
Intermittent Hypobaric Hypoxic Preconditioning Provides Neuroprotection by Increasing Antioxidant Activity, Erythropoietin Expression and Preventing Apoptosis and Astrogliosis in the Brain of Adult Rats Exposed to Acute Severe Hypoxia


 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Oxidative Stress
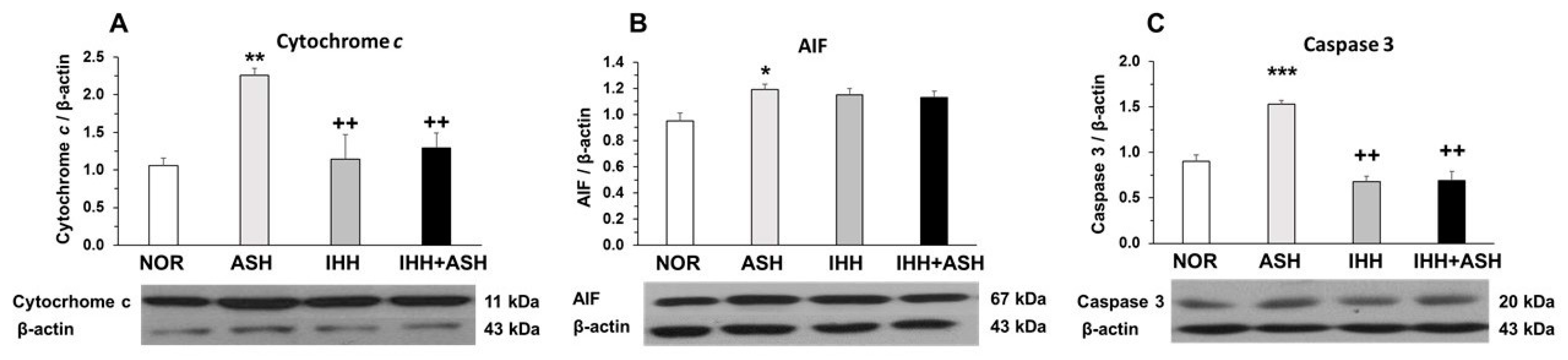
2.2. Mitochondrial Apoptosis
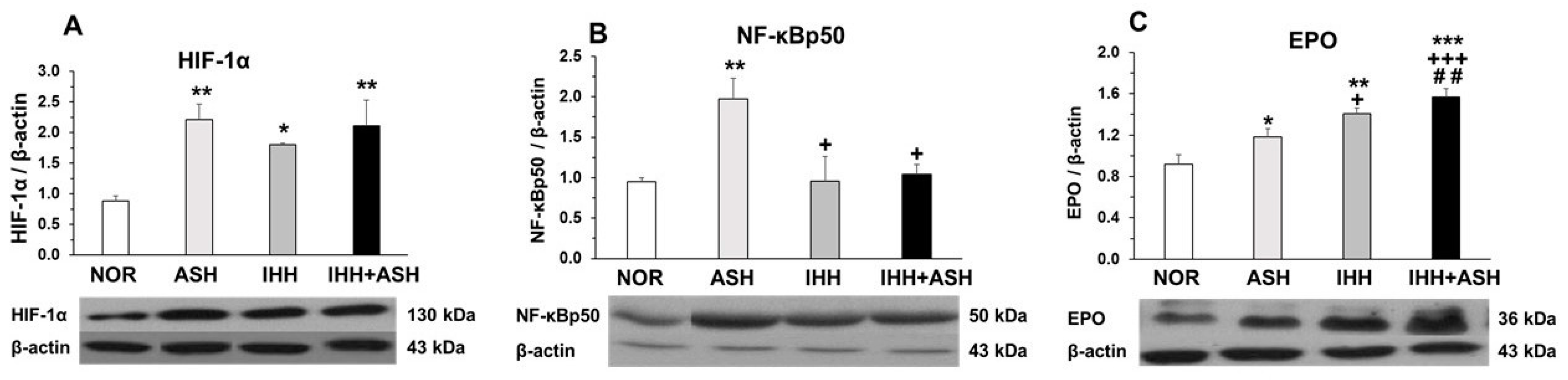
2.3. Transcriptional Factors in Response to Hypoxia
2.4. Erythropoietin (EPO) Expression in the Brain
2.5. Astrocytes
3. Discussion
3.1. Oxidative Stress
3.2. Mitochondrial Apoptosis
3.3. Transcriptional Factors in Response to Hypoxia
3.4. Erythropoietin (EPO) Expression in the Brain
3.5. Astrocytes
4. Materials and Methods
4.1. Animals and Experimental Groups
4.2. Oxidative Stress Assays
4.2.1. Measurement of Oxidant Activity in the Brain
4.2.2. Measurement of Antioxidant Activity in the Brain
4.3. Western Blotting
4.4. Immunohistochemistry
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIF | Apoptosis-inducing factor |
| AOPP | Advanced oxidation protein products |
| ASH | Acute severe hypoxia |
| CA1 | Hippocampus Cornu ammonis area 1 |
| CNS | Central nervous system |
| EPO | Erythropoietin |
| GFAP | Glial fibrillary acidic protein |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| GSH | Reduced glutathione |
| GSSG | Oxidized glutathione |
| HIF | Hypoxia-inducible factor |
| IHH | Intermittent hypobaric hypoxia |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NOR | Normoxic |
| NOx | Nitric oxide species |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| TBARS | Thiobarbituric acid reactive substances (lipid peroxidation) |
References
- Acker, T.; Acker, H. Cellular oxygen sensing need in CNS function: Physiological and pathological implications. J. Exp. Biol. 2004, 207, 3171–3188. [Google Scholar] [CrossRef] [Green Version]
- Maa, E.H. Hypobaric hypoxic cerebral insult: The neurological consequences of going higher. NeuroRehabilitation 2010, 26, 73–84. [Google Scholar] [CrossRef]
- Wilson, M.H.; Newman, S.; Imray, C.H. The cerebral effects of ascent to high altitudes. Lancet Neurol. 2009, 8, 175–191. [Google Scholar] [CrossRef]
- Francis, K.R.; Wei, L. Human embryonic stem cell neural differentiation and enhanced cell survival promoted by hypoxic preconditioning. Cell Death Dis. 2010, 1, e22. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.C.; Keith, B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008, 9, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.; Lizasoain, I.; Sobrino, T.; Vivancos, J.; Castillo, J. Ischemic preconditioning: A novel target for neuroprotective therapy. Cerebrovasc. Dis. 2006, 21, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Rybnikova, E.; Samoilov, M. Current insights into the molecular mechanisms of hypoxic pre- and postconditioning using hypobaric hypoxia. Front. Neurosci. 2015, 9, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, R.; Xu, H.; Lu, A.; Bernaudin, M.; Sharp, F.R. Hypoxia preconditioning in the brain. Dev. Neurosci. 2005, 27, 87–92. [Google Scholar] [CrossRef]
