PubChem Substance and Compound Databases—NCBI—NIH. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Nitrate (accessed on 18 December 2020).
- Bianco, C.L.; Toscano, J.P.; Bartberger, M.D.; Fukuto, J.M. The chemical biology of HNO signaling. Arch. Biochem. Biophys. 2017, 617, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, J.R., Jr. Historical origins of the discovery of mammalian nitric oxide (nitrogen monoxide) production/physiology/pathophysiology. Biochem. Pharmacol. 2020, 176, 113793. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N.; Chandra, M.; Tuteja, R.; Misra, M.K. Nitric oxide as a unique bioactive signaling messenger in physiology and pathophysiology. J. Biomed. Biotechnol. 2004, 4, 227–237. [Google Scholar] [CrossRef]
- Murad, F. Discovery of some of the biological effects of nitric oxide and its role in cell signaling. Biosci. Rep. 2004, 24, 452–474. [Google Scholar] [CrossRef]
- Murad, F. Nitric oxide signaling: Would you believe that a simple free radical could be a second messenger, autacoid, paracrine substance, neurotransmitter, and hormone? Recent Prog. Horm. Res. 1998, 53, 43–59. [Google Scholar] [PubMed]
- Patel, R.P.; McAndrew, J.; Sellak, H.; White, C.R.; Jo, H.; Freeman, B.A.; Darley-Usmar, V.M. Biological aspects of reactive nitrogen species. Biochim. Biophys. Acta 1999, 1411, 385–400. [Google Scholar] [CrossRef] [Green Version]
- Crow, J.P.; Beckman, J.S. The importance of superoxide in nitric oxide-dependent toxicity: Evidence for peroxynitrite-mediated injury. Adv. Exp. Med. Biol. 1996, 387, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr J. 2016, 15, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubanyi, G.M.; Ho, E.H.; Cantor, E.H.; Lumma, W.C.; Botelho, L.H. Cytoprotective function of nitric oxide: Inactivation of superoxide radicals produced by human leukocytes. Biochem. Biophys. Res. Commun. 1991, 181, 1392–1397. [Google Scholar] [CrossRef]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, V.B.; Eiserich, J.P.; Chumley, P.H.; Jablonsky, M.J.; Krishna, N.R.; Kirk, M.; Barnes, S.; Darley-Usmar, V.M.; Freeman, B.A. Nitration of unsaturated fatty acids by nitric oxide-derived reactive nitrogen species peroxynitrite, nitrous acid, nitrogen dioxide, and nitronium ion. Chem. Res. Toxicol. 1999, 12, 83–92. [Google Scholar] [CrossRef]
- Wink, D.A.; Darbyshire, J.F.; Nims, R.W.; Saavedra, J.E.; Ford, P.C. Reactions of the bioregulatory agent nitric oxide in oxygenated aqueous media: Determination of the kinetics for oxidation and nitrosation by intermediates generated in the NO/O2 reaction. Chem. Res. Toxicol. 1993, 6, 23–27. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Psychol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Griffith, O.W.; Stuehr, D.J. Nitric oxide synthases: Properties and catalytic mechanism. Annu. Rev. Physiol. 1995, 57, 707–736. [Google Scholar] [CrossRef]
- GeneCards—Human Genes|Gene Database|Gene Search. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=NOS1 (accessed on 4 December 2020).
- GeneCards—Human Genes|Gene Database|Gene Search. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=NOS3 (accessed on 4 December 2020).
- Knott, A.B.; Bossy-Wetzel, E. Nitric oxide in health and disease of the nervous system. Antioxid. Redox Signal. 2009, 11, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Fish, J.E.; Marsden, P.A. Endothelial nitric oxide synthase: Insight into cell-specific gene regulation in the vascular endothelium. Cell Mol. Life Sci. 2006, 63, 144–162. [Google Scholar] [CrossRef]
- Sumpio, B.E.; Riley, J.T.; Dardik, A. Cells in focus: Endothelial cell. Int. J. Biochem. Cell Biol. 2002, 34, 1508–1512. [Google Scholar] [CrossRef]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hevel, J.M.; White, K.A.; Marietta, M.A. Purification of the inducible murine macrophage nitric oxide synthase. Identification as a flavoprotein. J. Biol. Cherm. 1991, 266, 22789–22791. [Google Scholar] [PubMed]
- Karupiah, G.; Xie, Q.W.; Buller, M.L.; Nathan, C.; Duarte, C.; MacMicking, J.D. Inhibition of viral replication by interferon-y-induced nitric oxide synthase. Science 1993, 261, 1445–1448. [Google Scholar] [CrossRef] [PubMed]
- GeneCards—Human Genes|Gene Database|Gene Search. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=NOS2 (accessed on 4 December 2020).
