
Discovery of a Necroptosis Inhibitor Improving Dopaminergic Neuronal Loss after MPTP Exposure in Mice

 ,
,  , , , ,
, , , , 
Abstract
:1. Introduction
2. Results
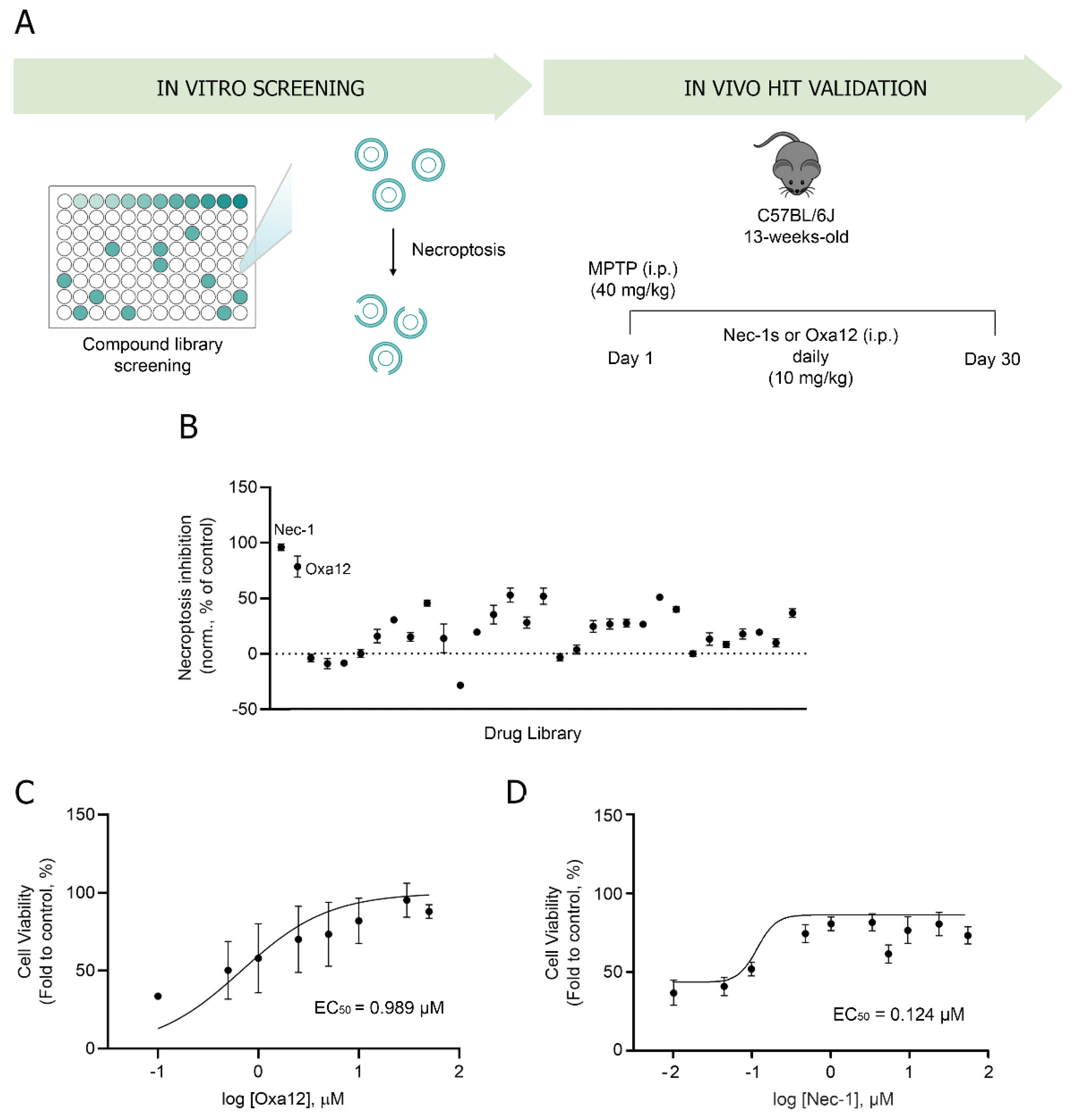
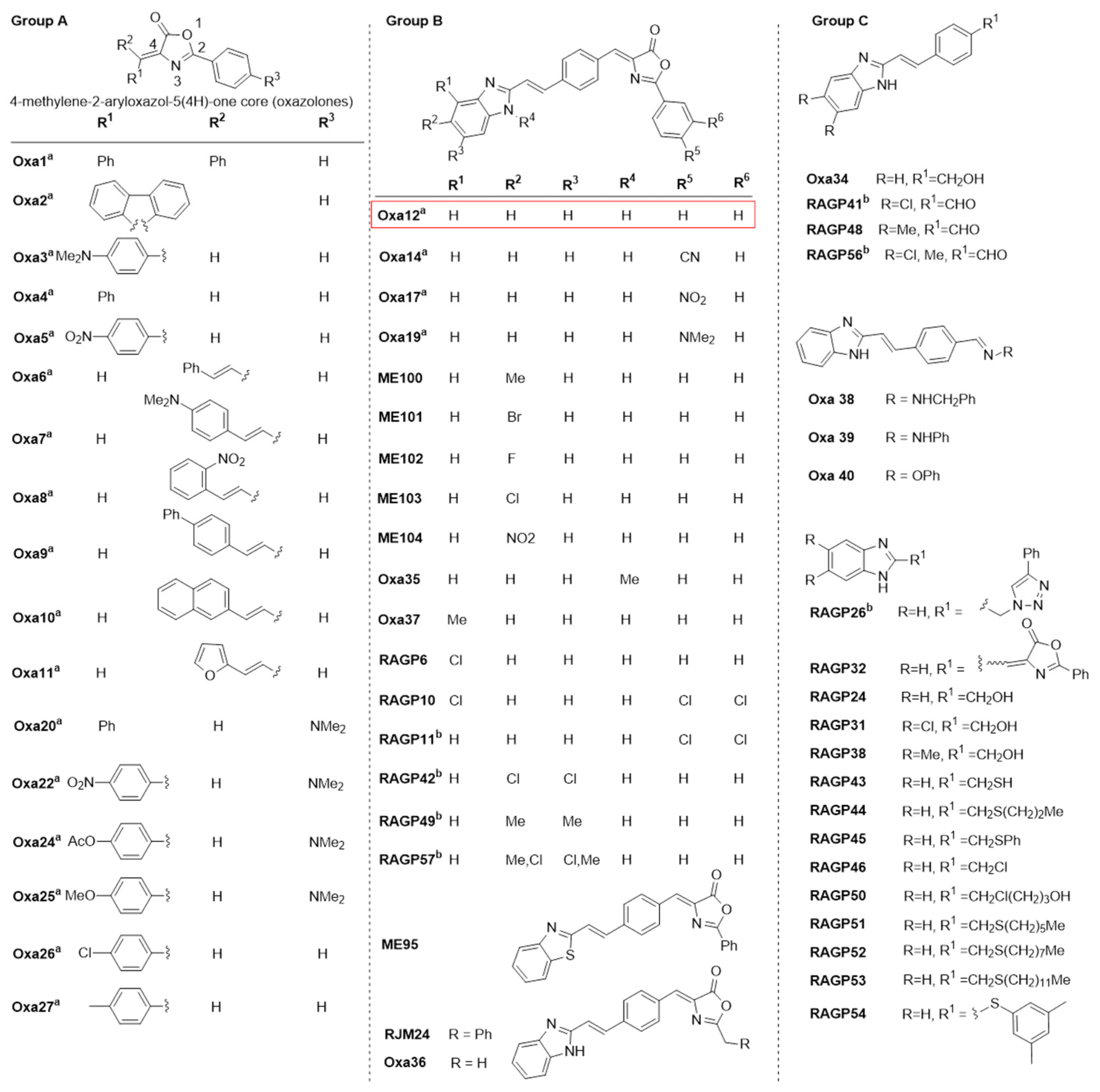
2.1. Phenotypic Screening for Hit Selection
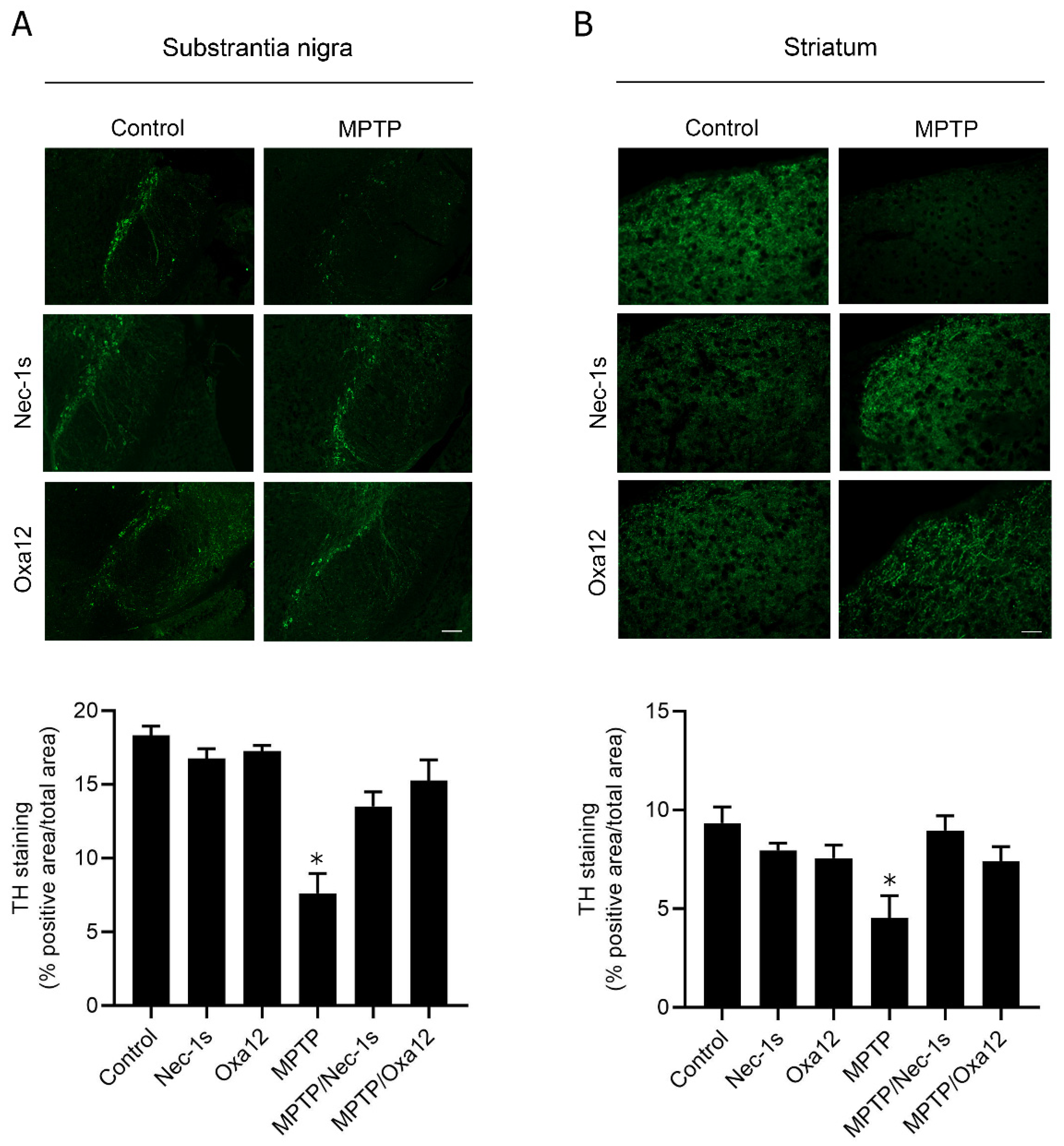
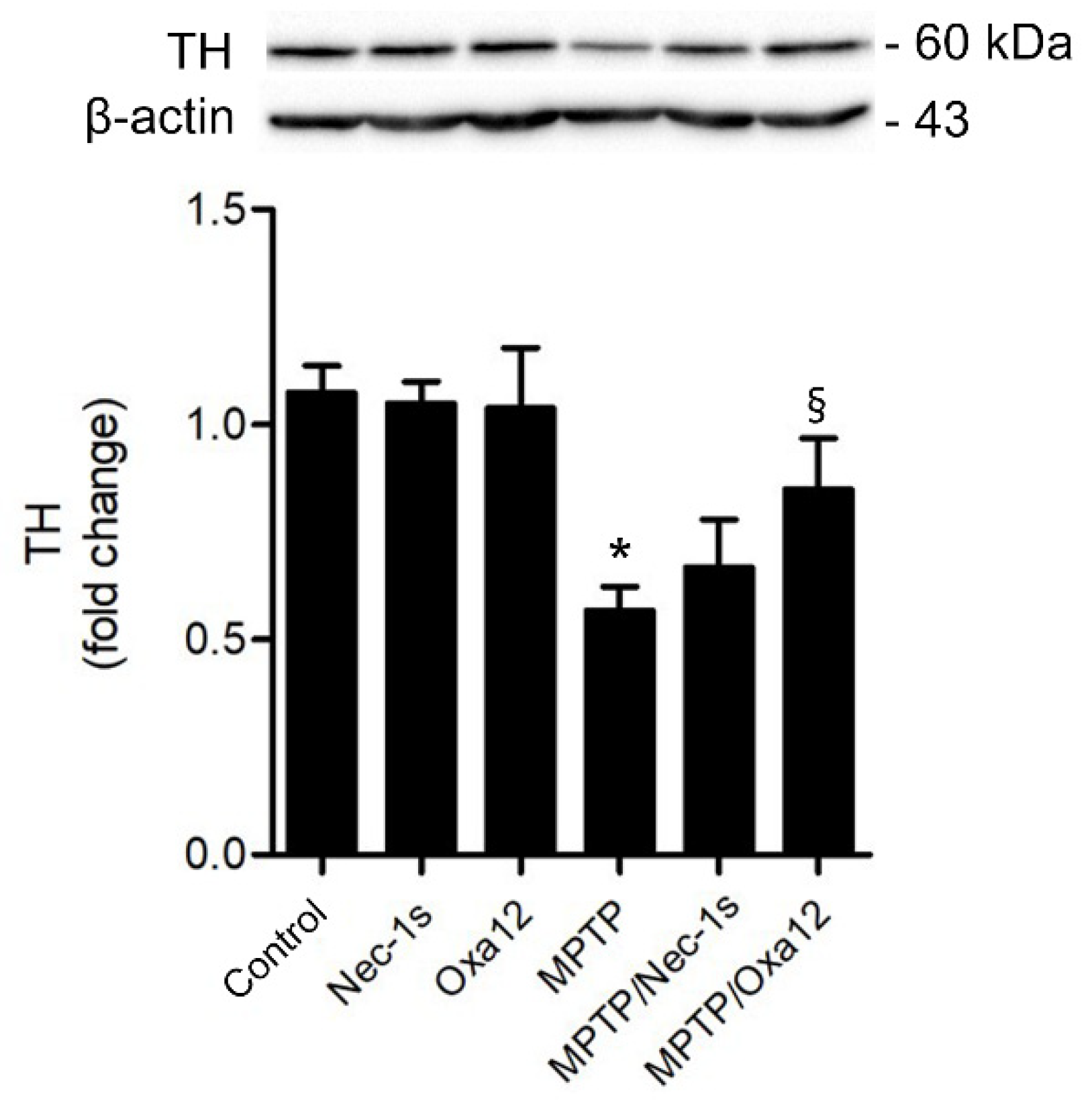
2.2. In Vivo Efficacy of Oxa12 in the Sub-Acute MPTP Mouse Model
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Chemical Synthesis and Analysis
4.3. Screening of Necroptosis Inhibitors
4.4. EC50 Determination
4.5. RIP1 and RIP3 Kinase Activity Assays
4.6. Microsomal Stability Assay
4.7. MPTP Mouse Model
4.8. Immunohistochemistry
4.9. Image Analysis
4.10. Protein Isolation
4.11. Western Blot
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 2013, 136, 2419–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, O.A.; Malagelada, C.; Greene, L.A. Cell death pathways in Parkinson’s disease: Proximal triggers, distal effectors, and final steps. Apoptosis 2009, 14, 478–500. [Google Scholar] [CrossRef] [PubMed]
- Venderova, K.; Park, D.S. Programmed cell death in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009365. [Google Scholar] [CrossRef]
- Conrad, M.; Angeli, J.P.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug. Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, M.C.; Preukschat, D.; Welz, P.S.; van Loo, G.; Ermolaeva, M.A.; Bloch, W.; Haase, I.; Pasparakis, M. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 2011, 35, 572–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootjans, S.T.