
Beneficial Effects of Metformin on the Central Nervous System, with a Focus on Epilepsy and Lafora Disease


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Proposed Mechanism of Action of Metformin to Lower Glucose Levels
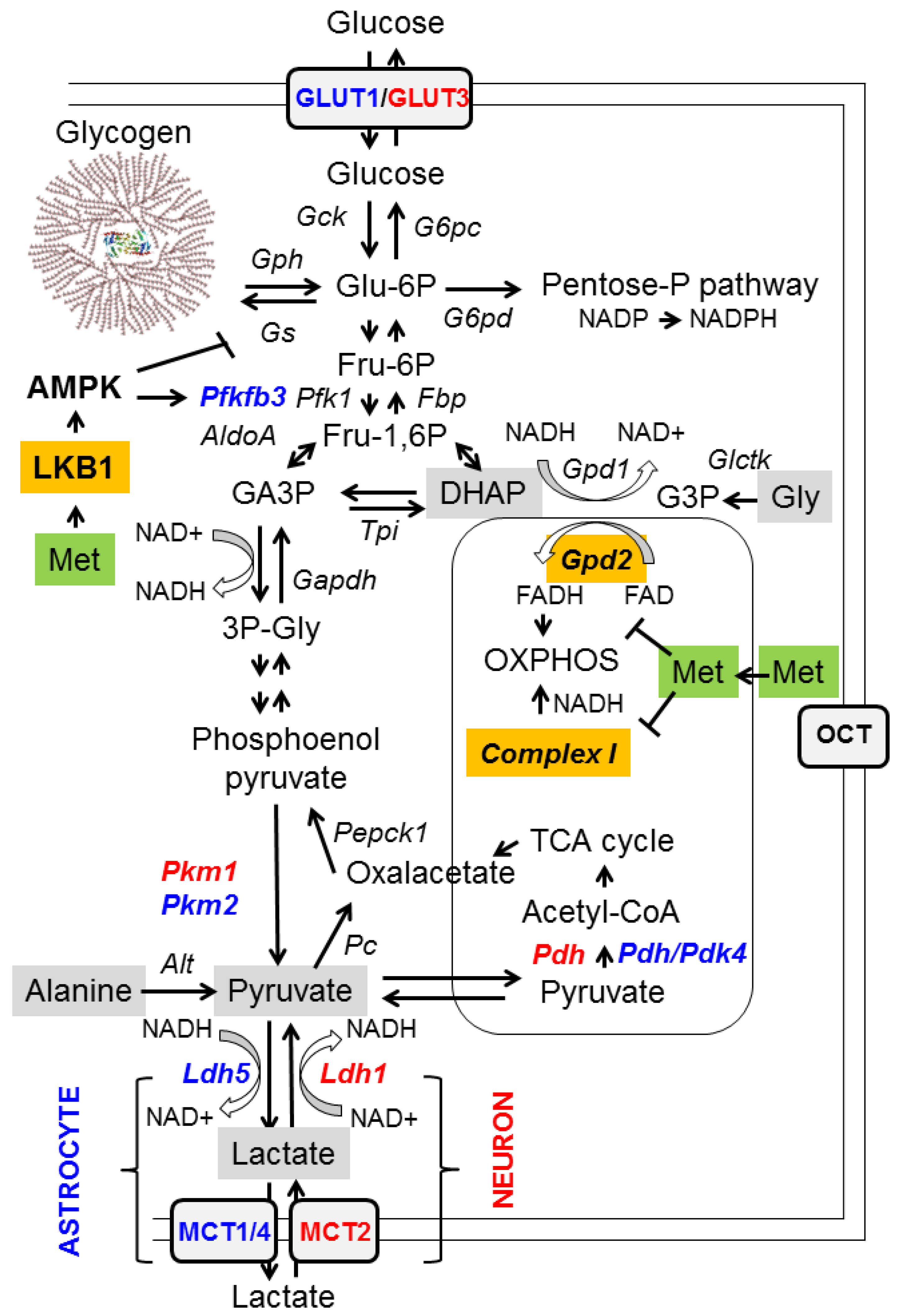
2.1. Inhibition of Mitochondrial Glycerol-3-Phosphate Dehydrogenase (GPD2)
- (a)
- At the transcriptional level: metformin prevents cAMP responsive binding (CREB)-mediated transcription of the gluconeogenic glucose 6-phosphatase (G6PC) and phosphoenolpyruvate carboxykinase 1 (PEPCK1) genes. This is an indirect effect due to the inhibition of mitochondrial complex I by metformin (see below), leading to an increase in AMP levels, which inhibits adenylate cyclase and thus leads to a decrease in levels of cAMP, a mediator of CREB-dependent transcription. In addition, metformin has been proposed to activate AMP-activated protein kinase (AMPK), which has a negative effect on the transcriptional regulation of gluconeogenesis genes, among others [4] (see below);
- (b)
- Reducing the availability of gluconeogenic substrates: hepatic gluconeogenesis depends on the availability of appropriate substrates, such as glycerol, lactate, pyruvate, alanine, and dihydroxyacetone phosphate (DHAP), in order to convert them to glucose (Figure 1, grey boxes). Glycerol and DHAP are mutually interconnected, since glycerol is converted to glycerol 3-P (G3P) by glycerate kinase (Glctk) and then G3P is converted to DHAP by mitochondrial glycerol-3-phosphate dehydrogenase (GPD2). Metformin has been shown to inhibit mitochondrial GPD2 at regular concentrations (50–100 μM) (reviewed in [1,2,3]; Figure 1, orange box). Therefore, after treatment with metformin, the levels of DHAP are reduced, and this leads to a decrease in the flux of gluconeogenesis. As a consequence of this inhibition, G3P and glycerol accumulate in hepatocytes (Figure 1).