- Lechner, M.; Lirk, P.; Rieder, J. Inducible nitric oxide synthase (iNOS) in tumor biology: The two sides of the same coin. Semin. Cancer Biol. 2005, 15, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Vanninia, F.; Kashfia, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Fischmann, T.O.; Hruza, A.; Niu, X.D.; Fossetta, J.D.; Lunn, C.A.; Dolphin, E.; Prongay, A.J.; Reichert, P.; Lundell, D.J.; Narula, S.K.; et al. Structural characterization of nitric oxide synthase isoforms reveals striking active-site conservation. Nat. Struct. Biol. 1999, 6, 233–242. [Google Scholar] [CrossRef]
- Mehl, M.; Daiber, A.; Herold, S.; Shoun, H.; Ullrich, V. Peroxynitrite reaction with heme proteins. Nitric Oxide 1999, 3, 142–152. [Google Scholar] [CrossRef]
- Roman, L.J.; Martasek, P.; Masters, B.S. Intrinsic and extrinsic modulation of nitric oxide synthase activity. Chem. Rev. 2002, 102, 1179–1190. [Google Scholar] [CrossRef]
- Garcin, E.D.; Bruns, C.M.; Lloyd, S.J.; Hosfield, D.J.; Tiso, M.; Gachhui, R.; Stuehr, D.J.; Tainer, J.A.; Getzoff, E.D. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J. Biol. Chem. 2004, 279, 37918–37927. [Google Scholar] [CrossRef] [Green Version]
- Lowenstein, C.J.; Padalko, E. iNOS (NOS2) at a glance. J. Cell Sci. 2004, 117, 2865–3286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.A.; Tang, S.S.; Loscalzo, J. S-Nitrosothiols and the S-nitrosoproteome of the cardiovascular system. Antioxid. Redox Signal. 2013, 18, 270–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahani, N.; Sawa, A. Protein S-nitrosylation: Role for nitric oxide signaling in neuronal death. Biochim. Biophys. Acta 2012, 1820, 736–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Soud, H.M.; Wang, J.; Rousseau, D.L.; Fukuto, J.M.; Ignarro, L.J.; Stuehr, D.J. Neuronal nitric oxide synthase self-inactivates by forming a ferrous-nitrosyl complex during aerobic catalysis. J. Biol. Chem. 1995, 270, 22997–23006. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.M., Jr. Enzymes of arginine metabolism. J. Nutr. 2004, 134, 2743S–2747S. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Mattioli, T.A.; Stuehr, D.J.; Santolini, J. Activation of peroxynitrite by inducible nitric-oxide synthase. A direct source of nitrative stress. J. Biol. Chem. 2007, 282, 14101–14112. [Google Scholar] [CrossRef] [Green Version]
- Negrerie, M.; Berka, V.; Vos, M.H.; Liebl, U.; Lambry, J.-C.; Tsai, A.-L.; Martin, J.-L. Geminate recombination of nitric oxide to endothelial nitric oxide-synthase and mechanistic implications. J. Biol. Chem. 1999, 274, 24694–24702. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention (US); National Center for Chronic Disease Prevention and Health Promotion (US); Office on Smoking and Health (US). How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General; Centers for Disease Control and Prevention (US): Atlanta, GA, USA, 2010.
