
Neuroprotective Studies of Evodiamine in an Okadaic Acid-Induced Neurotoxicity


Abstract
:1. Introduction
2. Results
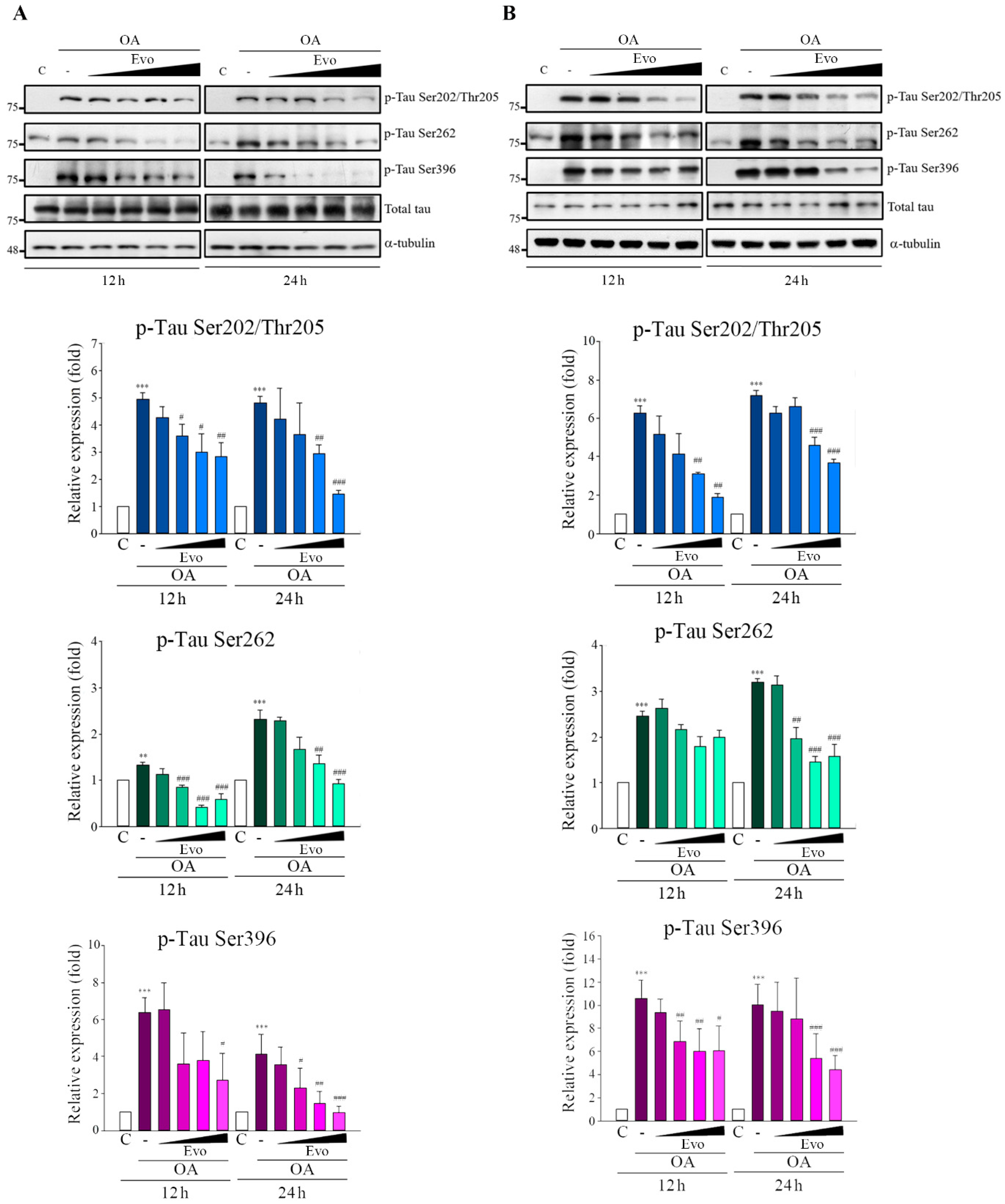
2.1. Evodiamine Significantly Inhibited Hyperphosphorylation of Tau
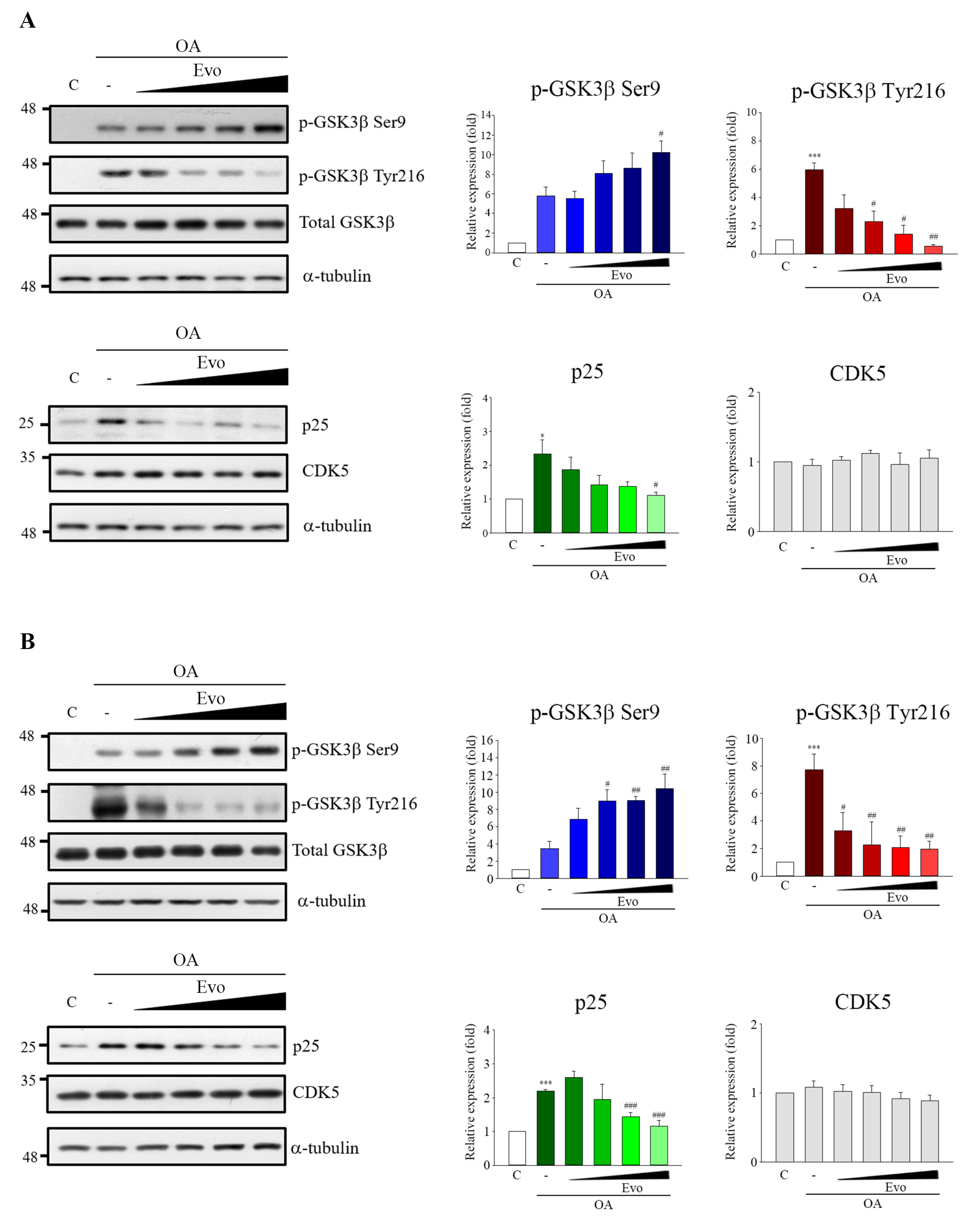
2.2. Studies on Mechanism Underlying Phosphorylated Tau Inhibition in Response to Evodiamine
2.3. Neuroprotective Effects of Evodiamine
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. PP2A Activity
4.4. Estimation of Lipid Peroxidation
4.5. Estimation of Glutathione
4.6. Flow Cytometry
4.7. Subcellular Fractionation
4.8. Immunoblot Analyses
4.9. Surgery and Microinjection for Intracerebroventricular (ICV) Administration of Okadaic Acid
4.10. Analysis of Cognitive Dysfunction
4.11. Morris Water Maze
4.12. Magnetic Resonance Imaging (MRI)
4.13. Passive Avoidance Test
4.14. Immunohistochemical Staining
4.15. Data Analysis and Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- World Health Organization. Updated September 2019. Available online: https://www.who.int/features/factfiles/dementia/en/ (accessed on 1 March 2021).