2.2. Inhibition of Mitochondrial Complex I of the Respiratory Chain and Activation of AMP-Activated Protein Kinase (AMPK)
2.3. Effects of Metformin on Glucose Metabolism in the Brain
2.4. Metformin Ameliorates Oxidative Stress
2.5. Metformin and Neuroinflammation
3. Metformin as a Neuroprotective Agent in Different Neurological Disorders
3.1. Alzheimer’s Disease
3.2. Parkinson’s Disease
3.3. Huntington’s Disease
3.4. Multiple Sclerosis
3.5. Epilepsy
4. Metformin and Lafora Disease
4.1. Clinical Aspects of Lafora Disease
4.2. Animal Models of Lafora Disease
4.3. Pharmacological Interventions in Animal Models of Lafora Disease
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- LaMoia, T.E.; Shulman, G.I. Cellular and molecular mechanisms of metformin action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Demare, S.; Kothari, A.; Calcutt, N.A.; Fernyhough, P. Metformin as a potential therapeutic for neurological disease: Mobilizing ampk to repair the nervous system. Expert Rev. Neurother. 2021, 21, 45–63. [Google Scholar] [CrossRef]
- Wang, D.S.; Jonker, J.W.; Kato, Y.; Kusuhara, H.; Schinkel, A.H.; Sugiyama, Y. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J. Pharmacol. Exp. Ther. 2002, 302, 510–515. [Google Scholar] [CrossRef] [Green Version]
- Alnouti, Y.; Petrick, J.S.; Klaassen, C.D. Tissue distribution and ontogeny of organic cation transporters in mice. Drug Metab. Dispos. 2006, 34, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Russo, E.; Pellino, G.; D’Angelo, S.; Chiaravalloti, A.; De Sarro, G.; Manfredini, R.; De Giorgio, R. Metformin and autoimmunity: A “new deal” of an old drug. Front. Immunol. 2018, 9, 1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, E.M.; Mendoza, S.; Hamer, T.; Sosa, F.; Glueck, C.J. Metformin therapy in polycystic ovary syndrome reduces hyperinsulinemia, insulin resistance, hyperandrogenemia, and systolic blood pressure, while facilitating normal menses and pregnancy. Metabolism 1994, 43, 647–654. [Google Scholar] [CrossRef]
- Lexis, C.P.; van der Horst-Schrivers, A.N.; Lipsic, E.; Valente, M.A.; Muller Kobold, A.C.; de Boer, R.A.; van Veldhuisen, D.J.; van der Harst, P.; van der Horst, I.C. The effect of metformin on cardiovascular risk profile in patients without diabetes presenting with acute myocardial infarction: Data from the glycometabolic intervention as adjunct to primary coronary intervention in st elevation myocardial infarction (gips-iii) trial. BMJ Open Diabetes Res. Care 2015, 3, e000090. [Google Scholar]
- Petrie, J.R.; Chaturvedi, N.; Ford, I.; Brouwers, M.; Greenlaw, N.; Tillin, T.; Hramiak, I.; Hughes, A.D.; Jenkins, A.J.; Klein, B.E.K.; et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (removal): A double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.W.; He, S.J.; Feng, X.; Cheng, J.; Luo, Y.T.; Tian, L.; Huang, Q. Metformin: A review of its potential indications. Drug Des. Devel. Ther. 2017, 11, 2421–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [Green Version]
- Li, D. Metformin as an antitumor agent in cancer prevention and treatment. J. Diabetes 2011, 3, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a tool to target aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piskovatska, V.; Storey, K.B.; Vaiserman, A.M.; Lushchak, O. The use of metformin to increase the human healthspan. Adv. Exp. Med. Biol. 2020, 1260, 319–332. [Google Scholar]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 2020, 32, 44–55.e46. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The effects of metformin on gut microbiota and the immune system as research frontiers. Diabetologia 2017, 60, 1662–1667. [Google Scholar] [CrossRef] [Green Version]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The therapeutic potential of metformin in neurodegenerative diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex i. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Carling, D. Amp-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- Greer, E.L.; Dowlatshahi, D.; Banko, M.R.; Villen, J.; Hoang, K.; Blanchard, D.; Gygi, S.P.; Brunet, A. An ampk-foxo pathway mediates longevity induced by a novel method of dietary restriction in c. Elegans. Curr. Biol. 2007, 17, 1646–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase lkb1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [Green Version]