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.S.; Chen, P.; Wang, W.X.; Lin, C.C.; Zhou, Y.; Yu, L.H.; Lin, Y.X.; Xu, Y.F.; Kang, D.Z. RIP1/RIP3/MLKL mediates dopaminergic neuron necroptosis in a mouse model of Parkinson disease. Lab. Investig. 2020, 100, 503–511. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofferson, D.E.; Li, Y.; Hitomi, J.; Zhou, W.; Upperman, C.; Zhu, H.; Gerber, S.A.; Gygi, S.; Yuan, J. A novel role for RIP1 kinase in mediating TNFα production. Cell Death Dis. 2012, 3, e320. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Japtag, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Declercq, W.; Vanden Berghe, T.; Vandenabeele, P. RIP kinases at the crossroads of cell death and survival. Cell 2009, 138, 229–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, T.; Peng, W.; Liu, Y.; Yan, C.; Maki, J.; Degterev, A.; Yuan, J.; Shi, Y. Structural basis of RIP1 inhibition by necrostatins. Structure 2013, 21, 493–499. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef]
- Teng, X.; Degterev, A.; Japtag, P.; Xing, X.; Choi, S.; Denu, R.; Yuan, J.; Cuny, G.D. Structure-activity relationship study of novel necroptosis inhibitors. Bioorganic Med. Chem. Lett. 2005, 15, 5039–5044. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.T.; Peng, W.; Liu, Y.; Yan, C.; Maki, J.; Degterev, A.; Yuan, J.; Shi, Y. zVAD-induced necroptosis in L929 cells depends on autocrine production of TNFα mediated by the PKC-MAPKs-AP-1 pathway. Cell Death Differ. 