- Roy, B.; Lepoivre, M.; Henry, Y.; Fontecave, M. Inhibition of ribonucleotide reductase by nitric oxide derived from thionitrites: Reversible modifications of both subunits. Biochemistry 1995, 34, 5411–5418. [Google Scholar] [CrossRef]
- Kröncke, K.D.; Fehsel, K.; Kolb-Bachofen, V. Inducible nitric oxide synthase in human diseases. Clin. Exp. Immunol. 1998, 113, 147–156. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, D.Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric. Oxide 2009, 20, 223–230. [Google Scholar] [CrossRef]
- Vane, J.R.; Mitchell, J.A.; Appleton, I.; Tomlinson, A.; Bishop-Bailey, D.; Croxtall, J.; Willoughby, D.A. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc. Natl. Acad. Sci. USA 1994, 91, 2046–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.W. Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 2001, 1, 1397–1406. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt signaling and redox metabolism in cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function--role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef]
- Muriel, P.; Pérez-Rojas, J.M. Nitric oxide inhibits mitochondrial monoamine oxidase activity and decreases outer mitochondria membrane fluidity. Comp. Biochem. Physiol. Toxicol. Pharmacol. 2003, 136, 191–197. [Google Scholar] [CrossRef]
- van’t Hof, R.J.; Hocking, L.; Wright, P.K.; Ralston, S.H. Nitric oxide is a mediator of apoptosis in the rheumatoid joint. Rheumatology 2000, 39, 1004–1008. [Google Scholar] [CrossRef] [Green Version]
- Garg, N.; Syngle, A.; Krishan, P. Nitric oxide: Link between inflammation and endothelial dysfunction in rheumatoid arthritis. Int. J. Angiol. 2017, 26, 165–169. [Google Scholar] [CrossRef]
- Elizalde, M.; Rydén, M.; van Harmelen, V.; Eneroth, P.; Gyllenhammar, H.; Holm, C.; Ramel, S.; Ölund, A.; Arner, P.; Andersson, K. Expression of nitric oxide synthases in subcutaneous adipose tissue of nonobese and obese humans. J. Lipid Res. 2000, 41, 1244–1251. [Google Scholar] [PubMed]
- Yu, Y.; Park, S.J.; Beyak, M.J. Inducible nitric oxide synthase-derived nitric oxide reduces vagal satiety signalling in obese mice. J. Physiol. 2019, 597, 1487–1502. [Google Scholar] [CrossRef]
- Udi, S.; Hinden, L.; Ahmad, M.; Drori, A.; Iyer, M.R.; Cinar, R.; Herman-Edelstein, M.; Tam, J. Dual inhibition of cannabinoid CB1 receptor and inducible NOS attenuates obesity-induced chronic kidney disease. Br. J. Pharmacol. 2020, 177, 110–127. [Google Scholar] [CrossRef] [Green Version]
- Pawlik, A.; Błaszczyk, H.; Rać, M.; Maciejewska-Skrendo, A.; Safranow, K.; Dziedziejko, V. NOS3 gene rs1799983 and rs2070744 polymorphisms in patients with unstable angina. J. Vasc. Res. 2020, 57, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Miranda, J.A.; Belo, V.A.; Souza-Costa, D.C.; Lanna, C.M.; Tanus-Santos, J.E. eNOS polymorphism associated with metabolic syndrome in children and adolescents. Mol. Cell Biochem. 2013, 372, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, T.G.; Tibana, R.A.; Nascimento, D.D.; de Sousa, N.M.; de Souza, V.C.; Vieira, D.C.; Nóbrega, O.T.; de Almeida, J.A.; Navalta, J.; Prestes, J. Endothelial nitric oxide synthase Glu298Asp gene polymorphism influences body composition and biochemical parameters but not the nitric oxide response to eccentric resistance exercise in elderly obese women. Clin. Physiol. Funct. Imaging. 2016, 36, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Mezghenna, K.; Pomiès, P.; Chalançon, A.; Castex, F.; Leroy, J.; Niclauss, N.; Nadal, B.; Cambier, L.; Cazevieille, C.; Petit, P.; et al. Increased neuronal nitric oxide synthase dimerisation is involved in rat and human pancreatic beta cell hyperactivity in obesity. Diabetologia 2011, 54, 2856–2866. [Google Scholar] [CrossRef] [Green Version]
- Henningsson, R.; Salehi, A.; Lundquist, I. Role of nitric oxide synthase isoforms in glucose-stimulated insulin release. Am. J. Physiol. Cell Physiol. 2002, 283, C296–C304. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Feltstrom, J.; Lundquist, I.; Salehi, A. Glucose stimulates the expression and activities of nitric oxide synthases in incubated rat islets: An effect counteracted by GLP-1 through the cyclic AMP/PKA pathway. Cell Tissue Res. 2005, 319, 221–230. [Google Scholar] [CrossRef]