- Bolanos, J.P. Bioenergetics and redox adaptations of astrocytes to neuronal activity. J. Neurochem. 2016, 139 (Suppl. 2), 115–125. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Berthier, A.; Paya, M.; Garcia-Cabrero, A.M.; Ballester, M.I.; Heredia, M.; Serratosa, J.M.; Sanchez, M.P.; Sanz, P. Pharmacological interventions to ameliorate neuropathological symptoms in a mouse model of lafora disease. Mol. Neurobiol. 2016, 53, 1296–1309. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, N.; Iannantuoni, F.; Gruevska, A.; Muntane, J.; Rocha, M.; Victor, V.M. Mechanisms of action of metformin in type 2 diabetes: Effects on mitochondria and leukocyte-endothelium interactions. Redox Biol. 2020, 34, 101517. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Andreasson, K.I.; Bachstetter, A.D.; Colonna, M.; Ginhoux, F.; Holmes, C.; Lamb, B.; Landreth, G.; Lee, D.C.; Low, D.; Lynch, M.A.; et al. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J. Neurochem. 2016, 138, 653–693. [Google Scholar] [CrossRef] [PubMed]
- Sanz, P.; Garcia-Gimeno, M.A. Reactive glia inflammatory signaling pathways and epilepsy. Int. J. Mol. Sci. 2020, 21, 4096. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Ye, S.; Wang, S.; Sun, W.; Hu, Y. Metformin inhibits nuclear factor-kappab activation and inflammatory cytokines expression induced by high glucose via adenosine monophosphate-activated protein kinase activation in rat glomerular mesangial cells in vitro. Chin. Med. J. 2014, 127, 1755–1760. [Google Scholar] [PubMed]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.; Savinko, T.; Wong, A.K.; et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowicz-Piasecka, M.; Sikora, J.; Szydlowska, A.; Skupien, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin—A future therapy for neurodegenerative diseases: Theme: Drug discovery, development and delivery in alzheimer’s disease guest editor: Davide brambilla. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Hung, T.H.; Lee, C.Y.; Wang, L.F.; Wu, C.H.; Ke, C.H.; Chen, S.F. Berberine protects against neuronal damage via suppression of glia-mediated inflammation in traumatic brain injury. PLoS ONE 2014, 9, e115694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Tang, H.; Li, W.; Gong, Y.; Li, S.; Huang, J.; Fang, Y.; Yuan, W.; Liu, Y.; Wang, S.; et al. Ampk and its activator berberine in the treatment of neurodegenerative diseases. Curr. Pharm. Des. 2020, 26, 5054–5066. [Google Scholar] [CrossRef]
- Giri, S.; Nath, N.; Smith, B.; Viollet, B.; Singh, A.K.; Singh, I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: A possible role of amp-activated protein kinase. J. Neurosci. 2004, 24, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhun, B.S.; Jin, Q.; Oh, Y.T.; Kim, S.S.; Kong, Y.; Cho, Y.H.; Ha, J.; Baik, H.H.; Kang, I. 5-aminoimidazole-4-carboxamide riboside suppresses lipopolysaccharide-induced tnf-alpha production through inhibition of phosphatidylinositol 3-kinase/akt activation in raw 264.7 murine macrophages. Biochem. Biophys. Res. Commun. 2004, 318, 372–380. [Google Scholar] [CrossRef]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. Amp-activated protein kinase inhibits nf-kappab signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.M.; Nicol, C.J.; Cheng, Y.C.; Lin, K.H.; Chen, Y.L.; Pei, D.; Lin, C.H.; Shih, Y.N.; Yen, C.H.; Chen, S.J.; et al. Metformin activation of ampk suppresses age-induced inflammatory response in hnscs. Exp. Cell Res. 2017, 352, 75–83. [Google Scholar] [CrossRef]
- Bayliss, J.A.; Lemus, M.B.; Santos, V.V.; Deo, M.; Davies, J.S.; Kemp, B.E.; Elsworth, J.D.; Andrews, Z.B. Metformin prevents nigrostriatal dopamine degeneration independent of ampk activation in dopamine neurons. PLoS ONE 2016, 11, e0159381. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Cui, W.; Chen, S.; Shao, Z.; Li, Y.; Wang, W.; Mao, L.; Li, J.; Mei, X. Metformin alleviates high glucose-induced er stress and inflammation by inhibiting the interaction between caveolin1 and ampkalpha in rat astrocytes. Biochem. Biophys. Res. Commun. 2021, 534, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, J.L.; Hellberg, K.; Luo, E.C.; Van Nostrand, E.L.; Dayn, A.; Yu, J.; Shokhirev, M.N.; Dayn, Y.; Yeo, G.W.; Shaw, R.J. Ampk regulation of raptor and tsc2 mediate metformin effects on transcriptional control of anabolism and inflammation. Genes Dev. 2020, 34, 1330–1344. [Google Scholar] [CrossRef] [PubMed]
