
Inflammation-Induced Tumorigenesis and Metastasis

 , and
, and
Abstract
:1. Introduction
2. Triggers of Inflammation during Tumorigenesis
2.1. Inflammation Caused by Chronic Infection and Autoimmunity
2.2. Inflammation Caused by Environmental and Lifestyle Factors
2.3. Aging-Associated Inflammation
2.4. Cancer-Elicited Inflammation
2.5. Inflammation Caused by Cancer Therapy
3. Cell Types Engaged in Cancer-Associated Inflammation
3.1. Myeloid Cells
3.2. Lymphocytes
3.3. Tumor Endothelial Cells
3.4. Cancer-Associated Fibroblasts
4. Multifaceted Role of Inflammation during Tumorigenesis
4.1. Role of Inflammation in Tumor Initiation
4.2. Role of Inflammation in Tumor Promotion
4.3. Role of Inflammation in Tumor Progression and Metastasis
5. Tumor-Promoting Inflammatory Signaling
6. Role of Inflammation in Cancer Types
6.1. Esophageal Cancer
6.2. Gastric Cancer
6.3. Colorectal Cancer
6.4. Liver Cancer
6.5. Pancreatic Cancer
6.6. Lung Cancer
6.7. Prostate Cancer
6.8. Breast Cancer
6.9. Hematological Malignancies
7. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Fujiki, H. Gist of Dr. Katsusaburo Yamagiwa’s papers entitled “Experimental study on the pathogenesis of epithelial tumors” (I to VI reports). Cancer Sci. 2014, 105, 143–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar] [CrossRef] [Green Version]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Dvorak, H.F. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhou, L.; Liu, L.; Hou, Y.; Xiong, M.; Yang, Y.; Hu, J.; Chen, K. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat. Commun. 2020, 11, 5077. [Google Scholar] [CrossRef]
- Izar, B.; Tirosh, I.; Stover, E.H.; Wakiro, I.; Cuoco, M.S.; Alter, I.; Rodman, C.; Leeson, R.; Su, M.J.; Shah, P.; et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 2020, 26, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Karin, M. Tumor-Elicited Inflammation and Colorectal Cancer. Adv. Cancer Res. 2015, 128, 173–196. [Google Scholar]
- Rositch, A.F. Global burden of cancer attributable to infections: The critical role of implementation science. Lancet Glob. Health 2020, 8, e153–e154. [Google Scholar] [CrossRef]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef]
- Nanki, K.; Fujii, M.; Shimokawa, M.; Matano, M.; Nishikori, S.; Date, S.; Takano, A.; Toshimitsu, K.; Ohta, Y.; Takahashi, S.; et al. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature 2020, 577, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, N.; Yoshida, K.; Uchino, M.; Kihara, T.; Akaki, K.; Inoue, Y.; Kawada, K.; Nagayama, S.; Yokoyama, A.; Yamamoto, S.; et al. Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature 2020, 577, 260–265. [Google Scholar] [CrossRef]
- Takahashi, H.; Ogata, H.; Nishigaki, R.; Broide, D.H.; Karin, M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell 2010, 17, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadariya, Y.; Menges, C.W.; Talarchek, J.; Cai, K.Q.; Klein-Szanto, A.J.; Pietrofesa, R.A.; Christofidou-Solomidou, M.; Cheung, M.; Mossman, B.T.; Shukla, A.; et al. Inflammation-Related IL1β/IL1R Signaling Promotes the Development of Asbestos-Induced Malignant Mesothelioma. Cancer Prev. Res. 2016, 9, 406–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Olson, O.C.; Bhardwaj, P.; Walsh, L.A.; Akkari, L.; Quick, M.L.; Chen, I.C.; Wendel, N.; Ben-Chetrit, N.; Walker, J.; et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat. Cell Biol. 2017, 19, 974–987. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Dannenberg, A.J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 2019, 15, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Jordan, B.F. Gut microbiota-mediated inflammation in obesity: A link with gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Wada, S.; Yang, S.; Gosis, B.; Zeng, X.; Zhang, Z.; Shen, Y.; Lee, G.; Arany, Z.; Rabinowitz, J.D. The small intestine shields the liver from fructose-induced steatosis. Nat. Metab. 2020, 2, 586–593. [Google Scholar] [CrossRef]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 2, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Nagai, N.; Kudo, Y.; Aki, D.; Nakagawa, H.; Taniguchi, K. Immunomodulation by Inflammation during Liver and Gastrointestinal Tumorigenesis and Aging. Int. J. Mol. Sci. 2021, 22, 2238. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef] [Green Version]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; Coffelt, S.B.; Duits, D.E.M.; van Miltenburg, M.H.; Slagter, M.; de Rink, I.; Henneman, L.; Kas, S.M.; Prekovic, S.; Hau, C.S.; et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 2019, 572, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.H.; Mohammed, Z.; Qayyum, T.; Horgan, P.G.; McMillan, D.C. The prognostic value of histological tumor necrosis in solid organ malignant disease: A systematic review. Future Oncol. 2011, 7, 1223–1235. [Google Scholar] [CrossRef]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Roca, H.; Jones, J.D.; Purica, M.C.; Weidner, S.; Koh, A.J.; Kuo, R.; Wilkinson, J.E.; Wang, Y.; Daignault-Newton, S.; Pienta, K.J.; et al. Apoptosis-induced CXCL5 accelerates inflammation and growth of prostate tumor metastases in bone. J. Clin. Investig. 2018, 128, 248–266. [Google Scholar] [CrossRef] [PubMed]
- Gartung, A.; Yang, J.; Sukhatme, V.P.; Bielenberg, D.R.; Fernandes, D.; Chang, J.; Schmidt, B.A.; Hwang, S.H.; Zurakowski, D.; Huang, S.; et al. Suppression of chemotherapy-induced cytokine/lipid mediator surge and ovarian cancer by a dual COX-2/sEH inhibitor. Proc. Natl. Acad. Sci. USA 2019, 116, 1698–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Kim, M.K.; Di Caro, G.; Wong, J.; Shalapour, S.; Wan, J.; Zhang, W.; Zhong, Z.; Sanchez-Lopez, E.; Wu, L.; et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 2014, 41, 1052–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henson, P.M. Cell Removal: Efferocytosis. Annu. Rev. Cell Dev. Biol. 2017, 33, 127–144. [Google Scholar] [CrossRef]
- Green, D.R. Ghostly metabolic messages from dying cells. Nature 2020, 580, 36–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef]
- Templeton, A.J.; McNamara, M.G.; Šeruga, B.; Vera-Badillo, F.E.; Aneja, P.; Ocaña, A.; Leibowitz-Amit, R.; Sonpavde, G.; Knox, J.J.; Tran, B.; et al. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2014, 106, dju124. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Lu, Z.; Zou, J.; Li, S.; Topper, M.J.; Tao, Y.; Zhang, H.; Jiao, X.; Xie, W.; Kong, X.; Vaz, M.; et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020, 579, 284–290. [Google Scholar] [CrossRef]