- Muniyappa, R.; Sowers, J.R. Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef]
- Kaneko, Y.K.; Ishikawa, T. Dual role of nitric oxide in pancreatic β-cells. J. Pharmacol. Sci. 2013, 123, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Balakirev, M.Y.; Khramtsov, V.V.; Zimmer, G. Modulation of the mitochondrial permeability transition by nitric oxide. Eur. J. Biochem. 1997, 246, 710–718. [Google Scholar] [CrossRef]
- Vieira, H.; Kroemer, G. Mitochondria as targets of apoptosis regulation by nitric oxide. IUBMB Life 2003, 55, 613–616. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes mellitus, mitochondrial dysfunction and Ca2+-dependent permeability transition pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef] [PubMed]
- Mosén, H.; Ostenson, C.G.; Lundquist, I.; Alm, P.; Henningsson, R.; Jimenez-Feltström, J.; Guenifi, A.; Efendic, S.; Salehi, A. Impaired glucose-stimulated insulin secretion in the GK rat is associated with abnormalities in islet nitric oxide production. Regul. Pept. 2008, 151, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.Y.; Tribble, N.D.; Kraft, C.A.; Markwardt, M.; Gloyn, A.L.; Rizzo, M.A. Naturally occurring glucokinase mutations are associated with defects in posttranslational S-nitrosylation. Mol. Endocrinol. 2010, 24, 171–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, S.; Ishikawa, T.; Yamamoto, Y.; Kaneko, Y.; Nakayama, K. Constitutive nitric oxide synthases in rat pancreatic islets: Direct imaging of glucose-induced nitric oxide production in beta-cells. Pflugers Arch. 2003, 447, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Tsuura, Y.; Ishida, H.; Hayashi, S.; Sakamoto, K.; Horie, M.; Seino, Y. Nitric oxide opens ATP-sensitive K+ channels through suppression of phosphofructokinase activity and inhibits glucose-induced insulin release in pancreatic beta cells. J. Gen. Physiol. 1994, 104, 1079–1098. [Google Scholar] [CrossRef]
- Klatt, P.; Heinzel, B.; John, M.; Kastner, M.; Böhme, E.; Mayer, B. Ca2+/calmodulin-dependent cytochrome c reductase activity of brain nitric oxide synthase. J. Biol. Chem. 1992, 267, 11374–11378. [Google Scholar] [PubMed]
- Gheibi, S.; Ghasemi, A. Insulin secretion: The nitric oxide controversy. EXCLI J. 2020, 19, 1227–1245. [Google Scholar] [CrossRef]
- Muhammed, S.J.; Lundquist, I.; Salehi, A. Pancreatic β-cell dysfunction, expression of iNOS and the effect of phosphodiesterase inhibitors in human pancreatic islets of type 2 diabetes. Diabetes Obes. Metab. 2012, 14, 1010–1019. [Google Scholar] [CrossRef]
- Bahadoran, Z.; Mirmiran, P.; Ghasemi, A. Role of nitric oxide in insulin secretion and glucose metabolism. Trends Endocrinol. Metab. 2020, 31, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Corbett, J.A.; Sweetland, M.A.; Wang, J.L.; Lancaster, J.R., Jr.; McDaniel, M.L. Nitric oxide mediatescytokine-induced inhibition of insulin secretion byhuman islets of Langerhans. Proc. Natl Acad. Sci. USA 1993, 90, 1731–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagiotidis, G.; Akesson, B.; Rydell, E.L.; Lundquist, I. Influence of nitric oxide synthase inhibition, nitric oxide and hydroperoxide on insulin release induced by various secretagogues. Br. J. Pharmacol. 1995, 114, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Song, E.H.; Lee, H.J.; Oh, Y.K.; Park, Y.S.; Park, J.-W.; Kim, B.J.; Kim, D.J.; Lee, I.; Song, J.; et al. Chronic ethanol consumption-induced pancreaticb-cell dysfunction and apoptosisthrough glucokinase nitration and its down-regulation. J. Biol. Chem. 2010, 285, 37251–37262. [Google Scholar] [CrossRef] [Green Version]
- Fioramonti, X.; Marsollier, N.; Song, Z.; Fakira, K.A.; Patel, R.M.; Brown, S.; Duparc, T.; Pica-Mendez, A.; Sanders, N.M.; Knauf, C.; et al. Ventromedial hypothalamic nitric oxide production is necessary for hypoglycemia detection and counterregulation. Diabetes 2010, 59, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Shankar, R.; Zhu, J.S.; Ladd, B.; Henry, D.; Shen, H.Q.; Baron, A.D. Central nervous system nitric oxide synthase activity regulates insulin secretion and insulin action. J. Clin. Investig. 1998, 102, 1403–1412. [Google Scholar] [CrossRef]