- Soo, S.K.; Rudich, P.D.; Traa, A.; Harris-Gauthier, N.; Shields, H.J.; Van Raamsdonk, J.M. Compounds that extend longevity are protective in neurodegenerative diseases and provide a novel treatment strategy for these devastating disorders. Mech. Ageing Dev. 2020, 190, 111297. [Google Scholar] [CrossRef] [PubMed]
- Orkaby, A.R.; Cho, K.; Cormack, J.; Gagnon, D.R.; Driver, J.A. Metformin vs sulfonylurea use and risk of dementia in us veterans aged >/= 65 years with diabetes. Neurology 2017, 89, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. Sirt1 deacetylase protects against neurodegeneration in models for alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef]
- DiTacchio, K.A.; Heinemann, S.F.; Dziewczapolski, G. Metformin treatment alters memory function in a mouse model of alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, S.P.; Jain, P.D.; Ghumatkar, P.J.; Tambe, R.; Sathaye, S. Neuroprotective effect of metformin in mptp-induced parkinson’s disease in mice. Neuroscience 2014, 277, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.S.; Choi, D.Y. Metformin lowers alpha-synuclein phosphorylation and upregulates neurotrophic factor in the mptp mouse model of parkinson’s disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Shaikh, M.F.; Othman, I. Emerging neuroprotective effect of metformin in parkinson’s disease: A molecular crosstalk. Pharmacol. Res. 2020, 152, 104593. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Sanchis, A.; Garcia-Gimeno, M.A.; Canada-Martinez, A.J.; Sequedo, M.D.; Millan, J.M.; Sanz, P.; Vazquez-Manrique, R.P. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zq175 mouse model of huntington disease. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Antel, J.P.; Lin, Y.H.; Cui, Q.L.; Pernin, F.; Kennedy, T.E.; Ludwin, S.K.; Healy, L.M. Immunology of oligodendrocyte precursor cells in vivo and in vitro. J. Neuroimmunol. 2019, 331, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, L.P.; Krotenko, N.V.; Grishko, E.V.; Krotenko, N.M.; Alifirova, V.M.; Ivanova, S.A. State of antioxidant system in patients with multiple sclerosis during therapy. Biomed. Khim. 2011, 57, 661–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziedzic, A.; Saluk-Bijak, J.; Miller, E.; Bijak, M. Metformin as a potential agent in the treatment of multiple sclerosis. Int. J. Mol. Sci. 2020, 21, 5957. [Google Scholar] [CrossRef] [PubMed]
- Paintlia, A.S.; Paintlia, M.K.; Mohan, S.; Singh, A.K.; Singh, I. Amp-activated protein kinase signaling protects oligodendrocytes that restore central nervous system functions in an experimental autoimmune encephalomyelitis model. Am. J. Pathol. 2013, 183, 526–541. [Google Scholar] [CrossRef] [Green Version]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef]
- Citraro, R.; Leo, A.; Constanti, A.; Russo, E.; De Sarro, G. Mtor pathway inhibition as a new therapeutic strategy in epilepsy and epileptogenesis. Pharmacol. Res. 2016, 107, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Wang, C.; Yao, Y.; Chen, L.; Liu, G.; Zhang, R.; Liu, Q.; Shi, F.D.; Hao, J. Mtorc1 pathway disruption ameliorates brain inflammation following stroke via a shift in microglia phenotype from m1 type to m2 type. FASEB J. 2016, 30, 3388–3399. [Google Scholar] [CrossRef] [Green Version]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of ampk, inhibits mtorc1 in a rag gtpase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Mehrabi, S.; Sanadgol, N.; Barati, M.; Shahbazi, A.; Vahabzadeh, G.; Barzroudi, M.; Seifi, M.; Gholipourmalekabadi, M.; Golab, F. Evaluation of metformin effects in the chronic phase of spontaneous seizures in pilocarpine model of temporal lobe epilepsy. Metab. Brain. Dis. 2018, 33, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Bojja, S.L.; Medhi, B.; Anand, S.; Bhatia, A.; Joshi, R.; Minz, R.W. Metformin ameliorates the status epilepticus- induced hippocampal pathology through possible mtor modulation. Inflammopharmacology 2021, 29, 137–151. [Google Scholar] [CrossRef]