- Zhou, J.; Nefedova, Y.; Lei, A.; Gabrilovich, D. Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells. Semin. Immunol. 2018, 35, 19–28. [Google Scholar] [CrossRef]
- Condamine, T.; Dominguez, G.A.; Youn, J.I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef]
- Kwak, T.; Wang, F.; Deng, H.; Condamine, T.; Kumar, V.; Perego, M.; Kossenkov, A.; Montaner, L.J.; Xu, X.; Xu, W.; et al. Distinct Populations of Immune-Suppressive Macrophages Differentiate from Monocytic Myeloid-Derived Suppressor Cells in Cancer. Cell Rep. 2020, 33, 108571. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Jiang, S.; Dong, C. A complex issue on CD4(+) T-cell subsets. Immunol. Rev. 2013, 252, 5–11. [Google Scholar] [CrossRef]
- Kryczek, I.; Lin, Y.; Nagarsheth, N.; Peng, D.; Zhao, L.; Zhao, E.; Vatan, L.; Szeliga, W.; Dou, Y.; Owens, S.; et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014, 40, 772–784. [Google Scholar] [CrossRef] [Green Version]
- Hibino, S.; Chikuma, S.; Kondo, T.; Ito, M.; Nakatsukasa, H.; Omata-Mise, S.; Yoshimura, A. Inhibition of Nr4a Receptors Enhances Antitumor Immunity by Breaking Treg-Mediated Immune Tolerance. Cancer Res. 2018, 78, 3027–3040. [Google Scholar] [CrossRef] [Green Version]
- Blatner, N.R.; Mulcahy, M.F.; Dennis, K.L.; Scholtens, D.; Bentrem, D.J.; Phillips, J.D.; Ham, S.; Sandall, B.P.; Khan, M.W.; Mahvi, D.M.; et al. Expression of RORγt marks a pathogenic regulatory T cell subset in human colon cancer. Sci. Transl. Med. 2012, 4, 164ra159. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.; Di Giovangiulio, M.; Stolfi, C.; Franzè, E.; Fehling, H.J.; Carsetti, R.; Giorda, E.; Colantoni, A.; Ortenzi, A.; Rugge, M.; et al. RORγt-Expressing Tregs Drive the Growth of Colitis-Associated Colorectal Cancer by Controlling IL6 in Dendritic Cells. Cancer Immunol. Res. 2018, 6, 1082–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desdín-Micó, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabandé-Rodríguez, E.; Blanco, E.M.; Alfranca, A.; Cussó, L.; Desco, M.; et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef]
- Kroemer, G.; Zitvogel, L. CD4(+) T Cells at the Center of Inflammaging. Cell Metab. 2020, 32, 4–5. [Google Scholar] [CrossRef]
- Wang, S.; Wu, P.; Chen, Y.; Chai, Y. Ambiguous roles and potential therapeutic strategies of innate lymphoid cells in different types of tumor. Oncol. Lett. 2020, 20, 1513–1525. [Google Scholar] [CrossRef]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef]
- Klose, C.S.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef]
- Van Beek, J.J.P.; Martens, A.W.J.; Bakdash, G.; de Vries, I.J.M. Innate Lymphoid Cells in Tumor Immunity. Biomedicines 2016, 4, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef]
- Hirota, K.; Semenza, G.L. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit. Rev. Oncol./Hematol. 2006, 59, 15–26. [Google Scholar] [CrossRef]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.J.; Hoos, A.; Bauer, J.; Boettcher, S.; Knust, M.; Weber, A.; Simonavicius, N.; Schneider, C.; Lang, M.; Stürzl, M.; et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell 2012, 22, 91–105. [Google Scholar] [CrossRef] [Green Version]
- Tavora, B.; Mederer, T.; Wessel, K.J.; Ruffing, S.; Sadjadi, M.; Missmahl, M.; Ostendorf, B.N.; Liu, X.; Kim, J.Y.; Olsen, O.; et al. Tumoural activation of TLR3-SLIT2 axis in endothelium drives metastasis. Nature 2020, 586, 299–304. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10, 4375. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, T.; Koiwa, M.; Yonemura, A.; Miyake, K.; Kariya, R.; Kubota, S.; Yokomizo-Nakano, T.; Yasuda-Yoshihara, N.; Uchihara, T.; Itoyama, R.; et al. Inflammation-driven senescence-associated secretory phenotype in cancer-associated fibroblasts enhances peritoneal dissemination. Cell Rep 2021, 34, 108779. [Google Scholar] [CrossRef]
- Ishimoto, T.; Miyake, K.; Nandi, T.; Yashiro, M.; Onishi, N.; Huang, K.K.; Lin, S.J.; Kalpana, R.; Tay, S.T.; Suzuki, Y.; et al. Activation of Transforming Growth Factor Beta 1 Signaling in Gastric Cancer-associated Fibroblasts Increases Their Motility, via Expression of Rhomboid 5 Homolog 2, and Ability to Induce Invasiveness of Gastric Cancer Cells. Gastroenterology 2017, 153, 191–204.e16. [Google Scholar] [CrossRef]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [Green Version]
- Bu, L.; Baba, H.; Yoshida, N.; Miyake, K.; Yasuda, T.; Uchihara, T.; Tan, P.; Ishimoto, T. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene 2019, 38, 4887–4901. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Baba, H.; Yasuda, T.; Uchihara, T.; Ishimoto, T. Functional diversity of cancer-associated fibroblasts in modulating drug resistance. Cancer Sci. 2020, 111, 3468–3477. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883.e5. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Xia, T.; Konno, H.; Konno, K.; Ruiz, P.; Barber, G.N. Inflammation-driven carcinogenesis is mediated through STING. Nat. Commun. 2014, 5, 5166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, 6428. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, H.; Ishikawa, T.; Yoshida, G.J.; Naoi, K.; Maeda, Y.; Naka, K.; Ju, X.; Yamada, Y.; Minamoto, T.; Mukaida, N.; et al. TNF-α/TNFR1 signaling promotes gastric tumorigenesis through induction of Noxo1 and Gna14 in tumor cells. Oncogene 2014, 33, 3820–3829. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Derynck, R.; Weinberg, R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef]
- Sistigu, A.; Di Modugno, F.; Manic, G.; Nisticò, P. Deciphering the loop of epithelial-mesenchymal transition, inflammatory cytokines and cancer immunoediting. Cytokine Growth Factor Rev. 2017, 36, 67–77. [Google Scholar] [CrossRef]
- Shang, S.; Ji, X.; Zhang, L.; Chen, J.; Li, C.; Shi, R.; Xiang, W.; Kang, X.; Zhang, D.; Yang, F.; et al. Macrophage ABHD5 Suppresses NFκB-Dependent Matrix Metalloproteinase Expression and Cancer Metastasis. Cancer Res. 2019, 79, 5513–5526. [Google Scholar]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Zhou, Z.; Xia, G.; Xiang, Z.; Liu, M.; Wei, Z.; Yan, J.; Chen, W.; Zhu, J.; Awasthi, N.; Sun, X.; et al. A C-X-C Chemokine Receptor Type 2-Dominated Cross-talk between Tumor Cells and Macrophages Drives Gastric Cancer Metastasis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3317–3328. [Google Scholar] [CrossRef] [Green Version]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Körner, C.; Ben-Baruch, A. Tumor-Stroma-Inflammation Networks Promote Pro-metastatic Chemokines and Aggressiveness Characteristics in Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, 6409. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Karin, M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Lee, C.K. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—Using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; He, L.; Regev, A.; Struhl, K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9453–9462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Wu, L.W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Moroishi, T.; de Jong, P.R.; Krawczyk, M.; Grebbin, B.M.; Luo, H.; Xu, R.H.; Golob-Schwarzl, N.; Schweiger, C.; Wang, K.; et al. YAP-IL-6ST autoregulatory loop activated on APC loss controls colonic tumorigenesis. Proc. Natl. Acad. Sci. USA 2017, 114, 1643–1648. [Google Scholar] [CrossRef] [Green Version]
