
Solvent-Induced Formation of Novel Ni(II) Complexes Derived from Bis-Thiosemicarbazone Ligand: An Insight from Experimental and Theoretical Investigations

 ,
, 
 , ,
, ,  ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
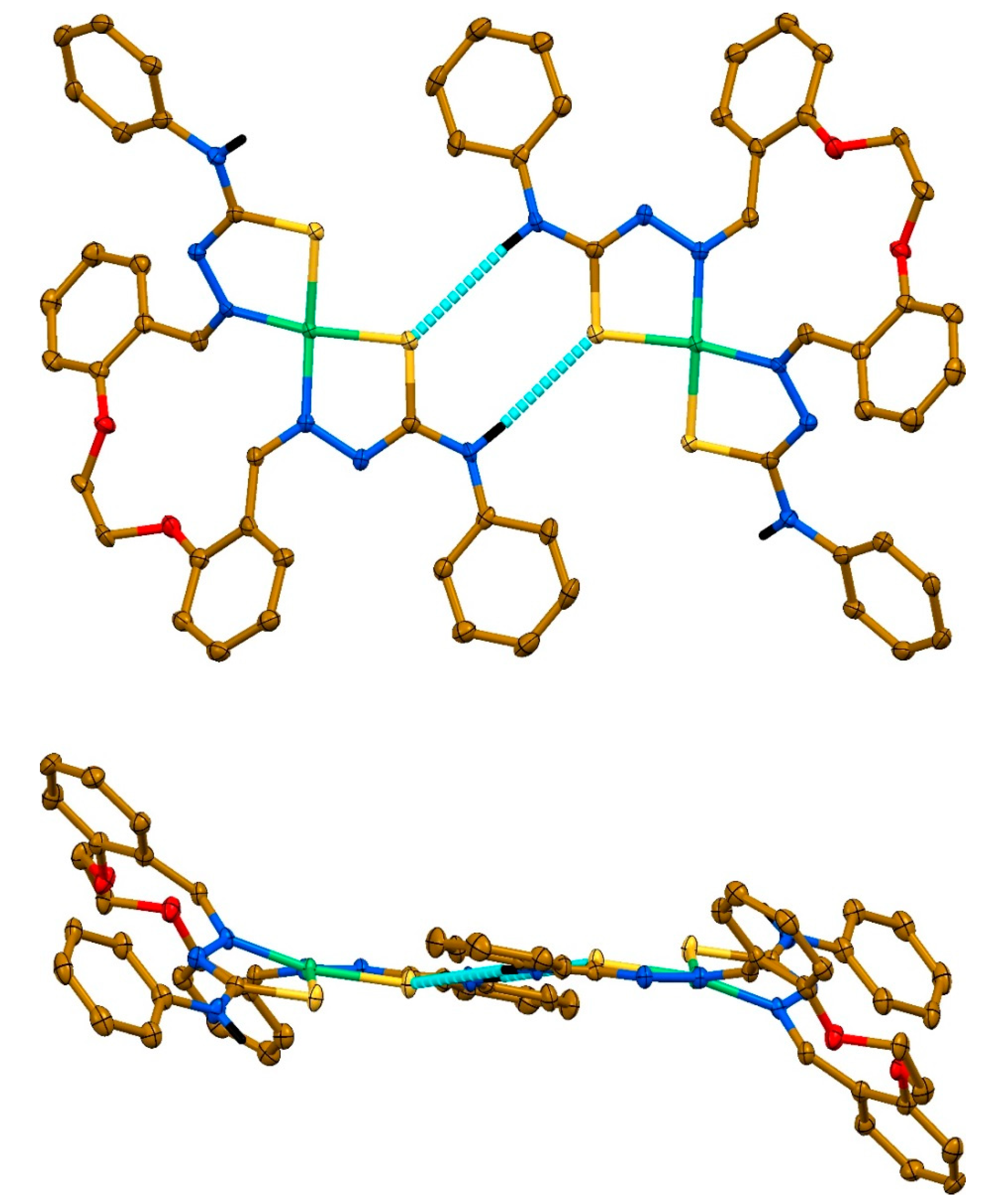
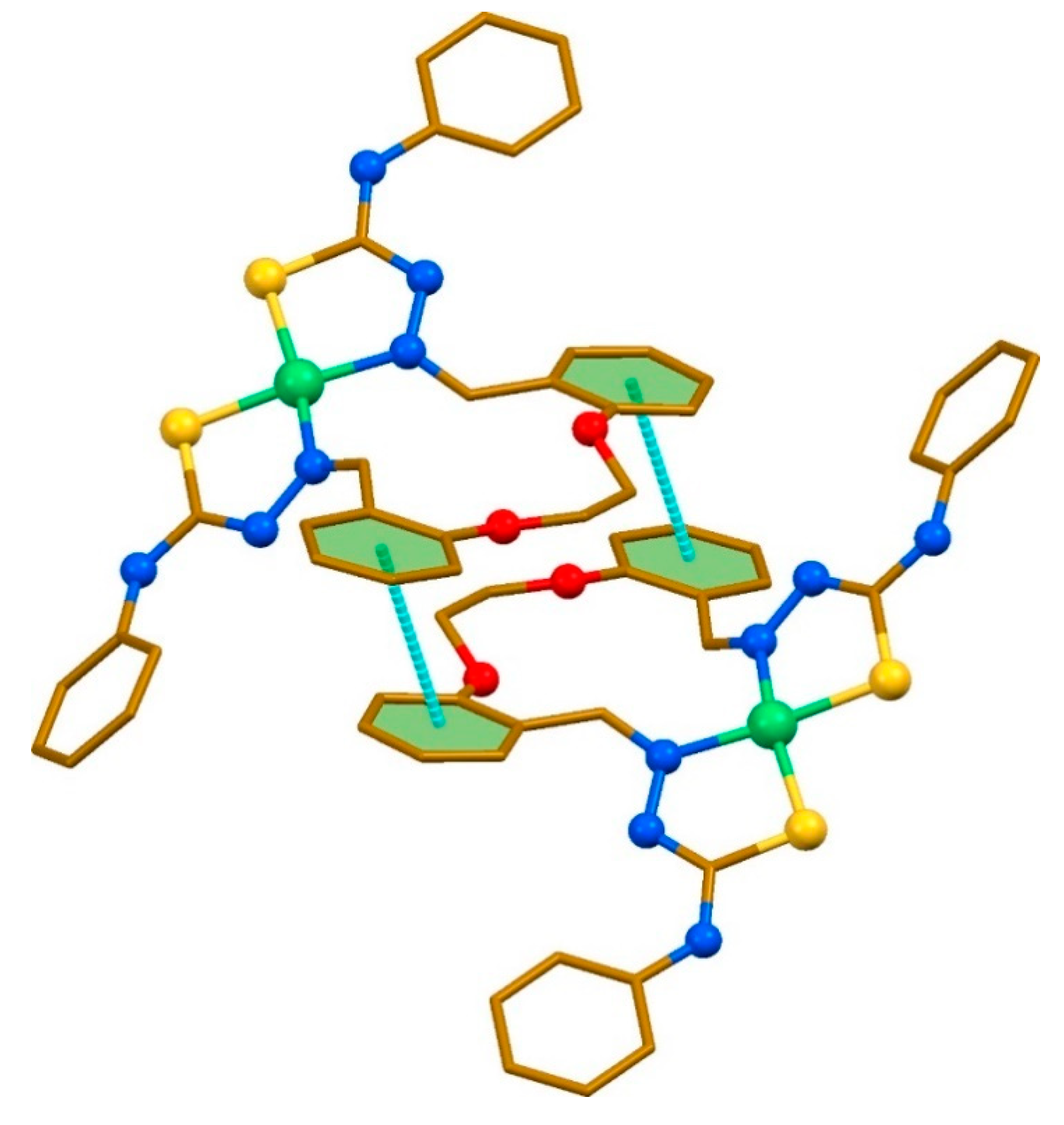
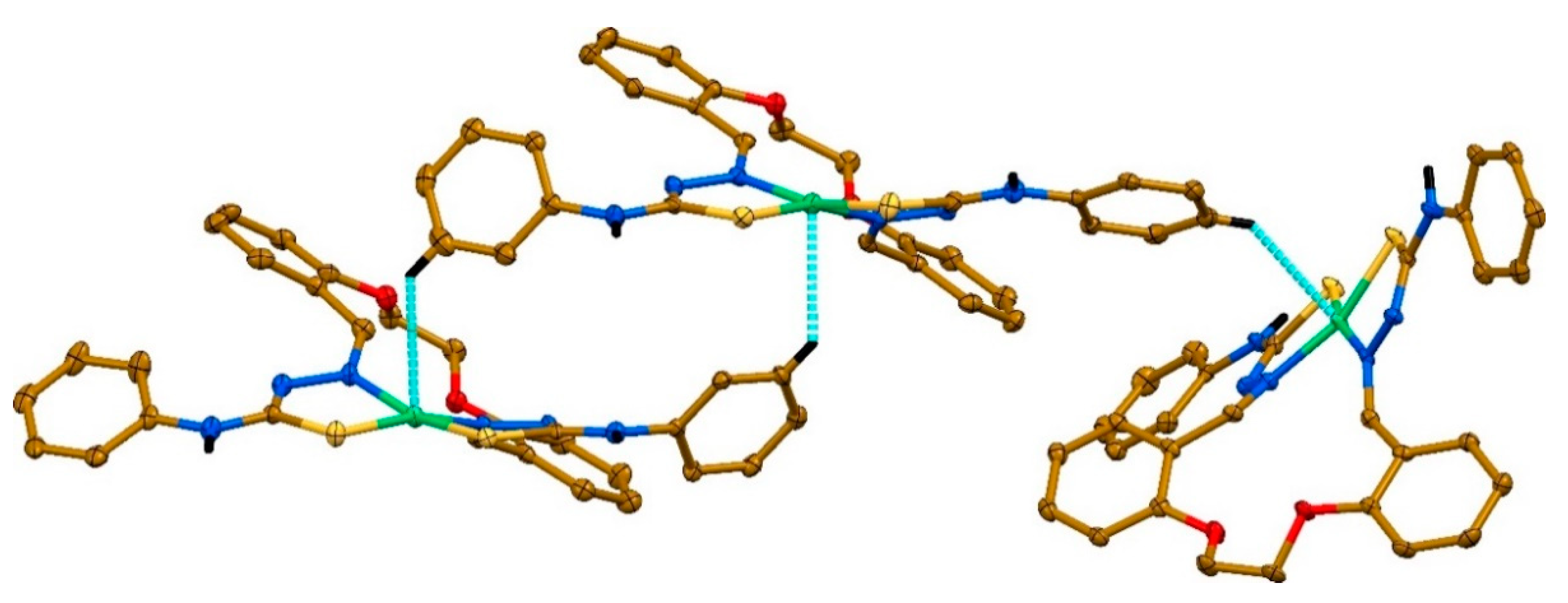
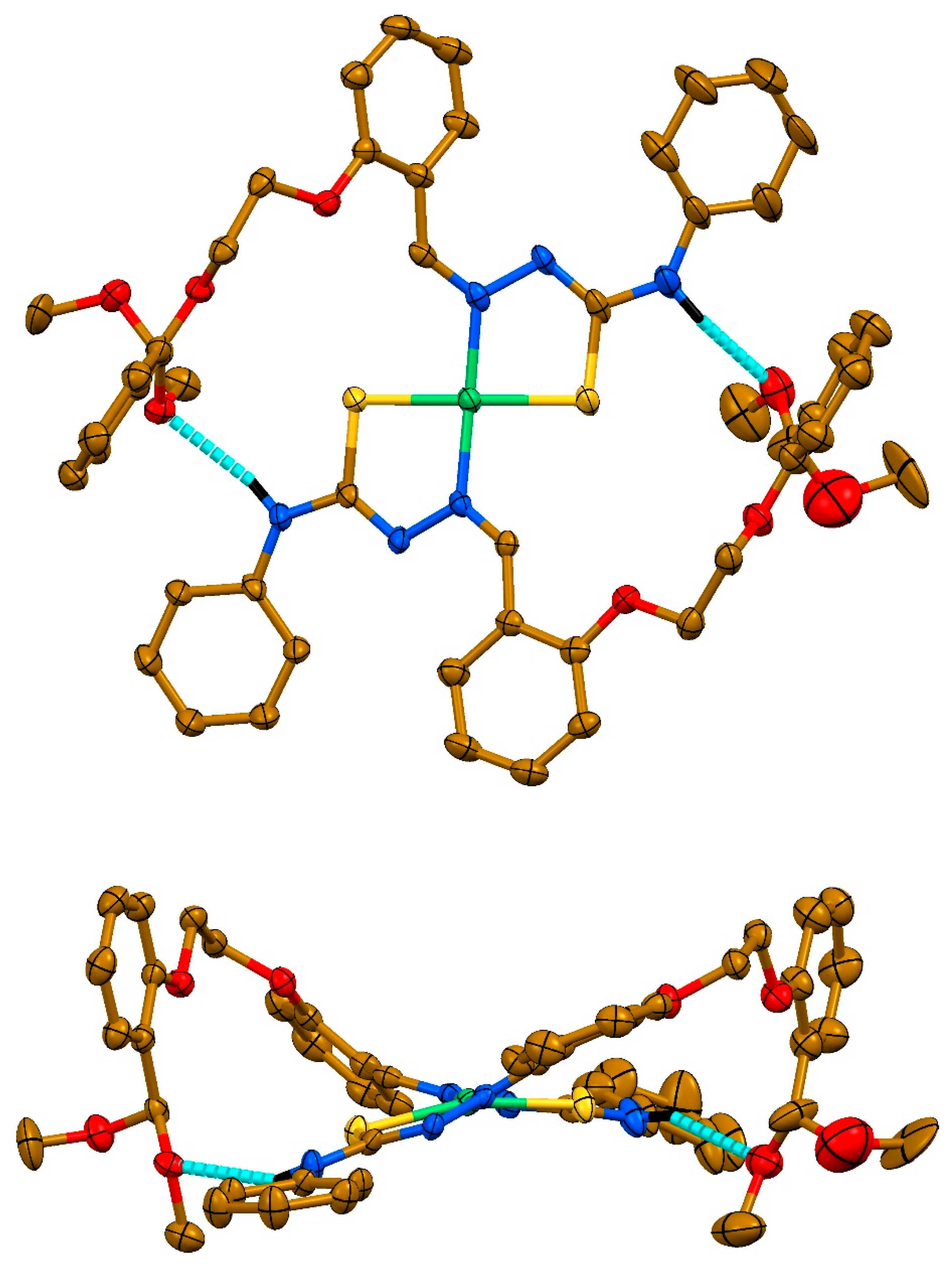
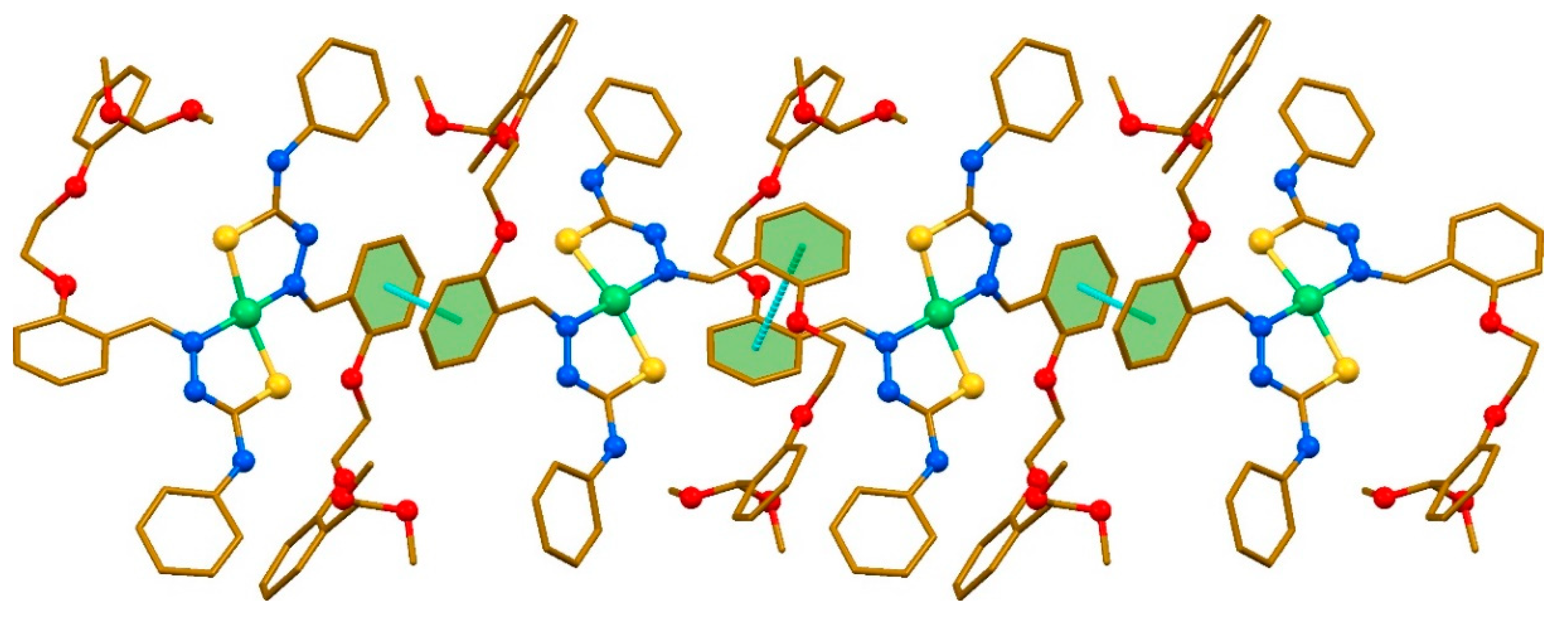
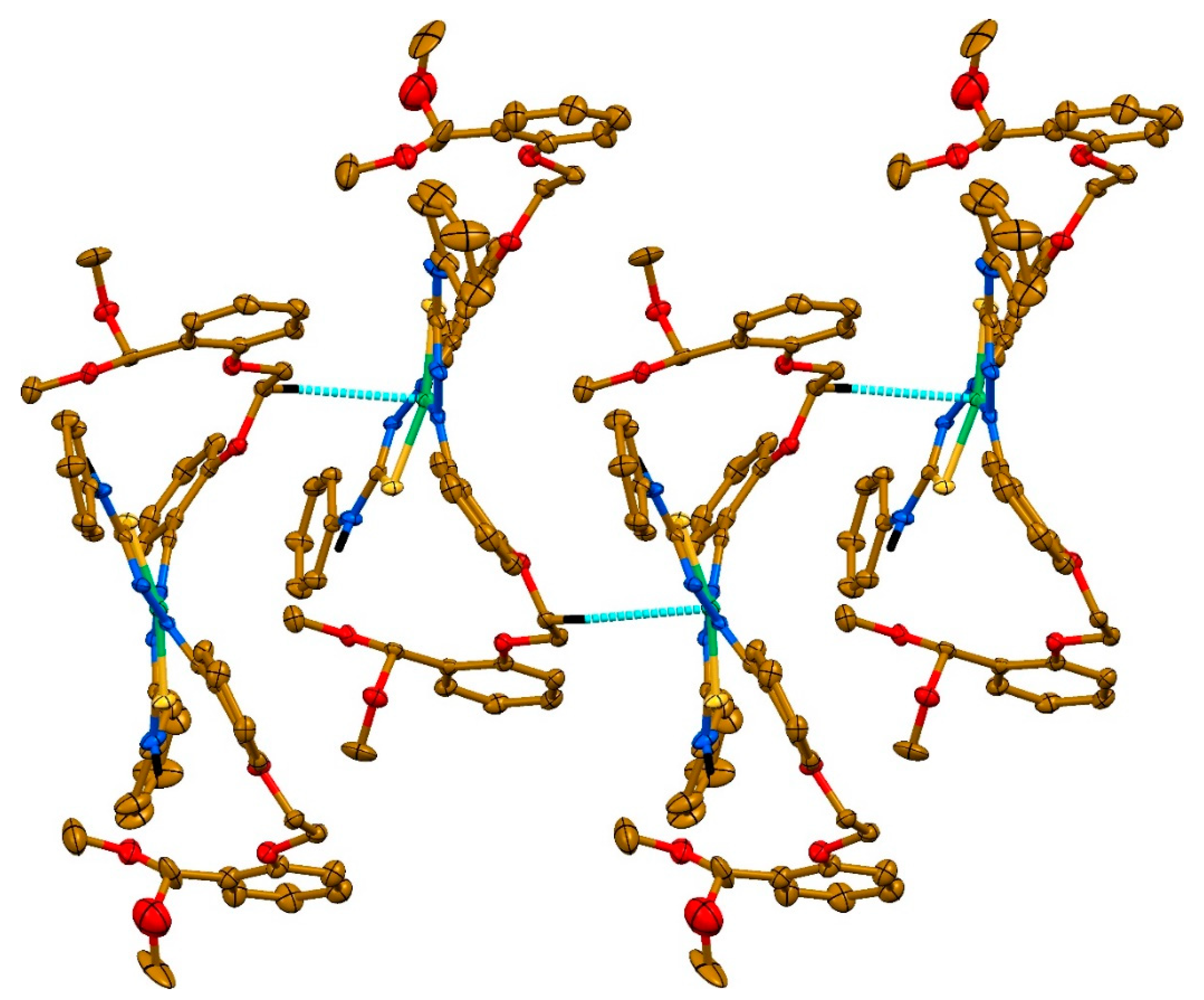
2. Results and discussion
3. Materials and Methods
3.1. Materials
3.2. Physical Measurements
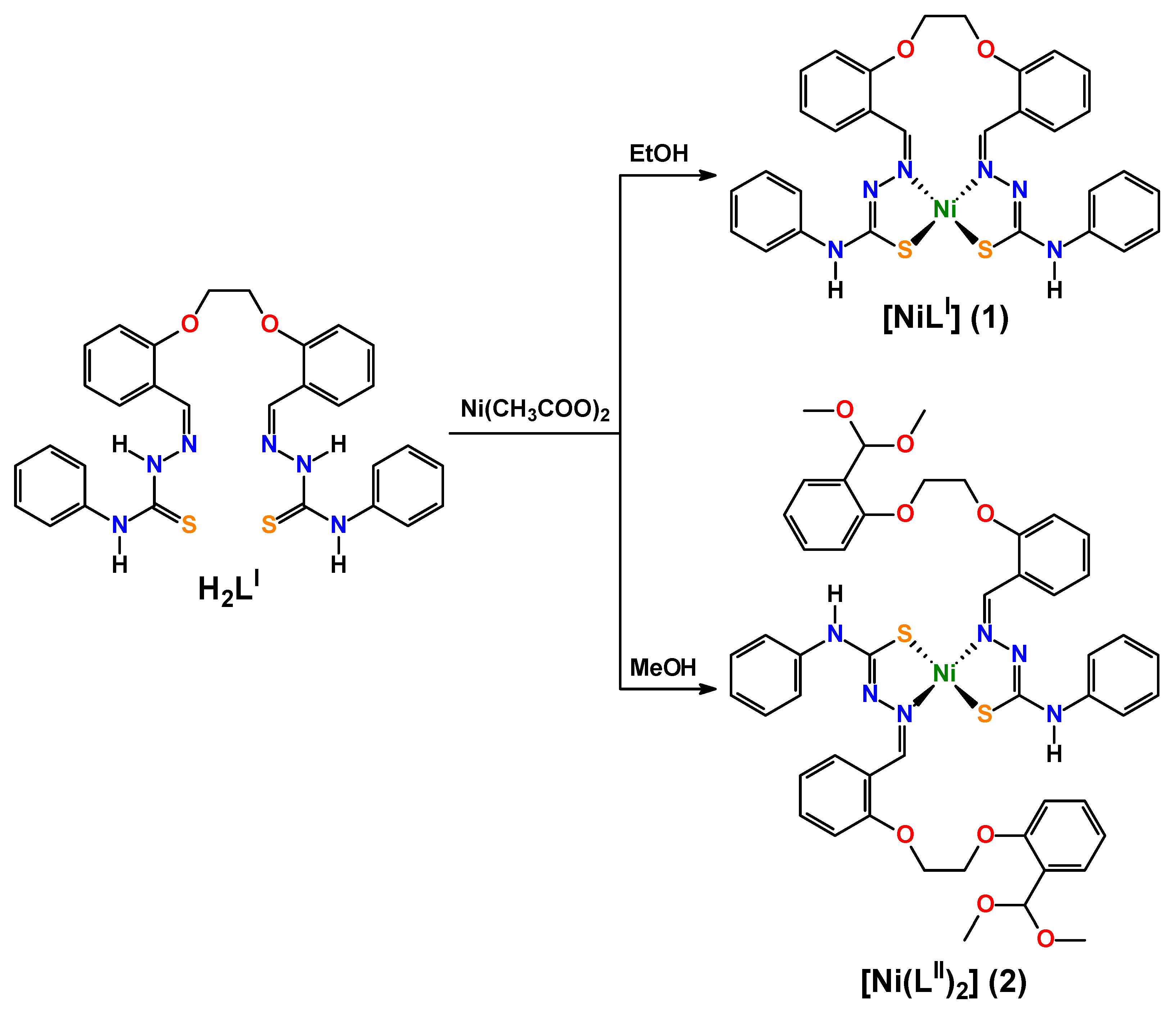
3.3. Synthesis of Complexes
3.4. Single-Crystal X-ray Diffraction
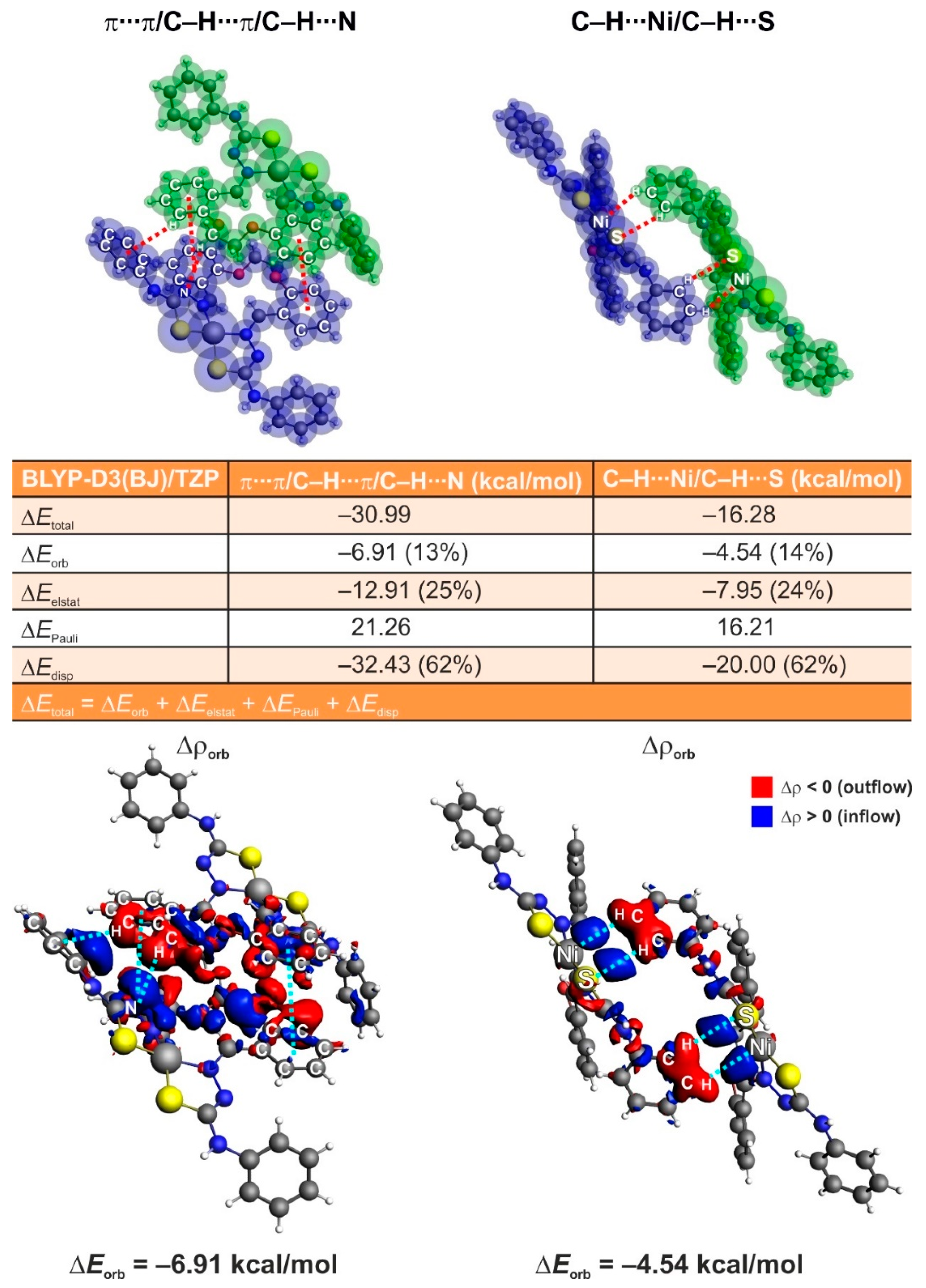
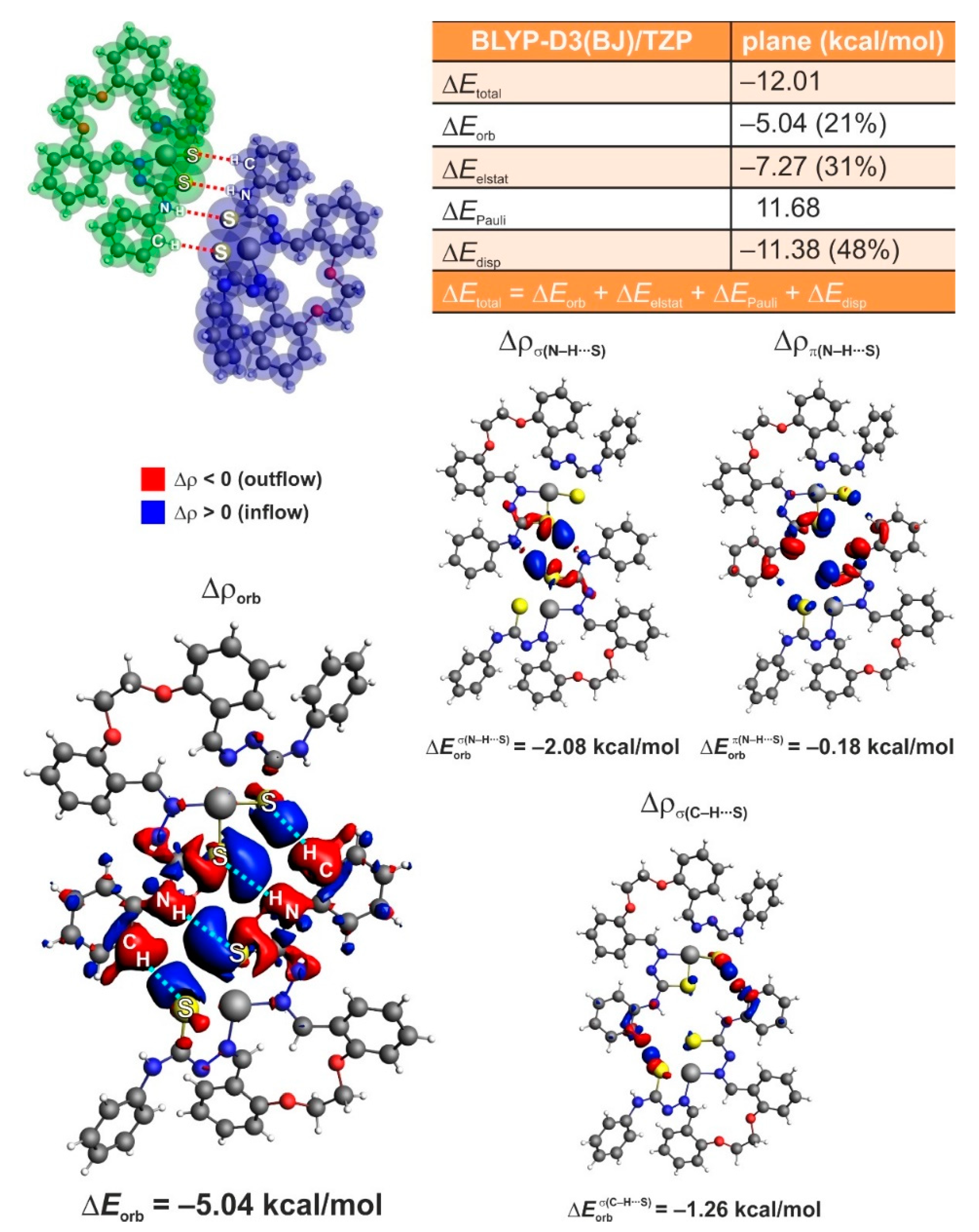
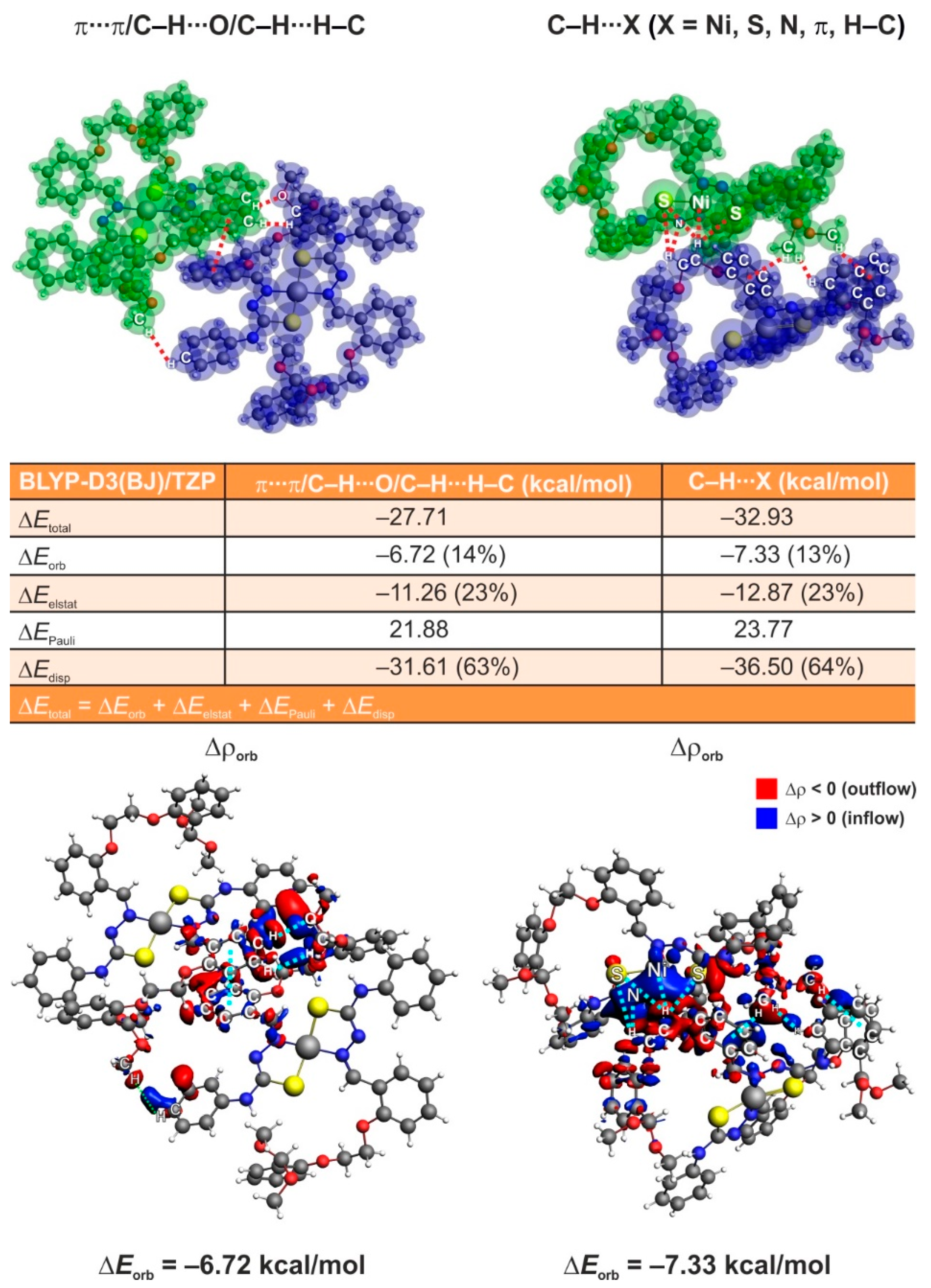
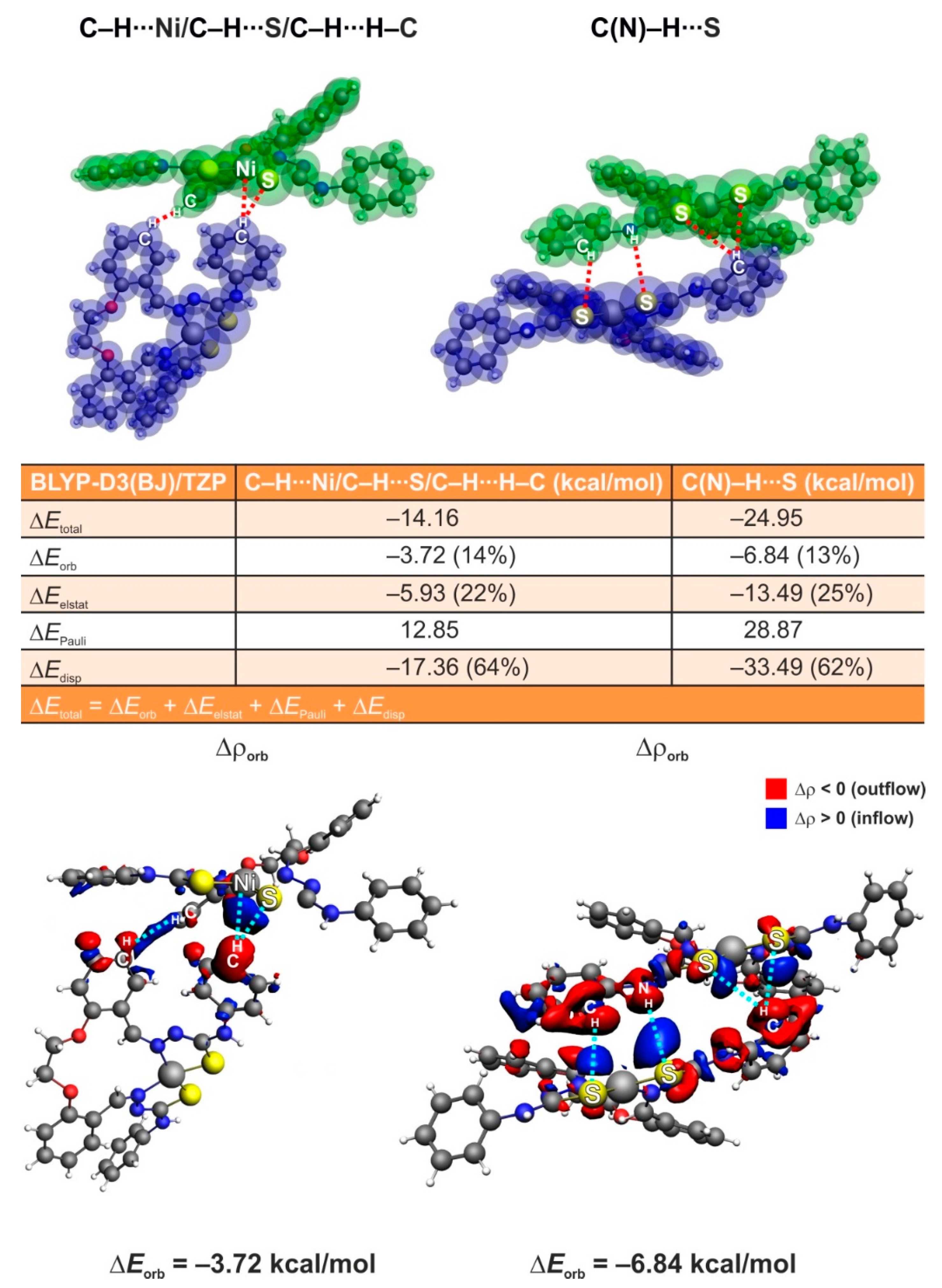
3.5. ETS-NOCV Studies
3.6. IQA studies
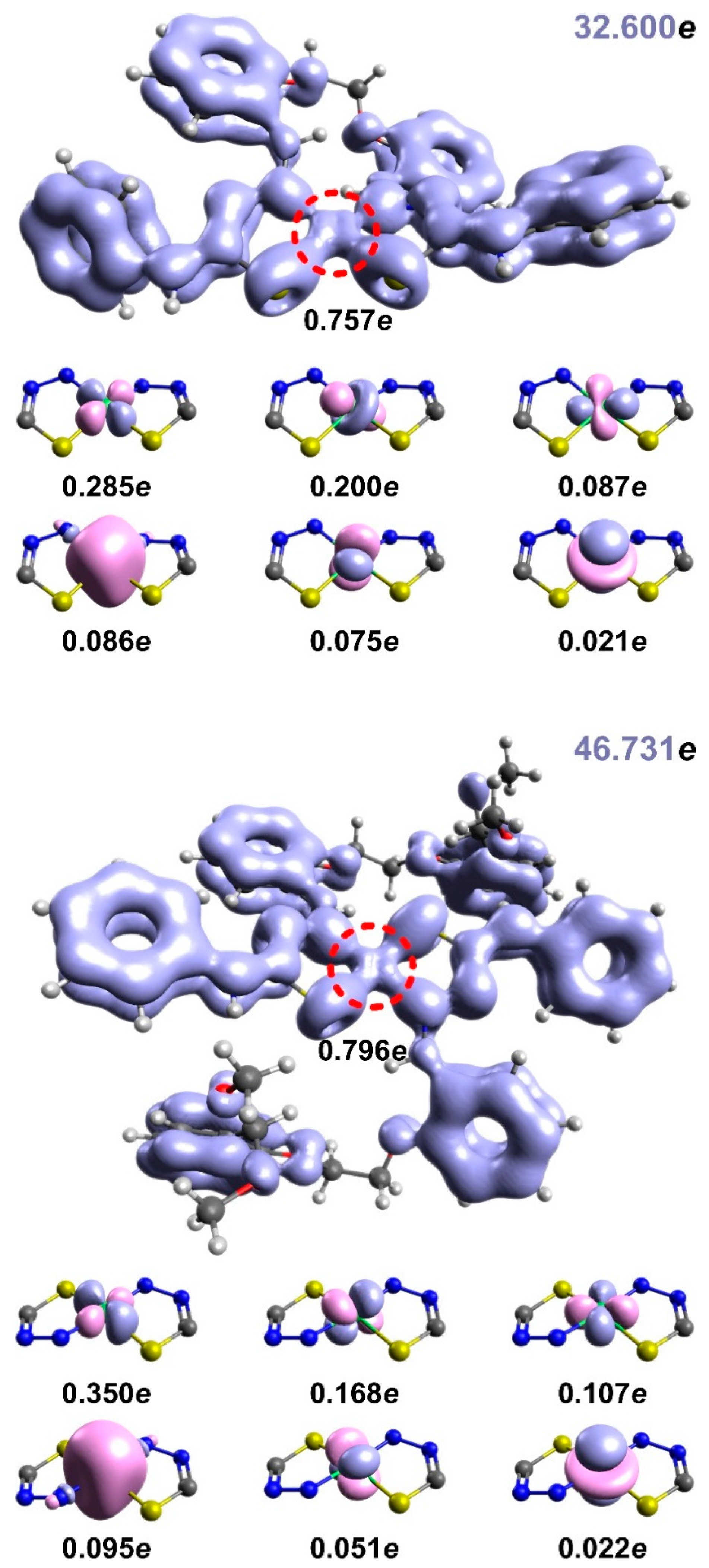
3.7. EDDB studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dilworth, J.R.; Hueting, R. Metal complexes of thiosemicarbazones for imaging and therapy. Inorg. Chim. Acta 2012, 389, 3–15. [Google Scholar] [CrossRef]