- Rubio Osornio, M.D.C.; Custodio Ramirez, V.; Calderon Gamez, D.; Paz Tres, C.; Carvajal Aguilera, K.G.; Phillips Farfan, B.V. Metformin plus caloric restriction show anti-epileptic effects mediated by mtor pathway inhibition. Cell Mol. Neurobiol. 2018, 38, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.A.E.; Abdel-Rahman, R.F.; Mahmoud, S.S.; Khattab, M.M.; Safar, M.M. Metformin and trimetazidine ameliorate diabetes-induced cognitive impediment in status epileptic rats. Epilepsy Behav. 2020, 104, 106893. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.R.; Xu, X.C.; Xu, F.; Zhang, W.L.; Liu, L.M.; Wang, W.P. Metformin protects against seizures, learning and memory impairments and oxidative damage induced by pentylenetetrazole-induced kindling in mice. Biochem. Biophys. Res. Commun. 2014, 448, 414–417. [Google Scholar] [CrossRef]
- Hussein, A.M.; Eldosoky, M.; El-Shafey, M.; El-Mesery, M.; Ali, A.N.; Abbas, K.M.; Abulseoud, O.A. Effects of metformin on apoptosis and alpha-synuclein in a rat model of pentylenetetrazole-induced epilepsy. Can. J. Physiol. Pharmacol. 2019, 97, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zheng, G.; Guo, H.; Shi, Z.N.; Jiang, J.; Wang, X.Y.; Yang, X.; Liu, X.Y. The effect of metformin treatment on endoplasmic reticulum (er) stress induced by status epilepticus (se) via the perk-eif2alpha-chop pathway. Bosn. J. Basic Med. Sci. 2018, 18, 49–54. [Google Scholar] [PubMed] [Green Version]
- Zeyghami, M.A.; Hesam, E.; Khadivar, P.; Hesam, H.K.; Ahmadnia, A.; Amini, A. Effects of atorvastatin and metformin on development of pentylenetetrazole-induced seizure in mice. Heliyon 2020, 6, e03761. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, B.; Zheng, F.; Li, Y.; Zhang, Y.; Hu, Y.; Wang, X. Chronic metformin treatment facilitates seizure termination. Biochem. Biophys. Res. Commun. 2017, 484, 450–455. [Google Scholar] [CrossRef]
- Vazifehkhah, S.; Khanizadeh, A.M.; Mojarad, T.B.; Nikbakht, F. The possible role of progranulin on anti-inflammatory effects of metformin in temporal lobe epilepsy. J. Chem. Neuroanat. 2020, 109, 101849. [Google Scholar] [CrossRef] [PubMed]
- Vazifehkhah, S.; Ali, M.K.; Babae, J.F.; Hashemi, P.; Alireza, M.S.; Nikbakht, F. Evaluation of the ameliorative effects of oral administration of metformin on epileptogenesis in the temporal lobe epilepsy model in rats. Life Sci. 2020, 257, 118066. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflugers. Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- Muraleedharan, R.; Gawali, M.V.; Tiwari, D.; Sukumaran, A.; Oatman, N.; Anderson, J.; Nardini, D.; Bhuiyan, M.A.N.; Tkac, I.; Ward, A.L.; et al. Ampk-regulated astrocytic lactate shuttle plays a non-cell-autonomous role in neuronal survival. Cell Rep. 2020, 32, 108092. [Google Scholar] [CrossRef] [PubMed]
- Nesci, V.; Russo, E.; Arcidiacono, B.; Citraro, R.; Tallarico, M.; Constanti, A.; Brunetti, A.; De Sarro, G.; Leo, A. Metabolic alterations predispose to seizure development in high-fat diet-treated mice: The role of metformin. Mol. Neurobiol. 2020, 57, 4778–4789. [Google Scholar] [CrossRef] [PubMed]
- Nandini, H.S.; Paudel, Y.N.; Krishna, K.L. Envisioning the neuroprotective effect of metformin in experimental epilepsy: A portrait of molecular crosstalk. Life Sci. 2019, 233, 116686. [Google Scholar]
- Yimer, E.M.; Surur, A.; Wondafrash, D.Z.; Gebre, A.K. The effect of metformin in experimentally induced animal models of epileptic seizure. Behav. Neurol. 2019, 2019, 6234758. [Google Scholar] [CrossRef]
- Pilon, G.; Dallaire, P.; Marette, A. Inhibition of inducible nitric-oxide synthase by activators of amp-activated protein kinase: A new mechanism of action of insulin-sensitizing drugs. J. Biol. Chem. 2004, 279, 20767–20774. [Google Scholar] [CrossRef] [Green Version]