- Li, S.; Hafeez, A.; Noorulla, F.; Geng, X.; Guo, G.; Ren, C.; Lu, G.; Zhao, H.; Ding, Y.; Ji, X. Preconditioning in neuroprotection: From hypoxia to ischemia. Prog. Neurobiol. 2017, 157, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirnagl, U.; Becker, K.; Meisel, A. Preconditioning and tolerance against cerebral ischaemia: From experimental strategies to clinical use. Lancet Neurol. 2009, 8, 398–412. [Google Scholar] [CrossRef] [Green Version]
- Sharp, F.R.; Ran, R.; Lu, A.; Tang, Y.; Strauss, K.I.; Glass, T.; Ardizzone, T.; Bernaudin, M. Hypoxic preconditioning protects against ischemic brain injury. NeuroRX 2004, 1, 26–35. [Google Scholar] [CrossRef]
- Rybnikova, E.A.; Khozhai, L.I.; Tyul’kova, E.I.; Glushchenko, T.S.; Sitnik, N.A.; PeltoHuikko, M.; Otellin, V.A.; Samoilov, M.O. Expression of early gene proteins, structural changes in brain neurons in hypobaric hypoxia, and the correcting effects of preconditioning. Neurosci. Behav. Physiol. 2005, 35, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Rybnikova, E.; Sitnik, N.; Gluschenko, T.; Tjulkova, E.; Samoilov, M.O. The preconditioning modified neuronal expression of apoptosis-related proteins of Bcl-2 superfamily following severe hypobaric hypoxia in rats. Brain Res. 2006, 1089, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Rybnikova, E.; Gluschenko, T.; Tulkova, E.; Churilova, A.; Jaroshevich, O.; Baranova, K.; Samoilov, M. Preconditioning induces prolonged expression of transcription factors pCREB and NF-kappa B in the neocortex of rats before and following severe hypobaric hypoxia. J. Neurochem. 2008, 106, 1450–1458. [Google Scholar]
- Stroev, S.A.; Gluschenko, T.S.; Tjulkova, E.I.; Rybnikova, E.A.; Samoilov, M.O.; Pelto-Huikko, M. The effect of preconditioning on the Cu, Zn superoxide dismutase expression and enzyme activity in rat brain at the early period after severe hypobaric hypoxia. Neurosci. Res. 2005, 53, 39–47. [Google Scholar] [CrossRef]
- Fan, X.; Wang, H.; Zhang, L.; Tang, J.; Qu, Y.; Mu, D. Neuroprotection of hypoxic/ischemic preconditioning in neonatal brain with hypoxic/ischemic injury. Rev. Neurosci. 2021, 32, 23–34. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, X.; Liang, J.; Han, S.; Wang, Y.; Yin, Y.; Luo, Y.; Li, J. Phosphorylation of p38 MAPK mediates hypoxic preconditioning-induced neuroprotection against cerebral ischemic injury via mitochondria translocation of Bcl-xL in mice. Brain Res. 2013, 1503, 78–88. [Google Scholar] [CrossRef]
- Bernaudin, M.; Tang, Y.; Reilly, M.; Petit, E.; Sharp, F.R. Brain genomic response following hypoxia and re-oxygenation in the neonatal rat-Identification of genes that might contribute to hypoxia-induced ischemic tolerance. J. Biol. Chem. 2002, 277, 39728–39778. [Google Scholar] [CrossRef] [Green Version]
- Rabie, T.; Marti, H.H. Brain protection by erythropoietin: A manifold task. Physiology 2008, 23, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Yenari, M.A.; Cheng, D.; Sapolsky, R.M.; Steinberg, G.K. Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase-3 activity. J. Neurochem. 2003, 85, 1026–1036. [Google Scholar] [CrossRef]