- Casas, J.S.; García-Tasende, M.S.; Sordo, J. Main group metal complexes of semicarbazones and thiosemicarbazones. A structural review. Coord. Chem. Rev. 2000, 209, 197–261. [Google Scholar] [CrossRef]
- Gómez Quiroga, A.; Navarro Ranninger, C. Contribution to the SAR field of metallated and coordination complexes: Studies of the palladium and platinum derivatives with selected thiosemicarbazones as antitumoral drugs. Coord. Chem. Rev. 2004, 248, 119–133. [Google Scholar] [CrossRef]
- Yu, Y.; Kalinowski, D.S.; Kovacevic, Z.; Siafakas, A.R.; Jansson, P.J.; Stefani, C.; Lovejoy, D.B.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Thiosemicarbazones from the old to new: Iron chelators that are more than just ribonucleotide reductase inhibitors. J. Med. Chem. 2009, 52, 5271–5294. [Google Scholar] [CrossRef]
- Kölmel, D.K.; Kool, E.T. Oximes and hydrazones in bioconjugation: Mechanism and catalysis. Chem. Rev. 2017, 117, 10358–10376. [Google Scholar] [CrossRef]
- Malik, M.A.; Dar, O.A.; Gull, P.; Wani, M.Y.; Hashmi, A.A. Heterocyclic schiff base transition metal complexes in antimicrobial and anticancer chemotherapy. Med. Chem. Commun. 2018, 9, 409–436. [Google Scholar] [CrossRef]