- Brueggeman, L.; Sturgeon, M.L.; Martin, R.M.; Grossbach, A.J.; Nagahama, Y.; Zhang, A.; Howard, M.A., 3rd; Kawasaki, H.; Wu, S.; Cornell, R.A.; et al. Drug repositioning in epilepsy reveals novel antiseizure candidates. Ann. Clin. Transl. Neurol. 2019, 6, 295–309. [Google Scholar] [CrossRef]
- Lafora, G.R.; Glueck, B. Beitrag zur histogpathologie der myoklonischen epilepsie. Gesamte Neurol. Psychiatr. 1911, 6, 1–14. [Google Scholar] [CrossRef]
- Harriman, D.G.; Millar, J.H.; Stevenson, A.C. Progressive familial myoclonic epilepsy in three families: Its clinical features and pathological basis. Brain 1955, 78, 325–349. [Google Scholar] [CrossRef]
- Schwarz, G.A.; Yanoff, M. Lafora’s disease. Distinct clinico-pathologic form of unverricht’s syndrome. Arch. Neurol. 1965, 12, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, S.; Austin, J.; Witmer, F.; Sakai, M. Studies in myoclonus epilepsy (lafora body form). I. Isolation and preliminary characterization of lafora bodies in two cases. Arch. Neurol. 1968, 19, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Austin, J.; Witmer, F.; Trueb, L. Studies in myoclonus epilepsy (lafora body form). Ii. Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology 1970, 20, 160–176. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; Karpati, G. Ultrastructural findings in lafora disease. Ann. Neurol. 1981, 10, 63–64. [Google Scholar] [CrossRef]
- Berkovic, S.F.; Andermann, F.; Carpenter, S.; Wolfe, L.S. Progressive myoclonus epilepsies: Specific causes and diagnosis. N. Engl. J. Med. 1986, 315, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, J.B. Corpora-amylacea and the family of polyglucosan diseases. Brain. Res. Brain. Res. Rev. 1999, 29, 265–295. [Google Scholar] [CrossRef]
- Raben, N.; Danon, M.; Lu, N.; Lee, E.; Shliselfeld, L.; Skurat, A.V.; Roach, P.J.; Lawrence, J.C., Jr.; Musumeci, O.; Shanske, S.; et al. Surprises of genetic engineering: A possible model of polyglucosan body disease. Neurology 2001, 56, 1739–1745. [Google Scholar] [CrossRef]
- Roach, P.J.; Depaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [CrossRef] [Green Version]
- Gentry, M.S.; Roma-Mateo, C.; Sanz, P. Laforin, a protein with many faces: Glucan phosphatase, adapter protein; et alii. FEBS J. 2013, 280, 525–537. [Google Scholar] [CrossRef] [Green Version]
- Duran, J.; Guinovart, J.J. Brain glycogen in health and disease. Mol. Asp. Med. 2015, 46, 70–77. [Google Scholar] [CrossRef]
- Brewer, M.K.; Putaux, J.L.; Rondon, A.; Uittenbogaard, A.; Sullivan, M.A.; Gentry, M.S. Polyglucosan body structure in lafora disease. Carbohydr. Polym. 2020, 240, 116260. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Delgado-Escueta, A.V.; Sakamoto, T.; Avila, M.R.; Machado-Salas, J.; Hoshii, Y.; Akagi, T.; Gomi, H.; Suzuki, T.; Amano, K.; et al. Targeted disruption of the epm2a gene causes formation of lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 2002, 11, 1251–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePaoli-Roach, A.A.; Tagliabracci, V.S.; Segvich, D.M.; Meyer, C.M.; Irimia, J.M.; Roach, P.J. Genetic depletion of the malin e3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J. Biol. Chem. 2010, 285, 25372–25381. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, J.; Wang, P.; Girard, J.M.; Ruggieri, A.; Wang, T.J.; Draginov, A.G.; Kameka, A.P.; Pencea, N.; Zhao, X.; Ackerley, C.A.; et al. Glycogen hyperphosphorylation underlies lafora body formation. Ann. Neurol. 2010, 68, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Valles-Ortega, J.; Duran, J.; Garcia-Rocha, M.; Bosch, C.; Saez, I.; Pujadas, L.; Serafin, A.; Canas, X.; Soriano, E.; Delgado-Garcia, J.M.; et al. Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of lafora disease. EMBO Mol. Med. 2011, 3, 667–681. [Google Scholar] [CrossRef]
- Criado, O.; Aguado, C.; Gayarre, J.; Duran-Trio, L.; Garcia-Cabrero, A.M.; Vernia, S.; San Millan, B.; Heredia, M.; Roma-Mateo, C.; Mouron, S.; et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum. Mol. Genet. 2012, 21, 1521–1533. [Google Scholar] [CrossRef]