- Chen, W.; Balland, E.; Cowley, M.A. Hypothalamic insulin resistance in obesity: Effects on glucose homeostasis. Neuroendocrinology 2017, 104, 364–381. [Google Scholar] [CrossRef]
- Dhananjayan, R.; Koundinya, K.S.; Malati, T.; Kutala, V.K. Endothelial dysfunction in type 2 diabetes mellitus. Indian. J. Clin. Biochem. 2016, 31, 372–379. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, Y.; Lerman, A. Endothelial dysfunction and coronary artery disease: Assessment, prognosis, and treatment. Coron. Artery Dis. 2014, 25, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Sansbury, B.E.; Hill, B.G. Regulation of obesity and insulin resistance by nitric oxide. Free Radic. Biol. Med. 2014, 73, 383–399. [Google Scholar] [CrossRef] [Green Version]
- Rippin, J.D.; Patel, A.; Belyaev, N.D.; Gill, G.V.; Barnett, A.H.; Bain, S.C. Nitric oxide synthase gene polymorphisms and diabetic nephropathy. Diabetologia 2003, 46, 426–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoukry, A.; Shalaby, S.M.; Abdelazim, S.; Abdelazim, M.; Ramadan, A.; Ismail, M.I.; Fouad, M. Endothelial nitric oxide synthase gene polymorphisms and the risk of diabetic nephropathy in type 2 diabetes mellitus. Genet. Test Mol. Biomark. 2012, 16, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.E.; Bryant, S.M.; Ashley, E.A.; Lygate, C.A.; Rakovic, S.; Wallis, H.L.; Neubauer, S.; Terrar, D.A.; Casadei, B. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ. Res. 2003, 92, e52–e59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.H.; Zhang, M.H.; Sears, C.E.; Emanuel, K.; Redwood, C.; El-Armouche, A.; Kranias, E.G.; Casadei, B. Reduced phospholamban phosphorylation is associated with impaired relaxation in left ventricular myocytes from neuronal NO synthase-deficient mice. Circ. Res. 2008, 102, 242–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, D.E.; Lu, X.; Lei, M.; Xiang, F.L.; Hammoud, L.; Jiang, M.; Wang, H.; Jones, D.L.; Sims, S.M.; Feng, Q. Neuronal nitric oxide synthase protects against myocardial infarction-induced ventricular arrhythmia and mortality in mice. Circulation 2009, 120, 1345–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schöneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef]
- Torres, J.; Darley-Usmar, V.; Wilson, M.T. Inhibition of cytochrome c oxidase in turnover by nitric oxide: Mechanism and implications for control of respiration. Biochem. J. 1995, 312, 169–173. [Google Scholar] [CrossRef] [Green Version]
- Welter, R.; Yu, L.; Yu, C.A. The effects of nitric oxide on electron transport complexes. Arch. Biochem. Biophys. 1996, 331, 9–14. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cochemé, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Strasen, J.; Ritter, O. Role of nNOS in cardiac ischemia-reperfusion injury. Trends Cardiovasc. Med. 2011, 21, 58–63. [Google Scholar] [CrossRef]
- Saraiva, R.M.; Minhas, K.M.; Raju, S.V.; Barouch, L.A.; Pitz, E.; Schuleri, K.H.; Vandegaer, K.; Li, D.; Hare, J.M. Deficiency of neuronal nitric oxide synthase increases mortality and cardiac remodeling after myocardial infarction: Role of nitroso-redox equilibrium. Circulation 2005, 112, 3415–3422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damy, T.; Ratajczak, P.; Shah, A.M.; Camors, E.; Marty, I.; Hasenfuss, G.; Marotte, F.; Samuel, J.L.; Heymes, C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet 2004, 363, 1365–1367. [Google Scholar] [CrossRef]
- Carnicer, R.; Crabtree, M.J.; Sivakumaran, V.; Casadei, B.; Kass, D.A. Nitric oxide synthases in heart failure. Antioxid. Redox Signal. 2013, 18, 1078–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.H.; Jin, C.Z.; Jang, J.H.; Wang, Y. Molecular mechanisms of neuronal nitric oxide synthase in cardiac function and pathophysiology. J. Physiol. 2014, 592, 3189–3200. [Google Scholar] [CrossRef]