- Hałdys, K.; Latajka, R. Thiosemicarbazones with tyrosinase inhibitory activity. Med. Chem. Commun. 2019, 10, 378–389. [Google Scholar] [CrossRef]
- Ong, Y.C.; Roy, S.; Andrews, P.C.; Gasser, G. Metal compounds against neglected tropical diseases. Chem. Rev. 2019, 119, 730–796. [Google Scholar] [CrossRef] [PubMed]
- Boros, E.; Packard, A.B. Radioactive transition metals for imaging and therapy. Chem. Rev. 2019, 119, 870–901. [Google Scholar] [CrossRef]
- Howard, K.C.; Dennis, E.K.; Watt, D.S.; Garneau-Tsodikova, S. A comprehensive overview of the medicinal chemistry of antifungal drugs: Perspectives and promise. Chem. Soc. Rev. 2020, 49, 2426–2480. [Google Scholar] [CrossRef]
- Sokolov, F.D.; Safin, D.A.; Bolte, M.; Shakirova, E.R.; Babashkina, M.G. New bifunctional N-thiophosphorylated thiourea and 2,5-dithiobiurea derivatives. Crystal structures of R[C(S)NHP(S)(OiPr)2]2 (R = –N(Ph)CH2CH2N(Ph)– and –NHNH–). Polyhedron 2008, 27, 3141–3145. [Google Scholar] [CrossRef]