- Kawazoe, T.; Saeki, H.; Oki, E.; Oda, Y.; Maehara, Y.; Mori, M.; Taniguchi, K. Autocrine Leukemia Inhibitory Factor Promotes Esophageal Squamous Cell Carcinoma Progression via Src Family Kinase-Dependent Yes-Associated Protein Activation. Mol. Cancer Res. 2020, 18, 1876–1888. [Google Scholar] [CrossRef]
- Short, M.W.; Burgers, K.G.; Fry, V.T. Esophageal Cancer. Am. Fam. Physician 2017, 95, 22–28. [Google Scholar]
- Lagergren, J.; Smyth, E.; Cunningham, D.; Lagergren, P. Oesophageal cancer. Lancet 2017, 390, 2383–2396. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, A.; Obi, K.O.; Rubenstein, J.H. The synergistic effects of alcohol and tobacco consumption on the risk of esophageal squamous cell carcinoma: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Enzinger, P.C.; Mayer, R.J. Esophageal cancer. N. Engl. J. Med. 2003, 349, 2241–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebhandl, S.; Huemer, M.; Greil, R.; Geisberger, R. AID/APOBEC deaminases and cancer. Oncoscience 2015, 2, 320–333. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, M.; Ying, S.; Zhang, C.; Lin, R.; Zheng, J.; Zhang, G.; Tian, D.; Guo, Y.; Du, C.; et al. Genetic Alterations in Esophageal Tissues From Squamous Dysplasia to Carcinoma. Gastroenterology 2017, 153, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Gao, F.X.; Wang, C.; Qin, M.; Han, F.; Xu, T.; Hu, Z.; Long, Y.; He, X.M.; Deng, X.; et al. IL-6 and IL-8 secreted by tumour cells impair the function of NK cells via the STAT3 pathway in oesophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 321. [Google Scholar] [CrossRef] [PubMed]
- Spechler, S.J. Carcinogenesis at the gastroesophageal junction: Free radicals at the frontier. Gastroenterology 2002, 122, 1518–1520. [Google Scholar] [CrossRef] [PubMed]
- Kawazoe, T.; Saeki, H.; Edahiro, K.; Korehisa, S.; Taniguchi, D.; Kudou, K.; Nakanishi, R.; Kubo, N.; Ando, K.; Nakashima, Y.; et al. A case of mixed adenoneuroendocrine carcinoma (MANEC) arising in Barrett’s esophagus: Literature and review. Surg. Case Rep. 2018, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Huo, X.; Zhang, X.; Yu, C.; Zhang, Q.; Cheng, E.; Wang, D.H.; Pham, T.H.; Spechler, S.J.; Souza, R.F. In oesophageal squamous cells exposed to acidic bile salt medium, omeprazole inhibits IL-8 expression through effects on nuclear factor-κB and activator protein-1. Gut 2014, 63, 1042–1052. [Google Scholar] [CrossRef]
- Mukaisho, K.I.; Kanai, S.; Kushima, R.; Nakayama, T.; Hattori, T.; Sugihara, H. Barretts’s carcinogenesis. Pathol. Int. 2019, 69, 319–330. [Google Scholar] [CrossRef]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, M.E.; Puccini, A.; Grothey, A.; Raghavan, D.; Goldberg, R.M.; Xiu, J.; Korn, W.M.; Weinberg, B.A.; Hwang, J.J.; Shields, A.F.; et al. Landscape of Tumor Mutation Load, Mismatch Repair Deficiency, and PD-L1 Expression in a Large Patient Cohort of Gastrointestinal Cancers. Mol. Cancer Res. 2018, 16, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Vivaldi, C.; Catanese, S.; Massa, V.; Pecora, I.; Salani, F.; Santi, S.; Lencioni, M.; Vasile, E.; Falcone, A.; Fornaro, L. Immune Checkpoint Inhibitors in Esophageal Cancers: Are We Finally Finding the Right Path in the Mist? Int. J. Mol. Sci. 2020, 21, 1658. [Google Scholar] [CrossRef] [Green Version]
- Pormohammad, A.; Mohtavinejad, N.; Gholizadeh, P.; Dabiri, H.; Salimi Chirani, A.; Hashemi, A.; Nasiri, M.J. Global estimate of gastric cancer in Helicobacter pylori-infected population: A systematic review and meta-analysis. J. Cell Physiol. 2019, 234, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.D.; Li, Y.; Zamble, D.B. Acid-responsive activity of the Helicobacter pylori metalloregulator NikR. Proc. Natl. Acad. Sci. USA 2018, 115, 8966–8971. [Google Scholar] [CrossRef] [Green Version]
- Ansari, S.; Yamaoka, Y. Survival of Helicobacter pylori in gastric acidic territory. Helicobacter 2017, 22, e12386. [Google Scholar] [CrossRef]
- Kusters, J.G.; van Vliet, A.H.M.; Kuipers, E.J. Pathogenesis of Helicobacter pylori Infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [Green Version]
- Bontems, P.; Aksoy, E.; Burette, A.; Segers, V.; Deprez, C.; Mascart, F.; Cadranel, S. NF-κB activation and severity of gastritis in Helicobacter pylori-infected children and adults. Helicobacter 2014, 19, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Byun, E.; Park, B.; Lim, J.W.; Kim, H. Activation of NF-κB and AP-1 Mediates Hyperproliferation by Inducing β-Catenin and c-Myc in Helicobacter pylori—Infected Gastric Epithelial Cells. Yonsei Med. J. 2016, 57, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, T.; Hattori, N. Molecular pathways: Involvement of Helicobacter pylori—Triggered inflammation in the formation of an epigenetic field defect, and its usefulness as cancer risk and exposure markers. Clin. Cancer Res. 2012, 18, 923–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimoto, T.; Izumi, D.; Watanabe, M.; Yoshida, N.; Hidaka, K.; Miyake, K.; Sugihara, H.; Sawayama, H.; Imamura, Y.; Iwatsuki, M.; et al. Chronic inflammation with Helicobacter pylori infection is implicated in CD44 overexpression through miR-328 suppression in the gastric mucosa. J. Gastroenterol. 2015, 50, 751–757. [Google Scholar] [CrossRef]
- Bernstein, C.; Nfonsam, V.; Prasad, A.R.; Bernstein, H. Epigenetic field defects in progression to cancer. World J. Gastrointest. Oncol. 2013, 5, 43–49. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Murphy, G.; Pfeiffer, R.; Camargo, M.C.; Rabkin, C.S. Meta-analysis shows that prevalence of Epstein-Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology 2009, 137, 824–833. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Li, J.; Hao, Y.; Nie, Y.; Li, Z.; Qian, M.; Liang, Q.; Yu, J.; Zeng, M.; Wu, K. Differentiated tumor immune microenvironment of Epstein-Barr virus-associated and negative gastric cancer: Implication in prognosis and immunotherapy. Oncotarget 2017, 8, 67094–67103. [Google Scholar] [CrossRef]