- Kar, R.; Kellogg, D.L., 3rd; Roman, L.J. Oxidative stress induces phosphorylation of neuronal NOS in cardiomyocytes through AMP-activated protein kinase (AMPK). Biochem. Biophys. Res. Commun. 2015, 459, 393–397. [Google Scholar] [CrossRef] [Green Version]
- Nishijima, Y.; Sridhar, A.; Bonilla, I.; Velayutham, M.; Khan, M.; Terentyeva, R.; Li, C.; Kuppusamy, P.; Elton, T.S.; Terentyev, D.; et al. Tetrahydrobiopterin depletion and NOS2 uncoupling contribute to heart failure-induced alterations in atrial electrophysiology. Cardiovasc. Res. 2011, 91, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lu, X.; Xiang, F.L.; Poelmann, R.E.; Gittenberger-de Groot, A.C.; Robbins, J.; Feng, Q. Nitric oxide synthase-3 deficiency results in hypoplastic coronary arteries and postnatal myocardial infarction. Eur. Heart J. 2014, 35, 920–931. [Google Scholar] [CrossRef] [Green Version]
- Levinsson, A.; Olin, A.C.; Björck, L.; Rosengren, A.; Nyberg, F. Nitric oxide synthase (NOS) single nucleotide polymorphisms are associated with coronary heart disease and hypertension in the INTERGENE study. Nitric. Oxide 2014, 39, 1–7. [Google Scholar] [CrossRef]
- Salimi, S.; Firoozrai, M.; Nourmohammadi, I.; Shabani, M.; Shafiee, S.M.; Mohebbi, A.; Tavilani, H. Lack of evidence for contribution of intron4a/b polymorphism of endothelial nitric oxide synthase (NOS3) gene to plasma nitric oxide levels. Acta Cardiol. 2008, 63, 229–234. [Google Scholar] [CrossRef]
- Chen, F.; Li, Y.M.; Yang, L.Q.; Zhong, C.G.; Zhuang, Z.X. Association of NOS2 and NOS3 gene polymorphisms with susceptibility to type 2 diabetes mellitus and diabetic nephropathy in the Chinese Han population. IUBMB Life 2016, 68, 516–525. [Google Scholar] [CrossRef] [Green Version]
- Garme, Y.; Moudi, M.; Saravani, R.; Galavi, H. Nitric oxide synthase 2 polymorphisms (rs2779248T/C and rs1137933C/T) and the risk of type 2 diabetes in zahedan, southeastern iran. Iran J. Public Health 2018, 47, 1734–1741. [Google Scholar] [PubMed]
- Möllsten, A.; Wessman, M.; Svensson, M.; Forsblom, C.; Parkkonen, M.; Brismar, K.; Groop, P.H.; Dahlquist, G. Glu298Asp and NOS4ab polymorphisms in diabetic nephropathy. Ann. Med. 2006, 38, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lin, Y.; Zhang, R. Associations between endothelial nitric oxide synthase gene polymorphisms and the risk of coronary artery disease: A systematic review and meta-analysis of 132 case-control studies. Eur. J. Prev. Cardiol. 2019, 26, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lynch, A.I.; Davis, B.R.; Ford, C.E.; Boerwinkle, E.; Eckfeldt, J.H.; Leiendecker-Foster, C.; Arnett, D.K. Pharmacogenetic association of NOS3 variants with cardiovascular disease in patients with hypertension: The GenHAT study. PLoS ONE 2012, 7, e34217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garvey, E.P.; Oplinger, J.A.; Tanoury, G.J.; Sherman, P.A.; Fowler, M.; Marshall, S.; Harmon, M.F.; Paith, J.E.; Furfine, E.S. Potent and selective inhibition of human nitric oxide synthases. Inhibition by non-amino acid isothioureas. J. Biol. Chem. 1994, 269, 26669–26676. [Google Scholar] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Garvey, E.P.; Oplinger, J.A.; Furfine, E.S.; Kiff, R.J.; Laszlo, F.; Whittle, B.J.; Knowles, R.G. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997, 272, 4959–4963. [Google Scholar] [CrossRef] [Green Version]
- Silverman, R.B. Design of selective neuronal nitric oxide synthase inhibitors for the prevention and treatment of neurodegenerative diseases. Acc. Chem. Res. 2009, 42, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Annedi, S.C. Cell-Permeable inhibitors of neuronal nitric oxide synthase open new prospects for the treatment of neurological disorders. J. Med. Chem. 2015, 58, 1064–1066. [Google Scholar] [CrossRef]
- Paige, J.S.; Jaffrey, S.R. Pharmacologic manipulation of nitric oxide signaling: Targeting NOS dimerization and protein-protein interactions. Curr. Top. Med. Chem. 2007, 7, 97–114. [Google Scholar] [CrossRef] [Green Version]