- Kenneth, N.S.; Rocha, S. Regulation of gene expression by hypoxia. Biochem. J. 2008, 414, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D´Ignazio, L.; Rocha, S. Hypoxia induced NF-κB. Cells 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melvin, A.; Mudie, S.; Rocha, S. Mechanism of hypoxia-induced NF-κB. Cell Cycle 2011, 10, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Kasymov, V.; Christie, I.; Sheikhbahaei, S.; Turovsky, E.; Marina, N.; Korsak, A.; Zwicker, J.; Teschemacher, A.G.; Ackland, G.L.; et al. Functional oxygen sensitivity of astrocytes. J. Neurosci. 2015, 35, 10460–10473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Coimbra-Costa, D.; Alva, N.; Duran, M.; Carbonell, T.; Rama, R. Oxidative stress and apoptosis after acute respiratory hypoxia and reoxygenation in rat brain. Redox Biol. 2017, 12, 216–225. [Google Scholar] [CrossRef]
- Maiti, P.; Singh, S.B.; Sharma, A.K.; Muthuraju, S.; Banerjee, P.K.; Ilavazhagan, G. Hypobaric hypoxia induces oxidative stress in rat brain. Neurochem. Int. 2006, 49, 709–716. [Google Scholar] [CrossRef]
- Peña, F.; Ramirez, J. Hypoxia-induced changes in neuronal network properties. Mol. Neurobiol. 2005, 32, 251–283. [Google Scholar] [CrossRef]
- Terraneo, L.; Paroni, R.; Bianciardi, P.; Giallongo, T.; Carelli, S.; Gorio, A.; Samaja, M. Brain adaptation to hypoxia and hyperoxia in mice. Redox Biol. 2017, 11, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H2O2 signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metabol. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, P.; Singh, S.B.; Mallick, B.; Muthuraju, S.; Ilavazhagan, G. High altitude memory impairment is due to neuronal apoptosis in hippocampus, cortex and striatum. J. Chem. Neuroanat. 2008, 36, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Franklin, J. Redox regulation of the intrinsic pathway in neuronal apoptosis. Antioxid. Redox Signal. 2011, 14, 1437–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwell, B. Free radicals and antioxidants—quo vadis? Trends Pharmacol. Sci. 2011, 32, 125–130. [Google Scholar] [CrossRef]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive oxygen species formation in the brain at different oxygen levels: The role of hypoxia inducible factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta 2010, 1797, 1171–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langley, B.; Ratan, R.R. Oxidative stress-induced death in the nervous system: Cell cycle dependent or independent? J. Neurosci. Res. 2004, 77, 621–629. [Google Scholar] [CrossRef]
- Öxler, E.M.; Dolga, A.; Culmsee, C. AIF depletion provides neuroprotection through a preconditioning effect. Apoptosis 2012, 17, 1027–1038. [Google Scholar] [CrossRef]
- Miyamoto, O.; Auer, R.N. Hypoxia, hyperoxia, ischemia, and brain necrosis. Neurology 2000, 54, 362–371. [Google Scholar] [CrossRef] [Green Version]
- Pearigen, P.; Gwinn, R.; Simon, R. The effects in vivo of hypoxia on brain injury. Brain Res. 1996, 725, 184–191. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1 alpha during hypoxia-A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Görlach, A.; Dimova, E.Y.; Petry, A.; Martínez-Ruiz, A.; Hernansanz-Agustín, P.; Rolo, A.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Culver, C.; Sundqvist, A.; Mudie, S.; Melvin, A.; Xirodimas, D.; Rocha, S. Mechanism of hypoxia-induced NF-κB. Mol. Cel. Biol. 2010, 30, 4901–4921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravati, A.; Ahlemeyer, B.; Becker, A.; Klumpp, S.; Krieglstein, J. Preconditioning-induced neuroprotection is mediated by reactive oxygen species and activation of the transcription factor nuclear factor-κB. J. Neurochem. 