- Ghasemi, F.; Tessier, T.M.; Gameiro, S.F.; Maciver, A.H.; Cecchini, M.J.; Mymryk, J.S. High MHC-II expression in Epstein-Barr virus-associated gastric cancers suggests that tumor cells serve an important role in antigen presentation. Sci. Rep. 2020, 10, 14786. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Song, X.; Xu, X.; Mou, Y. Cancer-Associated Fibroblasts Promote the Chemo-resistance in Gastric Cancer through Secreting IL-11 Targeting JAK/STAT3/Bcl2 Pathway. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2019, 51, 194–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Zhu, F.; Jin, S.; Tian, J. Interleukin-22 regulates gastric cancer cell proliferation through regulation of the JNK signaling pathway. Exp. Ther. Med. 2020, 20, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yang, X.; Li, J.; Lu, Z.; Li, X.; Yu, J.; Li, N. IL-22 promotes the migration and invasion of gastric cancer cells via IL-22R1/AKT/MMP-9 signaling. Int. J. Clin. Exp. Pathol. 2014, 7, 3694–3703. [Google Scholar] [PubMed]
- Fukui, H.; Zhang, X.; Sun, C.; Hara, K.; Kikuchi, S.; Yamasaki, T.; Kondo, T.; Tomita, T.; Oshima, T.; Watari, J.; et al. IL-22 produced by cancer-associated fibroblasts promotes gastric cancer cell invasion via STAT3 and ERK signaling. Br. J. Cancer 2014, 111, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; Boku, N.; Satoh, T.; Ryu, M.H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.S.; Muro, K.; Kang, W.; et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 2461–2471. [Google Scholar]
- Sexton, R.E.; Al Hallak, M.N.; Diab, M.; Azmi, A.S. Gastric cancer: A comprehensive review of current and future treatment strategies. Cancer Metastasis Rev. 2020, 39, 1179–1203. [Google Scholar] [CrossRef]
- Lichtenstern, C.R.; Ngu, R.K.; Shalapour, S.; Karin, M. Immunotherapy, Inflammation and Colorectal Cancer. Cells 2020, 9, 618. [Google Scholar] [CrossRef] [Green Version]
- Leslie, A.; Carey, F.A.; Pratt, N.R.; Steele, R.J. The colorectal adenoma-carcinoma sequence. Br. J. Surg. 2002, 89, 845–860. [Google Scholar] [CrossRef] [Green Version]
- Schell, M.J.; Yang, M.; Teer, J.K.; Lo, F.Y.; Madan, A.; Coppola, D.; Monteiro, A.N.; Nebozhyn, M.V.; Yue, B.; Loboda, A.; et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat. Commun. 2016, 7, 11743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itatani, Y.; Yamamoto, T.; Zhong, C.; Molinolo, A.A.; Ruppel, J.; Hegde, P.; Taketo, M.M.; Ferrara, N. Suppressing neutrophil-dependent angiogenesis abrogates resistance to anti-VEGF antibody in a genetic model of colorectal cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 21598–21608. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Luo, C.; Zhang, H. Early detection of ulcerative colitis-associated colorectal cancer. Gastroenterol. Rep. 2018, 6, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.C.; Stappenbeck, T.S. Genetics and Pathogenesis of Inflammatory Bowel Disease. Annu. Rev. Pathol. 2016, 11, 127–148. [Google Scholar] [CrossRef] [Green Version]
- Kameyama, H.; Nagahashi, M.; Shimada, Y.; Tajima, Y.; Ichikawa, H.; Nakano, M.; Sakata, J.; Kobayashi, T.; Narayanan, S.; Takabe, K.; et al. Genomic characterization of colitis-associated colorectal cancer. World J. Surg. Oncol. 2018, 16, 121. [Google Scholar] [CrossRef] [Green Version]
- Tariq, K.; Ghias, K. Colorectal cancer carcinogenesis: A review of mechanisms. Cancer Biol. Med. 2016, 13, 120–135. [Google Scholar] [CrossRef] [Green Version]
- Evrard, C.; Tachon, G.; Randrian, V.; Karayan-Tapon, L.; Tougeron, D. Microsatellite Instability: Diagnosis, Heterogeneity, Discordance, and Clinical Impact in Colorectal Cancer. Cancers 2019, 11, 1567. [Google Scholar] [CrossRef] [Green Version]
- Nakano, K.; Yamamoto, H.; Fujiwara, M.; Koga, Y.; Tsuruta, S.; Ihara, E.; Oki, E.; Nakamura, M.; Ogawa, Y.; Oda, Y. Clinicopathologic and Molecular Characteristics of Synchronous Colorectal Carcinoma With Mismatch Repair Deficiency. Am. J. Surg. Pathol. 2018, 42, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Maby, P.; Tougeron, D.; Hamieh, M.; Mlecnik, B.; Kora, H.; Bindea, G.; Angell, H.K.; Fredriksen, T.; Elie, N.; Fauquembergue, E.; et al. Correlation between Density of CD8+ T-cell Infiltrate in Microsatellite Unstable Colorectal Cancers and Frameshift Mutations: A Rationale for Personalized Immunotherapy. Cancer Res 2015, 75, 3446–3455. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Wang, Y.; Chlewicki, L.K.; Zhang, Y.; Guo, J.; Liang, W.; Wang, J.; Wang, X.; Fu, Y.X. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell 2016, 29, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massarweh, N.N.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. Cancer Control 2017, 24, 1073274817729245. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Hamaoka, M.; Kozaka, K.; Matsui, O.; Komori, T.; Matsubara, T.; Yoneda, N.; Yoshida, K.; Inoue, D.; Kitao, A.; Koda, W.; et al. Early detection of intrahepatic cholangiocarcinoma. Jpn. J. Radiol. 2019, 37, 669–684. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Toffanin, S.; Friedman, S.L.; Llovet, J.M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013, 144, 512–527. [Google Scholar] [CrossRef] [Green Version]
- Budhu, A.; Forgues, M.; Ye, Q.H.; Jia, H.L.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.X.; Tang, Z.Y.; et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, J.; Müller, M.; Baumann, N.; Reichert, M.; Heneweer, C.; Bolik, J.; Lücke, K.; Gruber, S.; Carambia, A.; Boretius, S.; et al. IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology 2017, 65, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Isomoto, H.; Kobayashi, S.; Werneburg, N.W.; Bronk, S.F.; Guicciardi, M.E.; Frank, D.A.; Gores, G.J. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology 2005, 42, 1329–1338. [Google Scholar] [CrossRef]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Saalim, M.; Resham, S.; Manzoor, S.; Ahmad, H.; Jaleel, S.; Ashraf, J.; Imran, M.; Naseem, S. IL-22: A promising candidate to inhibit viral-induced liver disease progression and hepatocellular carcinoma. Tumour Biol. 2016, 37, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Dandri, M. The Role of cccDNA in HBV Maintenance. Viruses 2017, 9, 156. [Google Scholar] [CrossRef] [Green Version]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64 (Suppl. S1), S84–S101. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]