- Kurz, A.; Perneczky, R. Novel insights for the treatment of Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 373–379. [Google Scholar] [CrossRef]
- Anand, A.; Patience, A.A.; Sharma, N.; Khurana, N. The present and future of pharmacotherapy of Alzheimer’s disease: A comprehensive review. Eur. J. Pharmacol. 2017, 815, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Fuchsberger, T.; Giraldo, E.; Viña, J. Molecular mechanisms linking amyloid-β toxicity and tau hyperphosphorylation in Alzheimer’s disease. Free Radic. Biol. Med. 2015, 83, 186–191. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Nakano, Y.; Kizaki, M.; Hoshikuma, K.; Yokoo, Y.; Kamiya, T. Capsaicin-like anti-obese activities of evodiamine from fruits of Evodia rutaecarpa, a vanilloid receptor agonist. Planta Med. 2001, 67, 628–633. [Google Scholar] [CrossRef] [Green Version]
- Lv, Q.; Xue, Y.; Li, G.; Zou, L.; Zhang, X.; Ying, M.; Wang, S.; Guo, L.; Gao, Y.; Li, G.; et al. Beneficial effects of evodiamine on P2X4-mediated inflammatory injury of human umbilical vein endothelial cells due to high glucose. Int. Immunopharmacol. 2015, 28, 1044–1049. [Google Scholar] [CrossRef]
- Iwaoka, E.; Wang, S.; Matsuyoshi, N.; Kogure, Y.; Aoki, S.; Yamamoto, S.; Noguchi, K.; Dai, Y. Evodiamine suppresses capsaicin-induced thermal hyperalgesia through activation and subsequent desensitization of the transient receptor potential V1 channels. J. Nat. Med. 2016, 70, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Y.; Chang, M.C.; Chen, C.S.; Lin, H.C.; Tsai, H.P.; Yang, C.C.; Yang, C.H.; Lin, C.M. Topoisomerase I inhibitor evodiamine acts as an antibacterial agent against drug-resistant Klebsiella pneumonia. Planta Med. 2013, 79, 27–29. [Google Scholar] [PubMed] [Green Version]
- Jiang, M.L.; Zhang, Z.X.; Li, Y.Z.; Wang, X.H.; Yan, W.; Gong, G.Q. Antidepressant-like effect of evodiamine on chronic unpredictable mild stress rats. Neurosci. Lett. 2015, 588, 154–158. [Google Scholar] [CrossRef]
- Zhao, T.; Zhang, X.; Zhao, Y.; Zhang, L.; Bai, X.; Zhang, J.; Zhao, X.; Chen, L.; Wang, L.; Cui, L. Pretreatment by evodiamine is neuroprotective in cerebral ischemia: Up-regulated pAkt, pGSK3β, down-regulated NF-κB expression, and ameliorated BBB permeability. Neurochem. Res. 2014, 39, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J.; Wang, C.; Li, Z.; Liu, X.; Zhang, J.; Lu, J.; Wang, D. Pharmacological basis for the use of evodiamine in Alzheimer’s disease: Antioxidation and antiapoptosis. Int. J. Mol. Sci. 2018, 19, 1527. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, C.; Liu, L.; Li, S. Protective effects of evodiamine in experimental paradigm of Alzheimer’s disease. Cog. Neurodyn. 2018, 12, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.M.; Huang, F.I.; Yang, C.R. Moscatilin ameliorates tau phosphorylation and cognitive deficits in Alzheimer’s disease models. J. Nat. Prod. 2019, 82, 1979–1988. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Yang, Y.F.; Yang, X.W. Blood-brain barrier permeability and neuroprotective effects of three main alkaloids from the fruits of Euodia rutaecarpa with MDCK-pHaMDR cell monolayer and PC12 cell line. Biomed. Pharmacother. 2018, 98, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased levels of active GSK-3β in Alzheimer’s disease and accumulation in argyrophilic grains and in neurons at different stages of neurofibrillary degeneration. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef]
- Das, T.K.; Jana, P.; Chakrabarti, S.K.; Abdul Hamid, M.R.W. Curcumin downregulates GSK3 and Cdk5 in scopolamine-induced Alzheimer’s disease rats abrogating Aβ40/42 and tau hyperphosphorylation. J. Alzheimer’s Dis. Rep. 2019, 3, 257–267. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.; Ayton, S.; Bush, A.I.; Adlard, P.A. GSK-3 in neurodegenerative diseases. Int. J. Alzheimer’s Dis. 2011, 2011, 189246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Sharmg, G.; Ishiguro, K.; Hisanaga, S.I. Phospho-tau bar code: Analysis of phosphoisotypes of tau and its application to tauopathy. Front. Neurosci. 2018, 12, 44. [Google Scholar] [CrossRef]