- Poulos, T.L.; Li, H. Structural basis for isoform-selective inhibition in nitric oxide synthase. Acc. Chem. Res. 2013, 46, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Leiper, J. Blocking NO synthesis: How, where and why? Nat. Rev. Drug Discov. 2002, 1, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.H.; Reynolds, H.R.; Stebbins, A.L.; Dzavik, V.; Harrington, R.A.; Van der Werf, F.; Hechman, J.S. Effect of tilarginine acetate in patients with acute myocardial infarction and cardiogenic shock: The TRIUMPH randomized controlled trial. J. Am. Med Assoc. 2007, 297, 1657–1666. [Google Scholar] [CrossRef]
- Wong, V.W.; Lerner, E. Nitric oxide inhibition strategies. Future Sci. OA 2015, 1, FSO35. [Google Scholar] [CrossRef]
- Dzavík, V.; Cotter, G.; Reynolds, H.R.; Alexander, J.H.; Ramanathan, K.; Stebbins, A.L.; Hathaway, D.; Farkouh, M.E.; Ohman, E.M.; Baran, D.A.; et al. Should we inhibit nitric Oxide synthase in Cardiogenic shocK 2 (SHOCK-2) investigators. Effect of nitric oxide synthase inhibition on haemodynamics and outcome of patients with persistent cardiogenic shock complicating acute myocardial infarction: A phase II dose-ranging study. Eur. Heart J. 2007, 28, 1109–1116. [Google Scholar] [CrossRef]
- Cottera, G.; Kaluskia, E.; Miloa, O.; Blatta, A.; Salaha, A.; Hendlera, A.; Krakovera, R.; Golickb, A.; Vereda, Z. LINCS: L-NAME (a NO synthase inhibitor) in the treatment of refractory cardiogenic shock: A prospective randomized study. Eur. Heart J. 2003, 24, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Yargiçoglu, P.; Yaraş, N.; Agar, A.; Gümüşlü, S.; Abidin, I.; Bilmen, S. Effects of N-nitro l-arginine methyl ester (l-NAME), a potent nitric oxide synthase inhibitor, on visual evoked potentials of rats exposed to different experimental stress models. Acta Physiol. Scand. 2004, 180, 307–316. [Google Scholar] [CrossRef]
- Sibal, L.; Agarwal, S.C.; Home, P.D.; Boger, R.H. The role of asymmetric dimethylarginine (ADMA) in endothelial dysfunction and cardiovascular disease. Curr. Cardiol. Rev. 2010, 6, 82–90. [Google Scholar] [CrossRef]
- Böger, R.H. Asymmetric dimethylarginine (ADMA) and cardiovascular disease: Insights from prospective clinical trials. Vasc. Med. 2005, 10, S19–S25. [Google Scholar] [CrossRef] [Green Version]
- Grover, R.; Zaccardelli, D.; Colice, G.; Guntupalli, K.; Watson, D.; Vincent, J.L. An open-label dose escalation study of the nitric oxide synthase inhibitor, N(G)-methyl-L-arginine hydrochloride (546C88), in patients with septic shock. Glaxo Wellcome International Septic Shock Study Group. Crit. Care Med. 1999, 27, 913–922. [Google Scholar] [CrossRef]
- Hauser, B.; Bracht, H.; Matejovic, M.; Radermacher, P.; Venkatesh, B. Nitric oxide synthase inhibition in sepsis? Lessons learned from large-animal studies. Anesth. Analg. 2005, 101, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Alabadí, J.A.; Thibault, J.L.; Pinard, E.; Seylaz, J.; Lasbennes, F. 7-Nitroindazole, a selective inhibitor of nNOS, increases hippocampal extracellular glutamate concentration in status epilepticus induced by kainic acid in rats. Brain Res. 1999, 839, 305–312. [Google Scholar] [CrossRef]
- Bush, M.A.; Pollack, G.M. Pharmacokinetics and pharmacodynamics of 7-nitroindazole, a selective nitric oxide synthase inhibitor, in the rat hippocampus. Pharm. Res. 2001, 18, 1607–1612. [Google Scholar] [CrossRef] [PubMed]
- Soliman, M.M. Effects of aminoguanidine, a potent nitric oxide synthase inhibitor, on myocardial and organ structure in a rat model of hemorrhagic shock. J. Emerg. Trauma. Shock. 2014, 7, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Ghosh, T.; Ghosh, R.; Gupta, P. Nitric oxide synthase inhibitor, aminoguanidine reduces intracerebroventricular colchicine induced neurodegeneration, memory impairments and changes of systemic immune responses in rats. J. Neuroimmunol. 2017, 303, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Salem, R.; Mebazaa, A. Nitric oxide inhibition rapidly increases blood pressure with no change in outcome in cardiogenic shock: The TRIUMPH trial. Crit. Care 2007, 11, 136. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Molecule Name | Summary Formula | Reactivity | Characteristics |
|---|---|---|---|