2011, 18, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.R.; Dionísio, P.A.; Brito, H.; Franco, L.; Rodrigues, C.A.B.; Guedes, R.C.; Afonso, C.A.M.; Amaral, J.D.; Rodrigues, C.M.P. Phenotypic screening identifies a new oxazolone inhibitor of necroptosis and neuroinflammation. Cell Death Discov. 2018, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyondrules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central nervous system multiparameter optimization desirable application in drug discovery. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef] [Green Version]
- Degterev, A.; Maki, J.L.; Yuan, J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2013, 20, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinson’s Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langston, J.W. The MPTP Story. J. Parkinson’s Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransom, B.R.; Kunis, D.M.; Irwin, I.; Langston, J.W. Astrocytes convert the parkinsonism inducing neurotoxin, MPTP, to its active metabolite, MPP+. Neurosci. Lett. 1987, 75, 323–328. [Google Scholar] [CrossRef]
- Schildknecht, S.; Pape, R.; Meiser, J.; Karreman, C.; Strittmatter, T.; Odermatt, M.; Cirri, E.; Friemel, A.; Ringwald, M.; Pasquarelli, N.; et al. Preferential Extracellular Generation of the Active Parkinsonian Toxin MPP+ by Transporter-Independent Export of the Intermediate MPDP+. Antioxid. Redox Signal. 2015, 23, 1001–1016. [Google Scholar] [CrossRef] [Green Version]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.A.B.; Mariz, I.F.A.; Maçôas, E.M.S.; Afonso, C.A.M.; Martinho, J.M.G. Unsaturated oxazolones as nonlinear fluorophores. Dyes Pigment. 