- Safin, D.A.; Bolte, M.; Shakirova, E.R.; Babashkina, M.G. The influence of the substituent [PhNHNH– and EtN(NH2)–] on the N-thiophosphorylated thiosemicarbazides RC(S)NHP(S)(OiPr)2 crystal design. Polyhedron 2009, 28, 501–504. [Google Scholar] [CrossRef]
- Safin, D.A.; Babashkina, M.G.; Bolte, M.; Klein, A. The influence of the spacer Z on N-phosphorylated bis-thioureas and 2,5-dithiobiurea Z[C(S)NHP(O)(OiPr)2]2 (Z = NHCH2CH2NH, NHC6H4-2-NH, NHNH) crystal design. Polyhedron 2009, 28, 1403–1408. [Google Scholar] [CrossRef]
- Safin, D.A.; Babashkina, M.G.; Bolte, M.; Klein, A. Synthesis of N-(thio)phosphorylated thiosemicarbazides RC(S)NHP(X)(OiPr)2 (X = S, R = NH2N(Me)–; X = O, R = NH2N(Me)–, PhNHNH–): Reaction of NH2N(Me)C(S)NHP(S)(OiPr)2 with acetone. Polyhedron 2009, 28, 2693–2697. [Google Scholar]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 9, 955–964. [Google Scholar] [CrossRef]
- Qiu, X.-H.; Wu, H.-Y. [2,2′-(Ethylenedioxy)dibenzaldehyde bis(thiosemicarbazone)]nickel(II) diperchlorate methanol disolvate. Acta Cryst. 2004, 60, m1151–m1152. [Google Scholar] [CrossRef]