| nitrosyl cation | NO+ | a strong oxidizing agent | Intermediate in the amine diazotization reaction |
| nitrosyl anion | NO− | a strong oxidizing agent | Participates in the nitrosylation reaction of metals, forming metal nitrosyl complexes |
| nitrogen dioxide | •NO2 | a strong oxidizing agent | A good oxidizer; it combusts, sometimes explosively, with many compounds, such as hydrocarbons |
| dinitrogen trioxide | N2O3 | a strong oxidizing agent | Partially dissociates into NO and NO2; vapors very toxic by inhalation; reactivity likely to resemble that of nitrogen dioxide |
| peroxynitrite | ONOO− | highly reactive; a very strong oxidant and nitrating agent | Essentially stable but its protonated form (ONOOH) decomposes rapidly via homolysis of the O-O bond to form about 28% free NO2 and OH radicals |
| nitrite | NO2− | very reactive; a member of reactive nitrogen species | A nitrogen oxoanion formed by loss of a proton from nitrous acid; used for NO measurement |
| nitrate | NO3- | very reactive; a member of reactive nitrogen species | A nitrogen oxoanion formed by loss of a proton from nitric acid; used for NO measurement |
| nitroxyl | HNO | very reactive towards nucleophiles, including thiols | A weak acid; can be formed as a short-lived intermediate in the solution phase |
| Polymorphism | Isoform | Location | Disease | References |
|---|---|---|---|---|
| rs3782218 | NOS1 | C2637T | ischemic heart disease, hypertension | [118] |
| rs2682826 | NOS1 | C276T | ischemic heart disease | [118] |
| rs2779248 | NOS2 | T278C | type 2 diabetes | [120,121] |
| rs1137933 | NOS2 | C231T | type 2 diabetes | [120,121] |
| Tandem repeat polymorphism (VNTR) | NOS3 | 27-bp VNTR | diabetic nephropathy, acute eccentric resistance exercise, metabolic syndrome, atherosclerosis, coronary artery disease | [101,102,119] |
| rs1799983 | NOS3 | G894T | diabetic nephropathy | [73,102,122] |
| rs2070744 | NOS3 | T786C | diabetic nephropathy | [71,72,102] |
| rs1549758 | NOS3 | C774T | ischemic heart disease, coronary artery disease | [118,123] |
| rs3918226 | NOS3 | C665T | hypertension | [118,124] |
| Inhibitor Name | Country/Continent | Application in Clinical Test/Research | References |
|---|---|---|---|
| Tilarginine (L-NMMA) | North America, Europe | Patients with cardiogenic shock; patients with breast cancer | [134,135,136] |
| N(G)-nitro-L-arginine methyl ester (L-NAME) | North America, Asia | Patients with cardiogenic shock; patients with septic shock | [137,138] |
| Asymmetric dimethylarginine (ADMA) | Europe | Possible use as a cardiovascular risk factor | [139,140] |
| N(G)-methyl-l-arginine hydrochloride | Europe | Used to restore the balance of vasomotor tone in patients with septic shock | [141,142] |
| 7-nitroindazole (7-NI) | Europe, North America | Anticonvulsive properties in seizure models in rodents | [143,144] |
| Aminoguanidine | Asia | Alleviation of graft-versus-host disease in mice; alleviation of the susceptibility of mice to bacterial infections | [145,146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Król, M.; Kepinska, M. Human Nitric Oxide Synthase—Its Functions, Polymorphisms, and Inhibitors in the Context of Inflammation, Diabetes and Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 56. https://doi.org/10.3390/ijms22010056
Król M, Kepinska M. Human Nitric Oxide Synthase—Its Functions, Polymorphisms, and Inhibitors in the Context of Inflammation, Diabetes and Cardiovascular Diseases. International Journal of Molecular Sciences. 2021; 22(1):56. https://doi.org/10.3390/ijms22010056
Chicago/Turabian StyleKról, Magdalena, and Marta Kepinska. 2021. "Human Nitric Oxide Synthase—Its Functions, Polymorphisms, and Inhibitors in the Context of Inflammation, Diabetes and Cardiovascular Diseases" International Journal of Molecular Sciences 22, no. 1: 56. https://doi.org/10.3390/ijms22010056
APA StyleKról, M., & Kepinska, M. (2021). Human Nitric Oxide Synthase—Its Functions, Polymorphisms, and Inhibitors in the Context of Inflammation, Diabetes and Cardiovascular Diseases. International Journal of Molecular Sciences, 22(1), 56. https://doi.org/10.3390/ijms22010056