- Sanchez-Elexpuru, G.; Serratosa, J.M.; Sanz, P.; Sanchez, M.P. 4-phenylbutyric acid and metformin decrease sensitivity to pentylenetetrazol-induced seizures in a malin knockout model of lafora disease. Neuroreport 2017, 28, 268–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkovic, S.F.; So, N.K.; Andermann, F. Progressive myoclonus epilepsies: Clinical and neurophysiological diagnosis. J. Clin. Neurophysiol. 1991, 8, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Serratosa, J.M. Idiopathic epilepsies with a complex mode of inheritance. Epilepsia 1999, 40 (Suppl. 3), 12–16. [Google Scholar] [CrossRef] [Green Version]
- Minassian, B.A. Lafora’s disease: Towards a clinical, pathologic, and molecular synthesis. Pediatr. Neurol. 2001, 25, 21–29. [Google Scholar] [CrossRef]
- Serratosa, J.M.; Delgado-Escueta, A.V.; Posada, I.; Shih, S.; Drury, I.; Berciano, J.; Zabala, J.A.; Antunez, M.C.; Sparkes, R.S. The gene for progressive myoclonus epilepsy of the lafora type maps to chromosome 6q. Hum. Mol. Genet. 1995, 4, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Minassian, B.A.; Lee, J.R.; Herbrick, J.A.; Huizenga, J.; Soder, S.; Mungall, A.J.; Dunham, I.; Gardner, R.; Fong, C.Y.; Carpenter, S.; et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 1998, 20, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Serratosa, J.M.; Gomez-Garre, P.; Gallardo, M.E.; Anta, B.; de Bernabe, D.B.; Lindhout, D.; Augustijn, P.B.; Tassinari, C.A.; Malafosse, R.M.; Topcu, M.; et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the lafora type (epm2). Hum. Mol. Genet. 1999, 8, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Young, E.J.; Ianzano, L.; Munteanu, I.; Zhao, X.; Christopoulos, C.C.; Avanzini, G.; Elia, M.; Ackerley, C.A.; Jovic, N.J.; et al. Mutations in nhlrc1 cause progressive myoclonus epilepsy. Nat. Genet. 2003, 35, 125–127. [Google Scholar] [CrossRef]
- Ganesh, S.; Agarwala, K.L.; Ueda, K.; Akagi, T.; Shoda, K.; Usui, T.; Hashikawa, T.; Osada, H.; Delgado-Escueta, A.V.; Yamakawa, K. Laforin, defective in the progressive myoclonus epilepsy of lafora type, is a dual-specificity phosphatase associated with polyribosomes. Hum. Mol. Genet. 2000, 9, 2251–2261. [Google Scholar] [CrossRef] [Green Version]
- Gentry, M.S.; Worby, C.A.; Dixon, J.E. Insights into lafora disease: Malin is an e3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. USA 2005, 102, 8501–8506. [Google Scholar] [CrossRef] [Green Version]
- Worby, C.A.; Gentry, M.S.; Dixon, J.E. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J. Biol. Chem. 2006, 281, 30412–30418. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Abad, C.; Gómez-Garre, P.; Gutiérrez-Delicado, E.; Saygi, S.; Michelucci, R.; Tassinari, C.A.; Rodriguez de Cordoba, S.; Serratosa, J.M. Lafora disease due to epm2b mutations. A clinical and genetic study. Neurology 2005, 64, 982–986. [Google Scholar] [CrossRef]
- Jara-Prado, A.; Ochoa, A.; Alonso, M.E.; Lima Villeda, G.A.; Fernandez-Valverde, F.; Ruano-Calderon, L.; Vargas-Canas, S.; Duron, R.M.; Delgado-Escueta, A.V.; Martinez-Juarez, I.E. Late onset lafora disease and novel epm2a mutations: Breaking paradigms. Epilepsy Res. 2014, 108, 1501–1510. [Google Scholar] [CrossRef]
- Garcia-Gimeno, M.A.; Knecht, E.; Sanz, P. Lafora disease: A ubiquitination-related pathology. Cells 2018, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Cabrero, A.M.; Marinas, A.; Guerrero, R.; de Cordoba, S.R.; Serratosa, J.M.; Sanchez, M.P. Laforin and malin deletions in mice produce similar neurologic impairments. J. Neuropathol. Exp. Neurol. 2012, 71, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cabrero, A.M.; Sanchez-Elexpuru, G.; Serratosa, J.M.; Sanchez, M.P. Enhanced sensitivity of laforin- and malin-deficient mice to the convulsant agent pentylenetetrazole. Front. Neurosci. 2014, 8, 291. [Google Scholar] [PubMed] [Green Version]
- Aguado, C.; Sarkar, S.; Korolchuk, V.I.; Criado, O.; Vernia, S.; Boya, P.; Sanz, P.; de Cordoba, S.R.; Knecht, E.; Rubinsztein, D.C. Laforin, the most common protein mutated in lafora disease, regulates autophagy. Hum. Mol. Genet. 2010, 19, 2867–2876. [Google Scholar] [CrossRef] [Green Version]