- Zhu, X.-H.; Chen, X.-F.; Liu, Y.-J.; Duan, C.-Y.; You, X.-Z.; Tian, Y.-P.; Xie, F.-X. 2,2′-Ethylenedioxydibenzaldehyde bis(thiosemicarbazone) bis(dimethyl sulfoxide). Acta Cryst. 1999, 55, 1175–1176. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer 17.5; University of Western Australia: Crawley, WA, Australia, 2017. [Google Scholar]
- Safin, D.A.; Mitoraj, M.P.; Robeyns, K.; Filinchuk, Y.; Velde, C.M.V. Luminescent mononuclear mixed ligand complexes of copper(I) with 5-phenyl-2,2′-bipyridine and triphenylphosphine. Dalton Trans. 2015, 44, 16824–16832. [Google Scholar] [CrossRef]
- Babashkina, M.G.; Robeyns, K.; Filinchuk, Y.; Safin, D.A. Detailed studies of the interaction of 3-chloroaniline with O,O′-diphenylphosphorylisothiocyanate. New J. Chem. 2016, 40, 1230–1236. [Google Scholar] [CrossRef]
- Safin, D.A.; Velde, C.M.V.; Babashkina, M.G.; Robeyns, K.; Filinchuk, Y. Mononuclear heteroleptic complexes of copper(i) with 5-phenyl-2,2′-bipyridine and triphenylphosphine: Crystal structures, Hirshfeld surface analysis and luminescence properties. New J. Chem. 2016, 40, 6156–6163. [Google Scholar] [CrossRef]