2001, 78, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Rybnikova, E.; Glushchenko, T.; Tyulkova, E.; Baranova, K.; Samoilov, M. Mild hypobaric hypoxia preconditioning up-regulates expression of transcription factors c-Fos and NGFI-A in rat neocortex and hippocampus. Neurosci. Res. 2009, 65, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Gilmore, T.D. Good cop, bad cop: The different faces of NF-κB. Cell Death Differ. 2006, 13, 759–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Semenza, G.L. Molecular basis of hypoxia-induced erythropoietin expression. Curr. Opin. Hematol. 1996, 3, 156–162. [Google Scholar] [CrossRef]
- Jelkmann, W. Erythropoietin: Structure, control of production, and function. Physiol. Rev. 1992, 72, 449–489. [Google Scholar] [CrossRef]
- Gassmann, M.; Heinicke, K.; Soliz, J.; Ogunshola, O.O. Non-erythroid functions of erythropoietin. Adv. Exp. Med. Biol. 2003, 543, 323–330. [Google Scholar]
- Noguchi, C.T.; Asavaritikrai, P.; Teng, R.; Jia, Y. Role of erythropoietin in the brain. Crit. Rev. Oncol. Hematol. 2007, 64, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Narasimhan, P.; Yu, F.S.; Chan, P.H. Neuroprotection by hypoxic preconditioning involves oxidative stress-mediated expression of hypoxia-inducible factor and erythropoietin. Stroke 2005, 36, 1264–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garzón, F.; Rodriguez, Y.; García, J.C.; Rama, R. Neuroprotective effects of neuroEPO using an in vitro model of stroke. Behav. Sci. 2018, 8, 26. [Google Scholar]
- Garzón, F.; Coimbra, D.; Parcerisas, A.; Rodriguez, Y.; García, J.C.; Soriano, E.; Rama, R. NeuroEPO preserves neurons from glutamate-induced excitotoxicity. J. Alzheimers Dis. 2018, 65, 1469–1483. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.S. Erythropoietin and oxidative stress. Curr. Neurovasc. Res. 2008, 5, 125–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brines, M.; Cerami, A. Emerging biological roles for erythropoietin in the nervous system. Nat. Rev. Neurosci. 2005, 6, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. Regeneration in nervous system with erythropoietin. Front. Biosci. 2016, 21, 561–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Pekny, M.; Pekna, M. Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef]
- Vetrovoi, O.V.; Rybnikova, E.A.; Glushchenko, T.S.; Samoilov, M.O. Effects of hypobaric hypoxia in various modes on expression of neurogenesis marker NeuroD2 in the dentate gyrus of rats hippocampus. Bull. Exp. Biol. Med. 2016, 160, 510–513. [Google Scholar] [CrossRef]
- Mihara, M.; Uchiyama, M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal. Biochem. 1978, 86, 271–278. [Google Scholar]
- Barsotti, A.; Fabbi, P.; Fedele, M.; Garibaldi, S.; Balbi, M.; Bezante, G.P.; Risso, D.; Indiveri, F.; Ghigliotti, G.; Brunelli, C. Role of advanced oxidation protein products and thiol ratio in patients with acute coronary syndromes. Clin. Biochem. 2011, 44, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Rama, R.; Dávalos, A. Nitric oxide-related brain damage in acute ischemic stroke. Stroke 2000, 31, 852–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alva, N.; Carbonell, T.; Palomeque, J. Hypothermic protection in an acute hypoxia model in rats: Acid-base and oxidant/antioxidant profiles. Resuscitation 2010, 81, 609–616. [Google Scholar] [CrossRef]