2013, 99, 642–652. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Grootjans, S.; Callewaert, N.; Takahashi, N. Necrostatin-1 blocks both RIPK1 and IDO: Consequences for the study of cell death in experimental disease models. Cell Death Differ. 2013, 20, 185–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, S.B.; Harris, P.; Nagilla, R.; Kasparcova, V.; Hoffman, S.; Swift, B.; Dare, L.; Schaeffer, M.; Capriotti, C.; Ouellette, M.; et al. Characterization of GSK’963: A structurally distinct, potent and selective inhibitor of RIP1 kinase. Cell Death Discov. 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, P.A.; Bandyopadhyay, D.; Berger, S.B.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Finger, J.N.; Hoffman, S.J.; Kahler, K.M.; et al. Discovery of Small Molecule RIP1 Kinase Inhibitors for the Treatment of Pathologies Associated with Necroptosis. ACS Med. Chem. Lett. 2013, 4, 1238–1243. [Google Scholar] [CrossRef] [Green Version]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Pong, K.; Doctrow, S.R.; Huffman, K.; Adinolfi, C.A.; Baudry, M. Attenuation of staurosporine-induced apoptosis, oxidative stress, and mitochondrial dysfunction by synthetic superoxide dismutase and catalase mimetics, in cultured cortical neurons. Exp. Neurol. 2001, 171, 84–97. [Google Scholar] [CrossRef]