- Safin, D.A.; Robeyns, K.; Babashkina, M.G.; Filinchuk, Y.; Rotaru, A.; Jureschi, C.; Mitoraj, M.P.; Hooper, J.; Brela, M.; Garcia, Y. Polymorphism driven optical properties of an anil dye. CrystEngComm 2016, 18, 7249–7259. [Google Scholar] [CrossRef]
- Safin, D.A.; Robeyns, K.; Garcia, Y. 1,2,4-Triazole-based molecular switches: Crystal structures, Hirshfeld surface analysis and optical properties. CrystEngComm 2016, 18, 7284–7296. [Google Scholar] [CrossRef]
- Safin, D.A.; Babashkina, M.G.; Mitoraj, M.P.; Kubisiak, P.; Robeyns, K.; Bolte, M.; Garcia, Y. An intermolecular pyrene excimer in the pyrene-labeled N-thiophosphorylated thiourea and its nickel(II) complex. Inorg. Chem. Front. 2016, 3, 1419–1431. [Google Scholar] [CrossRef]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A combined charge and energy decomposition scheme for bond analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef]
- Velde, G.T.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; Van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ziegler, T.; Atkins, A.J.; Autschbach, J.; Baseggio, O.; Bashford, D.; Bérces, A.; Bickelhaupt, F.M.; Bo, C.; Boerrigter, P.M.; et al. Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 5 January 2021).