- Dash, S.; Aydin, Y.; Widmer, K.E.; Nayak, L. Hepatocellular Carcinoma Mechanisms Associated with Chronic HCV Infection and the Impact of Direct-Acting Antiviral Treatment. J. Hepatocell Carcinoma 2020, 7, 45–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, K.; Roca Suarez, A.A.; Wrensch, F.; Baumert, T.F.; Lupberger, J. Hepatitis C Virus and Hepatocellular Carcinoma: When the Host Loses Its Grip. Int. J. Mol. Sci. 2020, 2, 3057. [Google Scholar] [CrossRef]
- Prieto, J.; Melero, I.; Sangro, B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 681–700. [Google Scholar] [CrossRef]
- Greten, T.F.; Wang, X.W.; Korangy, F. Current concepts of immune based treatments for patients with HCC: From basic science to novel treatment approaches. Gut 2015, 64, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Yoshizumi, T.; Yugawa, K.; Imai, D.; Yoshiya, S.; Takeishi, K.; Toshima, T.; Harada, N.; Ikegami, T.; Soejima, Y.; et al. Impact of Immune Response on Outcomes in Hepatocellular Carcinoma: Association With Vascular Formation. Hepatology 2020, 72, 1987–1999. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [Green Version]
- Cascinu, S.; Falconi, M.; Valentini, V.; Jelic, S. Pancreatic cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010, 21 (Suppl. S5), v55–v58. [Google Scholar] [CrossRef]
- Basturk, O.; Hong, S.M.; Wood, L.D.; Adsay, N.V.; Albores-Saavedra, J.; Biankin, A.V.; Brosens, L.A.; Fukushima, N.; Goggins, M.; Hruban, R.H.; et al. A Revised Classification System and Recommendations From the Baltimore Consensus Meeting for Neoplastic Precursor Lesions in the Pancreas. Am. J. Surg. Pathol. 2015, 39, 1730–1741. [Google Scholar] [CrossRef]
- Shen, R.; Wang, Q.; Cheng, S.; Liu, T.; Jiang, H.; Zhu, J.; Wu, Y.; Wang, L. The biological features of PanIN initiated from oncogenic Kras mutation in genetically engineered mouse models. Cancer Lett. 2013, 339, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Hosoda, W.; Chianchiano, P.; Griffin, J.F.; Pittman, M.E.; Brosens, L.A.; Noë, M.; Yu, J.; Shindo, K.; Suenaga, M.; Rezaee, N.; et al. Genetic analyses of isolated high-grade pancreatic intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations in TP53 and SMAD4. J. Pathol. 2017, 242, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Löhr, M.; Klöppel, G.; Maisonneuve, P.; Lowenfels, A.B.; Lüttges, J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: A meta-analysis. Neoplasia 2005, 7, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Loncle, C.; Molejon, M.I.; Lac, S.; Tellechea, J.I.; Lomberk, G.; Gramatica, L.; Fernandez Zapico, M.F.; Dusetti, N.; Urrutia, R.; Iovanna, J.L. The pancreatitis-associated protein VMP1, a key regulator of inducible autophagy, promotes Kras(G12D)-mediated pancreatic cancer initiation. Cell Death Dis. 2016, 7, e2295. [Google Scholar] [CrossRef] [Green Version]
- Bang, D.; Wilson, W.; Ryan, M.; Yeh, J.J.; Baldwin, A.S. GSK-3α promotes oncogenic KRAS function in pancreatic cancer via TAK1-TAB stabilization and regulation of noncanonical NF-κB. Cancer Discov. 2013, 3, 690–703. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhu, L.; Mundade, R.; Korc, M.; Loehrer, P.J.; Lu, T. Critical role of NF-κB in pancreatic cancer. Oncotarget 2014, 5, 10969–10975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goumas, F.A.; Holmer, R.; Egberts, J.H.; Gontarewicz, A.; Heneweer, C.; Geisen, U.; Hauser, C.; Mende, M.M.; Legler, K.; Röcken, C.; et al. Inhibition of IL-6 signaling significantly reduces primary tumor growth and recurrencies in orthotopic xenograft models of pancreatic cancer. Int J Cancer 2015, 137, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Hu, B.; Ni, J.; Wu, J.; Jiang, W.; Chen, C.; Yang, L.; Zeng, Y.; Wan, R.; Hu, G.; et al. Transcriptional repression of SOCS3 mediated by IL-6/STAT3 signaling via DNMT1 promotes pancreatic cancer growth and metastasis. J. Exp. Clin. Cancer Res. 2016, 35, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Salomon, B.L.; Leclerc, M.; Tosello, J.; Ronin, E.; Piaggio, E.; Cohen, J.L. Tumor Necrosis Factor α and Regulatory T Cells in Oncoimmunology. Front. Immunol. 2018, 9, 444. [Google Scholar] [CrossRef]
- Macherla, S.; Laks, S.; Naqash, A.R.; Bulumulle, A.; Zervos, E.; Muzaffar, M. Emerging Role of Immune Checkpoint Blockade in Pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, 3505. [Google Scholar] [CrossRef] [Green Version]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Karakasheva, T.A.; Lin, E.W.; Tang, Q.; Qiao, E.; Waldron, T.J.; Soni, M.; Klein-Szanto, A.J.; Sahu, V.; Basu, D.; Ohashi, S.; et al. IL-6 Mediates Cross-Talk between Tumor Cells and Activated Fibroblasts in the Tumor Microenvironment. Cancer Res. 2018, 78, 4957–4970. [Google Scholar] [CrossRef] [Green Version]
- Ebbing, E.A.; van der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.L.; et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Li, Z.; Zhu, Q.; Wen, W.; Wang, J.; Xu, J.; Wu, W.; Zhu, Y.; Xu, H.; Chen, L. miR-204-5p regulates cell proliferation, invasion, and apoptosis by targeting IL-11 in esophageal squamous cell carcinoma. J. Cell Physiol. 2020, 235, 3043–3055. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, D.H.; Yang, X.; Sun, Y.; Yang, C.S. Colitis-induced IL11 promotes colon carcinogenesis. Carcinogenesis 2020, 42, 557–569. [Google Scholar] [CrossRef]
- Perez, L.G.; Kempski, J.; McGee, H.M.; Pelzcar, P.; Agalioti, T.; Giannou, A.; Konczalla, L.; Brockmann, L.; Wahib, R.; Xu, H.; et al. TGF-β signaling in Th17 cells promotes IL-22 production and colitis-associated colon cancer. Nat. Commun. 2020, 11, 2608. [Google Scholar] [CrossRef]
- Jiang, R.; Sun, B. IL-22 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2021, 1290, 81–88. [Google Scholar]
- Yin, Z.; Ma, T.; Lin, Y.; Lu, X.; Zhang, C.; Chen, S.; Jian, Z. IL-6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J. Cell Biochem. 2018, 119, 9419–9432. [Google Scholar] [CrossRef]
- Li, H.P.; Zeng, X.C.; Zhang, B.; Long, J.T.; Zhou, B.; Tan, G.S.; Zeng, W.X.; Chen, W.; Yang, J.Y. miR-451 inhibits cell proliferation in human hepatocellular carcinoma through direct suppression of IKK-β. Carcinogenesis 2013, 34, 2443–2451. [Google Scholar] [CrossRef]
- Wörmann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Görgülü, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016, 151, 180–193.e12. [Google Scholar] [CrossRef] [Green Version]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tian, J.; Su, G.H.; Lin, J. Blocking IL-6/GP130 Signaling Inhibits Cell Viability/Proliferation, Glycolysis, and Colony Forming Activity in Human Pancreatic Cancer Cells. Curr. Cancer Drug Targets 2019, 19, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.T.; Fer, N.; Galeas, J.; Collisson, E.A.; Kim, S.E.; Sharib, J.; McCormick, F. Blockade of leukemia inhibitory factor as a therapeutic approach to KRAS driven pancreatic cancer. Nat. Commun. 2019, 10, 3055. [Google Scholar] [CrossRef] [PubMed]