- Dionísio, P.A.; Oliveira, S.R.; Gaspar, M.M.; Gama, M.J.; Castro-Caldas, M.; Amaral, J.D.; Rodrigues, C.M.P. Ablation of RIP3 protects from dopaminergic neurodegeneration in experimental Parkinson’s disease. Cell Death Dis. 2019, 10, 840. [Google Scholar] [CrossRef] [Green Version]
- Brito, H.; Marques, V.; Afonso, M.B.; Brown, D.G.; Börjesson, U.; Selmi, N.; Smith, D.M.; Roberts, I.O.; Fitzek, M.; Aniceto, N.; et al. Phenotypic high-throughput screening platform identifies novel chemotypes for necroptosis inhibition. Cell Death Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Di, L.; Kerns, E.H.; Li, S.Q.; Petusky, S.L. High throughtput microsomal stability assay for insoluble compounds. Int. J. Pharm. 2006, 317, 54–60. [Google Scholar] [CrossRef]
- Castro-Caldas, M.; Neves Carvalho, A.; Peixeiro, I.; Rodrigues, E.; Lechner, M.C.; Gama, M.J. GSTpi expression in MPTP-induced dopaminergic neurodegeneration of C57BL/6 mouse midbrain and striatum. J. Mol. Neurosci. 2009, 38, 114–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saporito, M.S.; Thomas, B.A.; Scott, R.W. MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J. Neurochem. 2000, 75, 1200–1208. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxa12 | Nec-1 | ||||||
|---|---|---|---|---|---|---|---|
| Physiochemical Descriptor | Individual Score | Score CNS MPO | Physiochemical Descriptor | Individual Score | Score CNS MPO | ||
| MW | 391.43 | 0.8 | 3.6 | MW | 259.33 | 1.0 | 5.5 |
| clogP | 5.29 | 0 | clogP | 1.66 | 1.0 | ||
| clogD7.4 | 5.69 | 0 | clogD7.4 | 1.66 | 1.0 | ||
| TPSA | 67.34 | 1.0 | TPSA | 48.13 | 1.0 | ||
| HBD | 1 | 0.8 | HBD | 2 | 0.5 | ||