- Stasyuk, O.A.; Sedlak, R.; Guerra, C.F.; Hobza, P. Comparison of the DFT-SAPT and canonical EDA schemes for the energy decomposition of various types of noncovalent interactions. J. Chem. Theory Comput. 2018, 14, 3440–3450. [Google Scholar] [CrossRef]
- Van der Lubbe, S.; Guerra, C.F. The nature of hydrogen bonds: A delineation of the role of different energy components on hydrogen bond strengths and lengths. Chem. Asian J. 2019, 14, 2760–2769. [Google Scholar]
- Van der Lubbe, S.C.C.; Zaccaria, F.; Sun, X.; Guerra, C.F. Secondary electrostatic interaction model revised: Prediction comes mainly from measuring charge accumulation in hydrogen-bonded monomers. J. Am. Chem. Soc. 2019, 141, 4878–4885. [Google Scholar] [CrossRef]
- Kurczab, R.; Mitoraj, M.P.; Michalak, A.; Ziegler, T. Theoretical analysis of the resonance assisted hydrogen bond based on the combined extended transition state method and natural orbitals for chemical valence scheme. J. Phys. Chem. A 2010, 114, 8581–8590. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, H.; Wu, W.; Mo, Y. A critical check for the role of resonance in intramolecular hydrogen bonding. Chem. Eur. J. 2017, 23, 16885–16891. [Google Scholar] [CrossRef]
- Blanco, M.A.; Martín Pendás, A.; Francisco, E. Interacting quantum atoms: A correlated energy decomposition scheme based on the quantum theory of atoms in molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Babashkina, M.G.; Robeyns, K.; Sagan, F.; Szczepanik, D.W.; Seredina, Y.V.; Garcia, Y.; Safin, D.A. Chameleon-like nature of anagostic interactions and its impact on metalloaromaticity in square-planar nickel complexes. Organometallics 2019, 38, 1973–1981. [Google Scholar] [CrossRef]
- Scherer, W.; Wolstenholme, D.J.; Herz, V.; Eickerling, G.; Bruck, A.; Benndorf, P.; Roesky, P.W. On the nature of agostic interactions in transition-metal amido complexes. Angew. Chem. Int. Ed. 2010, 49, 2242–2246. [Google Scholar] [CrossRef]
- Danovich, D.; Shaik, S.; Neese, F.; Echeverría, J.; Aullón, G.; Alvarez, S. Understanding the nature of the CH···HC interactions in Alkanes. J. Chem. Theory Comput. 2013, 9, 1977–1991. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.P.; Schreiner, P.R. London dispersion in molecular chemistry—Reconsidering steric effects. Angew. Chem. Int. Ed. 2015, 54, 12274–12296. [Google Scholar] [CrossRef]
- Cukrowski, I.; Sagan, F.; Mitoraj, M.P. On the stability of cis- and trans-2-butene isomers. An insight based on the FAMSEC, IQA, and ETS-NOCV Schemes. J. Comput. Chem. 2016, 37, 2783–2798. [Google Scholar] [CrossRef]
- Liptrot, D.J.; Power, P.P. London dispersion forces in sterically crowded inorganic and organometallic molecules. Nat. Rev. Chem. 2017, 1, 4. [Google Scholar] [CrossRef]
- Lu, Q.; Neese, F.; Bistoni, G. Formation of agostic structures driven by London dispersion. Angew. Chem. Int. Ed. 2018, 57, 4760–4764. [Google Scholar] [CrossRef]
- Sagan, F.; Mitoraj, M.P. Transition Metals in Coordination Environments: Computational Chemistry and Catalysis Viewpoints; Broclawik, E., Borowski, T., Radoń, M., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 65–89. [Google Scholar]
- Mitoraj, M.P.; Sagan, F.; Szczepanik, D.W.; Lange, J.H.D.; Ptaszek, A.L.; Van Niekerk, D.M.E.; Cukrowski, I. Origin of hydrocarbons stability from a computational perspective: A case study of ortho-xylene isomers. ChemPhysChem 2020, 21, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wu, W.; Mo, Y. A theoretical perspective of the agostic effect in early transition metal compounds. Coord. Chem. Rev. 2020, 419, 213401. [Google Scholar] [CrossRef]
- Szczepanik, D.W. A new perspective on quantifying electron localization and delocalization in molecular systems. Comput. Theor. Chem. 2016, 1080, 33–37. [Google Scholar] [CrossRef]
- Afkhami, F.A.; Mahmoudi, G.; Qu, F.; Gupta, A.; Zangrando, E.; Frontera, A.; Safin, D.A. Supramolecular architecture constructed from the hemidirected lead(II) complex with N′-(4-hydroxybenzylidene)isonicotinohydrazide. Inorg. Chim. Acta 2020, 502, 119350. [Google Scholar] [CrossRef]
- Project, C.C. The CCP4 suite: Programs for protein crystallography. Acta Cryst. 1994, 50, 760–763. [Google Scholar]
- Otwinowski, Z.; Minor, W. Macromolecular Crystallography, part A. In Processing of X-Ray Diffraction Data Collected in Oscillation Mode, Methods in Enzymology; Carter, C.W., Jr., Sweet, R.M., Eds.; Academic Press: London, UK, 1997; Volume 276, pp. 307–326. [Google Scholar]
- Bruker, S.S. Software Reference Manual Bruker AXS Inc; Raith GmbH, Hauert 18, 44227 Dortmund, Germany; Madison, WI, USA. 2000. Available online: http://research.physics.illinois.edu/bezryadin/labprotocol/e_LiNE%20Software%20Reference%20Manual.pdf (accessed on 17 March 2021).