- Xuan, X.; Zhou, J.; Tian, Z.; Lin, Y.; Song, J.; Ruan, Z.; Ni, B.; Zhao, H.; Yang, W. ILC3 cells promote the proliferation and invasion of pancreatic cancer cells through IL-22/AKT signaling. Clin. Transl. Oncol. 2020, 22, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Perusina Lanfranca, M.; Zhang, Y.; Girgis, A.; Kasselman, S.; Lazarus, J.; Kryczek, I.; Delrosario, L.; Rhim, A.; Koneva, L.; Sartor, M.; et al. Interleukin 22 Signaling Regulates Acinar Cell Plasticity to Promote Pancreatic Tumor Development in Mice. Gastroenterology 2020, 158, 1417–1432.e11. [Google Scholar] [CrossRef]
- Proctor, R.N. Tobacco and the global lung cancer epidemic. Nat. Rev. Cancer 2001, 1, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Houghton, A.M. Mechanistic links between COPD and lung cancer. Nat. Rev. Cancer 2013, 13, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Stampfli, M.R.; Anderson, G.P. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat. Rev. Immunol. 2009, 9, 377–384. [Google Scholar] [CrossRef]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, A.L.; Cox, T.R. The Role of the ECM in Lung Cancer Dormancy and Outgrowth. Front. Oncol. 2020, 10, 1766. [Google Scholar] [CrossRef]
- Chaudhri, V.K.; Salzler, G.G.; Dick, S.A.; Buckman, M.S.; Sordella, R.; Karoly, E.D.; Mohney, R.; Stiles, B.M.; Elemento, O.; Altorki, N.K.; et al. Metabolic alterations in lung cancer-associated fibroblasts correlated with increased glycolytic metabolism of the tumor. Mol. Cancer Res. 2013, 11, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Bermudez, A.; Laza-Briviesca, R.; Vicente-Blanco, R.J.; Garcia-Grande, A.; Coronado, M.J.; Laine-Menendez, S.; Alfaro, C.; Sanchez, J.C.; Franco, F.; Calvo, V.; et al. Cancer-associated fibroblasts modify lung cancer metabolism involving ROS and TGF-beta signaling. Free Radic. Biol. Med. 2019, 130, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Ho, C.C.; Chang, Y.L.; Chen, H.Y.; Lin, C.A.; Ling, T.Y.; Yu, S.L.; Yuan, S.S.; Chen, Y.J.; Lin, C.Y.; et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Yang, S.C.; Zhu, L.; Reckamp, K.; Gardner, B.; Baratelli, F.; Huang, M.; Batra, R.K.; Dubinett, S.M. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res. 2005, 65, 5211–5220. [Google Scholar] [CrossRef] [Green Version]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T Cells to protect tumour cells. Nat. Commun. 2018, 9, 948. [Google Scholar] [CrossRef]
- Ando, A.; Hashimoto, N.; Sakamoto, K.; Omote, N.; Miyazaki, S.; Nakahara, Y.; Imaizumi, K.; Kawabe, T.; Hasegawa, Y. Repressive role of stabilized hypoxia inducible factor 1alpha expression on transforming growth factor beta-induced extracellular matrix production in lung cancer cells. Cancer Sci. 2019, 110, 1959–1973. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Sakabe, T.; Sunaga, A.; Sakai, K.; Rivera, A.L.; Keene, D.R.; Sasaki, T.; Stavnezer, E.; Iannotti, J.; Schweitzer, R.; et al. Conversion of mechanical force into TGF-beta-mediated biochemical signals. Curr. Biol. 2011, 21, 933–941. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Nakata, M.; Hirami, Y.; Yukawa, T.; Maeda, A.; Tanemoto, K. Tumor-infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-ras-Mutant Lung Cancer. Cancer Res. 2016, 76, 3189–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef] [Green Version]
- Conlon, T.M.; John-Schuster, G.; Heide, D.; Pfister, D.; Lehmann, M.; Hu, Y.; Ertuz, Z.; Lopez, M.A.; Ansari, M.; Strunz, M.; et al. Inhibition of LTbetaR signalling activates WNT-induced regeneration in lung. Nature 2020, 588, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Cheng, J.; Yang, T.; Li, Y.; Zhu, B. EGFR-TKI down-regulates PD-L1 in EGFR mutant NSCLC through inhibiting NF-κB. Biochem. Biophys. Res. Commun. 2015, 463, 95–101. [Google Scholar] [CrossRef]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-kappaB in Cancer. Front. Immunol. 2020, 11, 584626. [Google Scholar] [CrossRef]
- Zelenay, S.; van der Veen, A.G.; Bottcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.; Lee, M.; Lee, D.; Ha, E.H.; Chun, E.M. Association of Long-term Use of Low-Dose Aspirin as Chemoprevention With Risk of Lung Cancer. JAMA Netw. Open 2019, 2, e190185. [Google Scholar] [CrossRef]
- Lucotti, S.; Cerutti, C.; Soyer, M.; Gil-Bernabe, A.M.; Gomes, A.L.; Allen, P.D.; Smart, S.; Markelc, B.; Watson, K.; Armstrong, P.C.; et al. Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J. Clin. Investig. 2019, 129, 1845–1862. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, F.R.; Al Olama, A.A.; Berndt, S.I.; Benlloch, S.; Ahmed, M.; Saunders, E.J.; Dadaev, T.; Leongamornlert, D.; Anokian, E.; Cieza-Borrella, C.; et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2018, 50, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Multigner, L.; Ndong, J.R.; Giusti, A.; Romana, M.; Delacroix-Maillard, H.; Cordier, S.; Jegou, B.; Thome, J.P.; Blanchet, P. Chlordecone exposure and risk of prostate cancer. J. Clin. Oncol. 2010, 28, 3457–3462. [Google Scholar] [CrossRef] [Green Version]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Sfanos, K.S.; De Marzo, A.M. Prostate cancer and inflammation: The evidence. Histopathology 2012, 60, 199–215. [Google Scholar] [CrossRef] [Green Version]
- Sfanos, K.S.; Yegnasubramanian, S.; Nelson, W.G.; De Marzo, A.M. The inflammatory microenvironment and microbiome in prostate cancer development. Nat. Rev. Urol. 2018, 15, 11–24. [Google Scholar] [CrossRef]
- Platz, E.A.; Kulac, I.; Barber, J.R.; Drake, C.G.; Joshu, C.E.; Nelson, W.G.; Lucia, M.S.; Klein, E.A.; Lippman, S.M.; Parnes, H.L.; et al. A Prospective Study of Chronic Inflammation in Benign Prostate Tissue and Risk of Prostate Cancer: Linked PCPT and SELECT Cohorts. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1549–1557. [Google Scholar] [CrossRef] [Green Version]
- De Bono, J.S.; Guo, C.; Gurel, B.; De Marzo, A.M.; Sfanos, K.S.; Mani, R.S.; Gil, J.; Drake, C.G.; Alimonti, A. Prostate carcinogenesis: Inflammatory storms. Nat. Rev. Cancer 2020, 20, 455–469. [Google Scholar] [CrossRef]
- Golombos, D.M.; Ayangbesan, A.; O’Malley, P.; Lewicki, P.; Barlow, L.; Barbieri, C.E.; Chan, C.; DuLong, C.; Abu-Ali, G.; Huttenhower, C.; et al. The Role of Gut Microbiome in the Pathogenesis of Prostate Cancer: A Prospective, Pilot Study. Urology 2018, 111, 122–128. [Google Scholar] [CrossRef]