- Ruiz-Montasell, B.; Aguado, F.; Majo, G.; Chapman, E.R.; Canals, J.M.; Marsal, J.; Blasi, J. Differential distribution of syntaxin isoforms 1A and 1B in the rat central nervous system. Eur. J. Neurosci. 1996, 8, 2544–2552. [Google Scholar] [CrossRef]
- Plá, V.; Paco, S.; Ghezali, G.; Ciria, V.; Pozas, E.; Ferrer, I.; Aguado, F. Secretory sorting receptors carboxypeptidase E and Secretogranin III in amyloid b-associated neural degeneration in Alzheimer’s disease. Brain Pathol. 2013, 23, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NOR | ASH | IHH | IHH+ASH | |
|---|---|---|---|---|
| Glutathione | ||||
| GSH (nmol·mg−1) | 1.60 ± 0.11 | 1.08 ± 0.04 ** | 1.26 ± 0.07 * | 1.19 ± 0.11 * |
| GSSG (nmol·mg−1) | 0.90 ± 0.10 | 1.11 ± 0.06 | 0.88 ± 0.02 | 1.04 ± 0.12 |
| GSH/GSSG | 1.63 ± 0.05 | 0.94 ± 0.10 *** | 1.43 ± 0.04 | 1.35 ± 0.13 |
| Enzymes | ||||
| GPx (µU·mg−1) | 15.4 ± 0.93 | 13.0 ± 0.49 * | 8.3 ± 0.93 *** +++ | 10.4 ± 0.62 *** ++ |
| GR (µU·mg−1) | 6.47 ± 0.50 | 4.28 ± 0.45 ** | 5.51 ± 0.28 + | 6.02 ± 0.53 ++ |
| SOD (U·mg−1) | 1.52 ± 0.12 | 1.10 ± 0.05 * | 1.30 ± 0.10 | 1.45 ± 0.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coimbra-Costa, D.; Garzón, F.; Alva, N.; Pinto, T.C.C.; Aguado, F.; Torrella, J.R.; Carbonell, T.; Rama, R. Intermittent Hypobaric Hypoxic Preconditioning Provides Neuroprotection by Increasing Antioxidant Activity, Erythropoietin Expression and Preventing Apoptosis and Astrogliosis in the Brain of Adult Rats Exposed to Acute Severe Hypoxia. Int. J. Mol. Sci. 2021, 22, 5272. https://doi.org/10.3390/ijms22105272
Coimbra-Costa D, Garzón F, Alva N, Pinto TCC, Aguado F, Torrella JR, Carbonell T, Rama R. Intermittent Hypobaric Hypoxic Preconditioning Provides Neuroprotection by Increasing Antioxidant Activity, Erythropoietin Expression and Preventing Apoptosis and Astrogliosis in the Brain of Adult Rats Exposed to Acute Severe Hypoxia. International Journal of Molecular Sciences. 2021; 22(10):5272. https://doi.org/10.3390/ijms22105272
Chicago/Turabian StyleCoimbra-Costa, Débora, Fernando Garzón, Norma Alva, Tiago C. C. Pinto, Fernando Aguado, Joan Ramon Torrella, Teresa Carbonell, and Ramón Rama. 2021. "Intermittent Hypobaric Hypoxic Preconditioning Provides Neuroprotection by Increasing Antioxidant Activity, Erythropoietin Expression and Preventing Apoptosis and Astrogliosis in the Brain of Adult Rats Exposed to Acute Severe Hypoxia" International Journal of Molecular Sciences 22, no. 10: 5272. https://doi.org/10.3390/ijms22105272
APA StyleCoimbra-Costa, D., Garzón, F., Alva, N., Pinto, T. C. C., Aguado, F., Torrella, J. R., Carbonell, T., & Rama, R. (2021). Intermittent Hypobaric Hypoxic Preconditioning Provides Neuroprotection by Increasing Antioxidant Activity, Erythropoietin Expression and Preventing Apoptosis and Astrogliosis in the Brain of Adult Rats Exposed to Acute Severe Hypoxia. International Journal of Molecular Sciences, 22(10), 5272. https://doi.org/10.3390/ijms22105272