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Gupta, A.N.; Kumar, V.; Singh, V.; Manar, K.K.; Drew, M.G.B.; Singh, N. Intermolecular anagostic interactions in group 10 metal dithiocarbamates. CrystEngComm. 2014, 16, 9299–9307. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1-I | 1-II | 1-III | 2 | |
|---|---|---|---|---|
| Bond lengths | ||||
| Ni–N | 1.914(3) 1.928(4) | 1.918(3) 1.923(3) | 1.916(3) 1.935(3) | 1.903(4) 1.907(4) |
| Ni–S | 2.1464(14) 2.1506(11) | 2.1502(13) 2.1631(12) | 2.1389(14) 2.1453(14) | 2.160(2) 2.161(2) |
| Bond angles | ||||
| N–Ni–N | 101.26(15) | 100.27(14) | 102.20(14) | 173.1(2) |
| N–Ni–Sendocyclic | 85.59(11) 86.62(10) | 86.14(11) 86.46(11) | 86.53(11) 87.04(11) | 85.94(19) 86.12(19) |
| N–Ni–Sexocyclic | 163.87(11) 168.87(12) | 163.98(11) 164.92(11) | 162.85(11) 163.23(11) | 94.90(19) 94.95(19) |
| S–Ni–S | 89.90(5) | 90.96(5) | 88.39(5) | 164.23(8) |
| Dihedral angle | ||||
| NiNNCS∙∙∙ NiNNCS | 20.51(14) | 20.42(15) | 22.02(15) | 22.3(2) |
| D–H∙∙∙A | d(D–H) | d(H∙∙∙A) | d(D∙∙∙A) | ∠(DHA) | |
|---|---|---|---|---|---|
| 1 | N1–H1N∙∙∙S1 | 0.88 | 2.80 | 3.672(4) | 175 |
| N12–H12N∙∙∙S4 | 0.88 | 2.79 | 3.482(3) | 136 | |
| 2 | N1–H1N∙∙∙O7 | 0.86(7) | 2.12(7) | 2.922(9) | 154(7) |
| N4–H4N∙∙∙O4 | 0.89(7) | 2.14(8) | 2.949(7) | 152(6) |
| Cg(I) | Cg(J) | d[Cg(I)–Cg(J)] | α | β | γ | Slippage | |
|---|---|---|---|---|---|---|---|
| 1 | C6H4 | C6H4 | 3.728(3) | 1.3(2) | 24.3 | 25.3 | 1.534 |
| C6H4 | C6H4 | 3.729(3) | 1.3(2) | 25.3 | 24.3 | 1.595 | |
| 2 | C6H4 | C6H4 | 3.853(4) | 12.4(4) | 13.8 | 16.3 | 0.918 |
| C6H4 | C6H4 | 3.854(4) | 12.4(4) | 16.3 | 13.8 | 1.083 |
| 1-I | 1-II | 1-III | 2 | |
|---|---|---|---|---|
| Bond lengths | ||||
| C–H | 0.95 | 0.95 | 0.95 | 0.99 |
| Ni∙∙∙H | 2.71 | 2.94 | 2.84 | 2.91 |
| Ni∙∙∙C | 3.504(5) | 3.657 | 3.563 | 3.713 |
| Bond angle | ||||
| Ni∙∙∙H–C | 142 | 133 | 133 | 139 |
 |  |  |  | |||||||||||||||||||||
| H | C | N | O | S | Ni | H | C | N | O | S | Ni | H | C | N | O | S | Ni | H | C | N | O | S | Ni | |
| Contacts (C, %) 1 | ||||||||||||||||||||||||
| H | 47.3 | – | – | – | – | – | 38.9 | – | – | – | – | – | 39.6 | – | – | – | – | – | 54.7 | – | – | – | – | – |
| C | 26.2 | 5.0 | – | – | – | – | 24.4 | 4.6 | – | – | – | – | 30.4 | 3.3 | – | – | – | – | 24.0 | 3.5 | – | – | – | – |
| N | 6.9 | 0.4 | 0.0 | – | – | – | 8.1 | 1.5 | 0.1 | – | – | – | 7.5 | 1.4 | 0.0 | – | – | – | 5.5 | 0.8 | 0.0 | – | – | – |
| O | 2.3 | 0.7 | 0.0 | 0.0 | – | – | 3.0 | 1.7 | 0.0 | 0.0 | – | – | 1.4 | 2.1 | 0.0 | 0.0 | – | – | 6.6 | 0.4 | 0.2 | 0.0 | – | – |
| S | 9.8 | 0.1 | 0.0 | 0.0 | 0.0 | – | 14.0 | 0.1 | 0.2 | 0.0 | 0.3 | – | 9.9 | 0.1 | 0.2 | 0.0 | 0.9 | – | 2.5 | 0.1 | 0.0 | 0.2 | 0.0 | – |
| Ni | 1.4 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 2.3 | 0.7 | 0.1 | 0.0 | 0.0 | 0.0 | 2.3 | 0.8 | 0.1 | 0.0 | 0.0 | 0.0 | 0.8 | 0.8 | 0.0 | 0.0 | 0.0 | 0.0 |
| Surface (S, %) | ||||||||||||||||||||||||
| 70.6 | 18.7 | 3.7 | 1.5 | 5.0 | 0.7 | 64.8 | 18.8 | 5.1 | 2.4 | 7.5 | 1.6 | 65.4 | 20.7 | 4.6 | 1.8 | 6.0 | 1.6 | 74.4 | 16.6 | 3.3 | 3.7 | 1.4 | 0.8 | |
| Random contacts (R, %) | ||||||||||||||||||||||||
| H | 49.8 | – | – | – | – | – | 42.0 | – | – | – | – | – | 42.8 | – | – | – | – | – | 55.4 | – | – | – | – | – |
| C | 26.4 | 3.5 | – | – | – | – | 24.4 | 3.5 | – | – | – | – | 27.1 | 4.3 | – | – | – | – | 24.7 | 2.8 | – | – | – | – |
| N | 5.2 | 1.4 | 0.1 | – | – | – | 6.6 | 1.9 | 0.3 | – | – | – | 6.0 | 1.9 | 0.2 | – | – | – | 4.9 | 1.1 | 0.1 | – | – | – |
| O | 2.1 | 0.6 | 0.1 | 0.0 | – | – | 3.1 | 0.9 | 0.2 | 0.1 | – | – | 2.4 | 0.7 | 0.2 | 0.0 | – | – | 5.5 | 1.2 | 0.2 | 0.1 | – | – |
| S | 7.1 | 1.9 | 0.4 | 0.2 | 0.3 | – | 9.7 | 2.8 | 0.8 | 0.4 | 0.6 | – | 7.8 | 2.5 | 0.6 | 0.2 | 0.4 | – | 2.1 | 0.5 | 0.1 | 0.1 | 0.0 | – |
| Ni | 1.0 | 0.3 | 0.1 | 0.0 | 0.1 | 0.0 | 2.1 | 0.6 | 0.2 | 0.1 | 0.2 | 0.0 | 2.1 | 0.7 | 0.1 | 0.1 | 0.2 | 0.0 | 1.2 | 0.3 | 0.1 | 0.1 | 0.0 | 0.0 |
| Enrichment (E) 2 | ||||||||||||||||||||||||
| H | 0.95 | – | – | – | – | – | 0.93 | – | – | – | – | – | 0.93 | – | – | – | – | – | 0.99 | – | – | – | – | – |
| C | 0.99 | 1.43 | – | – | – | – | 1.00 | 1.31 | – | – | – | – | 1.12 | 0.77 | – | – | – | – | 0.97 | 1.25 | – | – | – | – |
| N | 1.33 | 0.29 | – | – | – | – | 1.23 | 0.79 | – | – | – | – | 1.25 | 0.74 | – | – | – | – | 1.12 | 0.73 | – | – | – | – |
| O | 1.10 | – | – | – | – | – | 0.97 | – | – | – | – | – | 0.58 | – | – | – | – | – | 1.20 | 0.33 | – | – | – | – |
| S | 1.38 | 0.05 | – | – | – | – | 1.44 | 0.04 | – | – | – | – | 1.27 | 0.04 | – | – | – | – | 1.19 | – | – | – | – | – |
| Ni | 1.40 | – | – | – | – | – | 1.10 | – | – | – | – | – | 1.10 | – | – | – | – | – | 0.67 | – | – | – | – | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmoudi, G.; Babashkina, M.G.; Maniukiewicz, W.; Afkhami, F.A.; Nunna, B.B.; Zubkov, F.I.; Ptaszek, A.L.; Szczepanik, D.W.; Mitoraj, M.P.; Safin, D.A. Solvent-Induced Formation of Novel Ni(II) Complexes Derived from Bis-Thiosemicarbazone Ligand: An Insight from Experimental and Theoretical Investigations. Int. J. Mol. Sci. 2021, 22, 5337. https://doi.org/10.3390/ijms22105337
Mahmoudi G, Babashkina MG, Maniukiewicz W, Afkhami FA, Nunna BB, Zubkov FI, Ptaszek AL, Szczepanik DW, Mitoraj MP, Safin DA. Solvent-Induced Formation of Novel Ni(II) Complexes Derived from Bis-Thiosemicarbazone Ligand: An Insight from Experimental and Theoretical Investigations. International Journal of Molecular Sciences. 2021; 22(10):5337. https://doi.org/10.3390/ijms22105337
Chicago/Turabian StyleMahmoudi, Ghodrat, Maria G. Babashkina, Waldemar Maniukiewicz, Farhad Akbari Afkhami, Bharath Babu Nunna, Fedor I. Zubkov, Aleksandra L. Ptaszek, Dariusz W. Szczepanik, Mariusz P. Mitoraj, and Damir A. Safin. 2021. "Solvent-Induced Formation of Novel Ni(II) Complexes Derived from Bis-Thiosemicarbazone Ligand: An Insight from Experimental and Theoretical Investigations" International Journal of Molecular Sciences 22, no. 10: 5337. https://doi.org/10.3390/ijms22105337
APA StyleMahmoudi, G., Babashkina, M. G., Maniukiewicz, W., Afkhami, F. A., Nunna, B. B., Zubkov, F. I., Ptaszek, A. L., Szczepanik, D. W., Mitoraj, M. P., & Safin, D. A. (2021). Solvent-Induced Formation of Novel Ni(II) Complexes Derived from Bis-Thiosemicarbazone Ligand: An Insight from Experimental and Theoretical Investigations. International Journal of Molecular Sciences, 22(10), 5337. https://doi.org/10.3390/ijms22105337