- Knecht, E.; Aguado, C.; Sarkar, S.; Korolchuk, V.I.; Criado-Garcia, O.; Vernia, S.; Boya, P.; Sanz, P.; Rodriguez de Cordoba, S.; Rubinsztein, D.C. Impaired autophagy in lafora disease. Autophagy 2010, 6, 991–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knecht, E.; Criado-Garcia, O.; Aguado, C.; Gayarre, J.; Duran-Trio, L.; Garcia-Cabrero, A.M.; Vernia, S.; San Millan, B.; Heredia, M.; Roma-Mateo, C.; et al. Malin knockout mice support a primary role of autophagy in the pathogenesis of lafora disease. Autophagy 2012, 8, 701–703. [Google Scholar] [CrossRef] [Green Version]
- Puri, R.; Suzuki, T.; Yamakawa, K.; Ganesh, S. Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for lafora disease. Hum. Mol. Genet. 2012, 21, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Roma-Mateo, C.; Aguado, C.; Garcia-Gimenez, J.L.; Knecht, E.; Sanz, P.; Pallardo, F.V. Oxidative stress, a new hallmark in the pathophysiology of lafora progressive myoclonus epilepsy. Free Radic. Biol. Med. 2015, 88, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Lahuerta, M.; Aguado, C.; Sanchez-Martin, P.; Sanz, P.; Knecht, E. Degradation of altered mitochondria by autophagy is impaired in lafora disease. FEBS J. 2018, 285, 2071–2090. [Google Scholar] [CrossRef] [Green Version]
- Espinos, C.; Galindo, M.I.; Garcia-Gimeno, M.A.; Ibanez-Cabellos, J.S.; Martinez-Rubio, D.; Millan, J.M.; Rodrigo, R.; Sanz, P.; Seco-Cervera, M.; Sevilla, T.; et al. Oxidative stress, a crossroad between rare diseases and neurodegeneration. Antioxidants 2020, 9, 313. [Google Scholar] [CrossRef] [Green Version]
- Lahuerta, M.; Gonzalez, D.; Aguado, C.; Fathinajafabadi, A.; Garcia-Gimenez, J.L.; Moreno-Estelles, M.; Roma-Mateo, C.; Knecht, E.; Pallardo, F.V.; Sanz, P. Reactive glia-derived neuroinflammation: A novel hallmark in lafora progressive myoclonus epilepsy that progresses with age. Mol. Neurobiol. 2020, 57, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Elexpuru, G.; Serratosa, J.M.; Sanchez, M.P. Sodium selenate treatment improves symptoms and seizure susceptibility in a malin-deficient mouse model of lafora disease. Epilepsia 2017, 58, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Perez-Mediavilla, A.; Frechilla, D.; Del Rio, J.; Garcia-Osta, A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an alzheimer’s disease mouse model. Neuropsychopharmacology 2009, 34, 1721–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Bercury, K.; Cummiskey, J.; Luong, N.; Lebin, J.; Freed, C.R. Phenylbutyrate up-regulates the dj-1 protein and protects neurons in cell culture and in animal models of parkinson disease. J. Biol. Chem. 2011, 286, 14941–14951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Brenneisen, P.; Steinbrenner, H.; Sies, H. Selenium, oxidative stress, and health aspects. Mol. Asp. Med. 2005, 26, 256–267. [Google Scholar] [CrossRef]
- Rehni, A.K.; Singh, T.G. Selenium induced anticonvulsant effect: A potential role of prostaglandin e(1) receptor activation linked mechanism. J. Trace Elem. Med. Biol. 2013, 27, 31–39. [Google Scholar] [CrossRef]
- Bisulli, F.; Muccioli, L.; d’Orsi, G.; Canafoglia, L.; Freri, E.; Licchetta, L.; Mostacci, B.; Riguzzi, P.; Pondrelli, F.; Avolio, C.; et al. Treatment with metformin in twelve patients with lafora disease. Orphanet J. Rare Dis. 2019, 14, 149. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanz, P.; Serratosa, J.M.; Sánchez, M.P. Beneficial Effects of Metformin on the Central Nervous System, with a Focus on Epilepsy and Lafora Disease. Int. J. Mol. Sci. 2021, 22, 5351. https://doi.org/10.3390/ijms22105351
Sanz P, Serratosa JM, Sánchez MP. Beneficial Effects of Metformin on the Central Nervous System, with a Focus on Epilepsy and Lafora Disease. International Journal of Molecular Sciences. 2021; 22(10):5351. https://doi.org/10.3390/ijms22105351
Chicago/Turabian StyleSanz, Pascual, José Maria Serratosa, and Marina P. Sánchez. 2021. "Beneficial Effects of Metformin on the Central Nervous System, with a Focus on Epilepsy and Lafora Disease" International Journal of Molecular Sciences 22, no. 10: 5351. https://doi.org/10.3390/ijms22105351
APA StyleSanz, P., Serratosa, J. M., & Sánchez, M. P. (2021). Beneficial Effects of Metformin on the Central Nervous System, with a Focus on Epilepsy and Lafora Disease. International Journal of Molecular Sciences, 22(10), 5351. https://doi.org/10.3390/ijms22105351