| pKa | 5.34 | 1.0 | pKa | 0 | 1.0 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, S.R.; Dionísio, P.A.; Gaspar, M.M.; Ferreira, M.B.T.; Rodrigues, C.A.B.; Pereira, R.G.; Estevão, M.S.; Perry, M.J.; Moreira, R.; Afonso, C.A.M.; et al. Discovery of a Necroptosis Inhibitor Improving Dopaminergic Neuronal Loss after MPTP Exposure in Mice. Int. J. Mol. Sci. 2021, 22, 5289. https://doi.org/10.3390/ijms22105289
Oliveira SR, Dionísio PA, Gaspar MM, Ferreira MBT, Rodrigues CAB, Pereira RG, Estevão MS, Perry MJ, Moreira R, Afonso CAM, et al. Discovery of a Necroptosis Inhibitor Improving Dopaminergic Neuronal Loss after MPTP Exposure in Mice. International Journal of Molecular Sciences. 2021; 22(10):5289. https://doi.org/10.3390/ijms22105289
Chicago/Turabian StyleOliveira, Sara R., Pedro A. Dionísio, Maria M. Gaspar, Maria B. T. Ferreira, Catarina A. B. Rodrigues, Rita G. Pereira, Mónica S. Estevão, Maria J. Perry, Rui Moreira, Carlos A. M. Afonso, and et al. 2021. "Discovery of a Necroptosis Inhibitor Improving Dopaminergic Neuronal Loss after MPTP Exposure in Mice" International Journal of Molecular Sciences 22, no. 10: 5289. https://doi.org/10.3390/ijms22105289
APA StyleOliveira, S. R., Dionísio, P. A., Gaspar, M. M., Ferreira, M. B. T., Rodrigues, C. A. B., Pereira, R. G., Estevão, M. S., Perry, M. J., Moreira, R., Afonso, C. A. M., Amaral, J. D., & Rodrigues, C. M. P. (2021). Discovery of a Necroptosis Inhibitor Improving Dopaminergic Neuronal Loss after MPTP Exposure in Mice. International Journal of Molecular Sciences, 22(10), 5289. https://doi.org/10.3390/ijms22105289