- Banerjee, S.; Alwine, J.C.; Wei, Z.; Tian, T.; Shih, N.; Sperling, C.; Guzzo, T.; Feldman, M.D.; Robertson, E.S. Microbiome signatures in prostate cancer. Carcinogenesis 2019, 40, 749–764. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Tschismarov, R.; Pilz, A.; Straubinger, S.; Carotta, S.; McDowell, A.; Decker, T. Cutibacterium acnes Infection Induces Type I Interferon Synthesis Through the cGAS-STING Pathway. Front. Immunol. 2020, 11, 571334. [Google Scholar] [CrossRef] [PubMed]
- Sfanos, K.S.; Markowski, M.C.; Peiffer, L.B.; Ernst, S.E.; White, J.R.; Pienta, K.J.; Antonarakis, E.S.; Ross, A.E. Compositional differences in gastrointestinal microbiota in prostate cancer patients treated with androgen axis-targeted therapies. Prostate Cancer Prostatic Dis. 2018, 21, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, M.; Fujita, K.; Nonomura, N. Influence of Diet and Nutrition on Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.S.; Lee, Y.R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Egawa, S. Epidemiology of prostate cancer in Asian countries. Int. J. Urol. 2018, 25, 524–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Fujita, K.; Nojima, S.; Hayashi, Y.; Nakano, K.; Ishizuya, Y.; Wang, C.; Yamamoto, Y.; Kinouchi, T.; Matsuzaki, K.; et al. High-Fat Diet-Induced Inflammation Accelerates Prostate Cancer Growth via IL6 Signaling. Clin. Cancer Res. 2018, 24, 4309–4318. [Google Scholar] [CrossRef] [Green Version]
- Laurent, V.; Guerard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y.; et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230. [Google Scholar] [CrossRef]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and Prostate Cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Kobayashi, T.; Floc’h, N.; Kinkade, C.W.; Aytes, A.; Dankort, D.; Lefebvre, C.; Mitrofanova, A.; Cardiff, R.D.; McMahon, M.; et al. B-Raf activation cooperates with PTEN loss to drive c-Myc expression in advanced prostate cancer. Cancer Res. 2012, 72, 4765–4776. [Google Scholar] [CrossRef] [Green Version]
- Labbe, D.P.; Zadra, G.; Yang, M.; Reyes, J.M.; Lin, C.Y.; Cacciatore, S.; Ebot, E.M.; Creech, A.L.; Giunchi, F.; Fiorentino, M.; et al. High-fat diet fuels prostate cancer progression by rewiring the metabolome and amplifying the MYC program. Nat. Commun. 2019, 10, 4358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; MacLennan, G.T.; Fu, P.; Patel, J.; Marengo, S.R.; Resnick, M.I.; Gupta, S. Nuclear factor-kappaB/p65 (Rel A) is constitutively activated in human prostate adenocarcinoma and correlates with disease progression. Neoplasia 2004, 6, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambara, G.; Cesaris, P.; Nunzio, C.; Ziparo, E.; Tubaro, A.; Filippini, A.; Riccioli, A. Toll-like receptors in prostate infection and cancer between bench and bedside. J. Cell. Mol. Med. 2013, 17, 713–722. [Google Scholar] [CrossRef] [Green Version]
- Ou, T.; Lilly, M.; Jiang, W. The Pathologic Role of Toll-Like Receptor 4 in Prostate Cancer. Front. Immunol. 2018, 9, 1188. [Google Scholar] [CrossRef] [PubMed]
- Melis, M.H.M.; Nevedomskaya, E.; van Burgsteden, J.; Cioni, B.; van Zeeburg, H.J.T.; Song, J.Y.; Zevenhoven, J.; Hawinkels, L.; de Visser, K.E.; Bergman, A.M. The adaptive immune system promotes initiation of prostate carcinogenesis in a human c-Myc transgenic mouse model. Oncotarget 2017, 8, 93867–93877. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linch, M.; Goh, G.; Hiley, C.; Shanmugabavan, Y.; McGranahan, N.; Rowan, A.; Wong, Y.N.S.; King, H.; Furness, A.; Freeman, A.; et al. Intratumoural evolutionary landscape of high-risk prostate cancer: The PROGENY study of genomic and immune parameters. Ann Oncol 2017, 28, 2472–2480. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.J.; Neisen, J.; Messenger, J.; Waugh, D.J. Tumor-derived CXCL8 signaling augments stroma-derived CCL2-promoted proliferation and CXCL12-mediated invasion of PTEN-deficient prostate cancer cells. Oncotarget 2014, 5, 4895–4908. [Google Scholar] [CrossRef] [Green Version]
- Vidotto, T.; Melo, C.M.; Castelli, E.; Koti, M.; Dos Reis, R.B.; Squire, J.A. Emerging role of PTEN loss in evasion of the immune response to tumours. Br. J. Cancer 2020, 122, 1732–1743. [Google Scholar] [CrossRef] [PubMed]
- Guccini, I.; Revandkar, A.; D’Ambrosio, M.; Colucci, M.; Pasquini, E.; Mosole, S.; Troiani, M.; Brina, D.; Sheibani-Tezerji, R.; Elia, A.R.; et al. Senescence Reprogramming by TIMP1 Deficiency Promotes Prostate Cancer Metastasis. Cancer Cell 2021, 39, 68–82 e9. [Google Scholar] [CrossRef]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Mitri, D.; Mirenda, M.; Vasilevska, J.; Calcinotto, A.; Delaleu, N.; Revandkar, A.; Gil, V.; Boysen, G.; Losa, M.; Mosole, S.; et al. Re-education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer. Cell Rep. 2019, 28, 2156–2168 e5. [Google Scholar] [CrossRef] [Green Version]
- De Cicco, P.; Ercolano, G.; Ianaro, A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Front. Immunol. 2020, 11, 1680. [Google Scholar] [CrossRef]
- Krueger, T.E.; Thorek, D.L.J.; Meeker, A.K.; Isaacs, J.T.; Brennen, W.N. Tumor-infiltrating mesenchymal stem cells: Drivers of the immunosuppressive tumor microenvironment in prostate cancer? Prostate 2019, 79, 320–330. [Google Scholar] [CrossRef]
- Cha, H.R.; Lee, J.H.; Ponnazhagan, S. Revisiting Immunotherapy: A Focus on Prostate Cancer. Cancer Res. 2020, 80, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Calagua, C.; Russo, J.; Sun, Y.; Schaefer, R.; Lis, R.; Zhang, Z.; Mahoney, K.; Bubley, G.J.; Loda, M.; Taplin, M.E.; et al. Expression of PD-L1 in Hormone-naïve and Treated Prostate Cancer Patients Receiving Neoadjuvant Abiraterone Acetate plus Prednisone and Leuprolide. Clin. Cancer Res. 2017, 23, 6812–6822. [Google Scholar] [CrossRef] [Green Version]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef]