- Ravindran, J.; Gupta, N.; Agrawal, M.; Bala Bhaskar, A.S.; Lakshmana Rao, P.V. Modulation of ROS/MAPK signaling pathways by okadaic acid leads to cell death via, mitochondrial mediated caspase-dependent mechanism. Apoptosis 2011, 16, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Dou, F.; Netzer, W.J.; Tanemura, K.; Li, F.; Hartl, F.U.; Takashima, A.; Gouras, G.K.; Greengard, P.; Xu, H. Chaperones increase association of tau protein with microtubule. Proc. Natl. Acad. Sci. USA 2003, 100, 721–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ropele, S.; Schmidt, R.; Enzinger, C.; Windisch, M.; Martinez, N.P.; Fazekas, F. Longitudinal magnetization transfer imaging in mild to severe Alzheimer disease. Am. J. Neuroradiol. 2012, 33, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Meng, T.; Fu, S.; He, D.; Hu, G.; Gao, X.; Zhang, Y.; Huang, B.; Du, J.; Zhou, A.; Su, Y.; et al. Evodiamine inhibits lipopolysaccharide (LPS)-induced inflammation in BV-2 cells via regulating AKT/Nrf2/HO-1/NF-κB signaling axis. Cell. Mol. Neurobiol. 2021, 41, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Simpkins, J.W. An okadaic acid-induced model of tauopathy and cognitive deficiency. Brain Res. 2010, 1359, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.; Gómez de Barreda, E.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A possible link between beta amyloid peptide and tau protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef]
- Engel, T.; Lucas, J.J.; Gomez-Ramos, P.; Moran, M.A.; Avila, J.; Hernandez, F. Coexpression of FTDP-17 tau and GSK-3β in transgenic mice induce tau polymerization and neurodegeneration. Neurobiol. Aging 2006, 27, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Kanungo, J.; Zheng, Y.L.; Amin, N.D.; Pant, H.C. Targeting Cdk5 activity in neuronal degeneration and regeneration. Cell. Mol. Neurobiol. 2009, 29, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Hancock, J.T.; Desikan, R.; Neill, S.J. Role of reactive oxygen species in cell signaling pathways. Biochem. Soc. Trans. 2001, 29, 345–350. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group Treatment | Oral (d 3–30) a | ICV Injection (d 7–11) b |
|---|---|---|
| Control | Vehicle c | No treatment |
| Sham | Vehicle c | Sterile artificial cerebrospinal fluid (ACSF) 1 μL |
| Okadaic acid | Vehicle c | Okadaic acid 100 ng/1 μL ACSF |
| 50 mg/kg Evodiamine | 50 mg/kg evodiamine (vehicle) c | Okadaic acid 100 ng/1 μL ACSF |
| 100 mg/kg Evodiamine | 100 mg/kg evodiamine (vehicle) c | Okadaic acid 100 ng/1 μL ACSF |
| Donepezil | 2.5 mg/kg donepezil (saline) | Okadaic acid 100 ng/1 μL ACSF |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chou, C.-H.; Yang, C.-R. Neuroprotective Studies of Evodiamine in an Okadaic Acid-Induced Neurotoxicity. Int. J. Mol. Sci. 2021, 22, 5347. https://doi.org/10.3390/ijms22105347
Chou C-H, Yang C-R. Neuroprotective Studies of Evodiamine in an Okadaic Acid-Induced Neurotoxicity. International Journal of Molecular Sciences. 2021; 22(10):5347. https://doi.org/10.3390/ijms22105347
Chicago/Turabian StyleChou, Ching-Hsuan, and Chia-Ron Yang. 2021. "Neuroprotective Studies of Evodiamine in an Okadaic Acid-Induced Neurotoxicity" International Journal of Molecular Sciences 22, no. 10: 5347. https://doi.org/10.3390/ijms22105347
APA StyleChou, C. -H., & Yang, C. -R. (2021). Neuroprotective Studies of Evodiamine in an Okadaic Acid-Induced Neurotoxicity. International Journal of Molecular Sciences, 22(10), 5347. https://doi.org/10.3390/ijms22105347