- Lehman, H.L.; Dashner, E.J.; Lucey, M.; Vermeulen, P.; Dirix, L.; Van Laere, S.; van Golen, K.L. Modeling and characterization of inflammatory breast cancer emboli grown in vitro. Int. J. Cancer 2013, 132, 2283–2294. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde Dehydrogenase 1-Positive Cancer Stem Cells Mediate Metastasis and Poor Clinical Outcome in Inflammatory Breast Cancer. Clin. Cancer Res. 2009, 16, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Silvera, D.; Arju, R.; Darvishian, F.; Levine, P.H.; Zolfaghari, L.; Goldberg, J.; Hochman, T.; Formenti, S.C.; Schneider, R.J. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat. Cell Biol. 2009, 11, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.R.; Trenton, N.J.; Debeb, B.G.; Larson, R.; Ruffell, B.; Chu, K.; Hittelman, W.; Diehl, M.; Reuben, J.M.; Ueno, N.T.; et al. Mesenchymal stem cells and macrophages interact through IL-6 to promote inflammatory breast cancer in pre-clinical models. Oncotarget 2016, 7, 82482–82492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provance, O.K.; Lewis-Wambi, J. Deciphering the role of interferon alpha signaling and microenvironment crosstalk in inflammatory breast cancer. Breast Cancer Res. 2019, 21, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, N.; Wang, X.; Lim, B.; Krishnamurthy, S.; Alvarez, R.H.; Willey, J.S.; Parker, C.A.; Song, J.; Shen, Y.; Hu, J.; et al. Safety and Efficacy of Panitumumab Plus Neoadjuvant Chemotherapy in Patients With Primary HER2-Negative Inflammatory Breast Cancer. JAMA Oncol. 2018, 4, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Teplinsky, E.; Silvera, D.; Valeta-Magara, A.; Arju, R.; Giashuddin, S.; Sarfraz, Y.; Alexander, M.; Darvishian, F.; Levine, P.H.; et al. Hyperactivated mTOR and JAK2/STAT3 Pathways: Molecular Drivers and Potential Therapeutic Targets of Inflammatory and Invasive Ductal Breast Cancers After Neoadjuvant Chemotherapy. Clin. Breast Cancer 2016, 16, 113–122 e1. [Google Scholar] [CrossRef] [Green Version]
- Rios-Fuller, T.J.; Ortiz-Soto, G.; Lacourt-Ventura, M.; Maldonado-Martinez, G.; Cubano, L.A.; Schneider, R.J.; Martinez-Montemayor, M.M. Ganoderma lucidum extract (GLE) impairs breast cancer stem cells by targeting the STAT3 pathway. Oncotarget 2018, 9, 35907–35921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Wang, X.; Reyes, M.E.; Zhang, D.; Funakoshi, Y.; Trape, A.P.; Gong, Y.; Kogawa, T.; Eckhardt, B.L.; Masuda, H.; Pirman, D.A., Jr.; et al. EGFR signaling promotes inflammation and cancer stem-like activity in inflammatory breast cancer. Oncotarget 2017, 8, 67904–67917. [Google Scholar] [CrossRef] [Green Version]
- Reddy, J.P.; Atkinson, R.L.; Larson, R.; Burks, J.K.; Smith, D.; Debeb, B.G.; Ruffell, B.; Creighton, C.J.; Bambhroliya, A.; Reuben, J.M.; et al. Mammary stem cell and macrophage markers are enriched in normal tissue adjacent to inflammatory breast cancer. Breast Cancer Res. Treat. 2018, 171, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Morley, T.S.; Kim, M.; Clegg, D.J.; Scherer, P.E. Obesity and cancer--mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 2014, 10, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyengar, N.M.; Zhou, X.K.; Gucalp, A.; Morris, P.G.; Howe, L.R.; Giri, D.D.; Morrow, M.; Wang, H.; Pollak, M.; Jones, L.W.; et al. Systemic Correlates of White Adipose Tissue Inflammation in Early-Stage Breast Cancer. Clin. Cancer Res. 2016, 22, 2283–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Sanchez-Jimenez, F.; Perez-Perez, A.; de la Cruz-Merino, L.; Sanchez-Margalet, V. Obesity and Breast Cancer: Role of Leptin. Front. Oncol. 2019, 9, 596. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Morris, P.G.; Zhou, X.K.; Morrow, M.; Du, B.; Giri, D.; Kopelovich, L.; Hudis, C.A.; Dannenberg, A.J. Increased levels of COX-2 and prostaglandin E2 contribute to elevated aromatase expression in inflamed breast tissue of obese women. Cancer Discov. 2012, 2, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simone, V.; D’Avenia, M.; Argentiero, A.; Felici, C.; Rizzo, F.M.; De Pergola, G.; Silvestris, F. Obesity and Breast Cancer: Molecular Interconnections and Potential Clinical Applications. Oncologist 2016, 21, 404–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, S.; Catalano, S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat. Rev. Endocrinol. 2011, 8, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, R.R.; Xu, Y.; Guo, S.; Watters, A.; Zhou, W.; Leibovich, S.J. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFkappaB/HIF-1alpha activation. Cell. Signal. 2010, 22, 1350–1362. [Google Scholar] [CrossRef] [Green Version]
- Barone, I.; Giordano, C.; Bonofiglio, D.; Ando, S.; Catalano, S. Leptin, obesity and breast cancer: Progress to understanding the molecular connections. Curr. Opin. Pharm. 2016, 31, 83–89. [Google Scholar] [CrossRef]
- Giordano, C.; Chemi, F.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Cordella, A.; Campana, A.; Hashim, A.; Rizza, P.; et al. Leptin as a mediator of tumor-stromal interactions promotes breast cancer stem cell activity. Oncotarget 2016, 7, 1262–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caffa, I.; Spagnolo, V.; Vernieri, C.; Valdemarin, F.; Becherini, P.; Wei, M.; Brandhorst, S.; Zucal, C.; Driehuis, E.; Ferrando, L.; et al. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 2020, 583, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentoor, I.; Engelbrecht, A.M.; van Jaarsveld, P.J.; Nell, T. Chemoresistance: Intricate Interplay Between Breast Tumor Cells and Adipocytes in the Tumor Microenvironment. Front. Endocrinol. 2018, 9, 758. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 136–150 e5. [Google Scholar] [CrossRef] [Green Version]
- De Heer, E.C.; Jalving, M.; Harris, A.L. HIFs, angiogenesis, and metabolism: Elusive enemies in breast cancer. J. Clin. Investig. 2020, 130, 5074–5087. [Google Scholar] [CrossRef]
- Incio, J.; Ligibel, J.A.; McManus, D.T.; Suboj, P.; Jung, K.; Kawaguchi, K.; Pinter, M.; Babykutty, S.; Chin, S.M.; Vardam, T.D.; et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci. Transl. Med. 2018, 10, eaag0945. [Google Scholar] [CrossRef] [Green Version]
- Kantono, M.; Guo, B. Inflammasomes and Cancer: The Dynamic Role of the Inflammasome in Tumor Development. Front. Immunol. 2017, 8, 1132. [Google Scholar] [CrossRef] [Green Version]
- Kolb, R.; Phan, L.; Borcherding, N.; Liu, Y.; Yuan, F.; Janowski, A.M.; Xie, Q.; Markan, K.R.; Li, W.; Potthoff, M.J.; et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat. Commun. 2016, 7, 13007. [Google Scholar] [CrossRef]
- Kulkarni, A.; Bowers, L.W. The role of immune dysfunction in obesity-associated cancer risk, progression, and metastasis. Cell. Mol. Life Sci. 2021, 78, 3423–3442. [Google Scholar] [CrossRef]
- Gibson, J.T.; Orlandella, R.M.; Turbitt, W.J.; Behring, M.; Manne, U.; Sorge, R.E.; Norian, L.A. Obesity-Associated Myeloid-Derived Suppressor Cells Promote Apoptosis of Tumor-Infiltrating CD8 T Cells and Immunotherapy Resistance in Breast Cancer. Front. Immunol. 2020, 11, 590794. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonapace, L.; Coissieux, M.M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef]
- Castano, Z.; San Juan, B.P.; Spiegel, A.; Pant, A.; DeCristo, M.J.; Laszewski, T.; Ubellacker, J.M.; Janssen, S.R.; Dongre, A.; Reinhardt, F.; et al. IL-1beta inflammatory response driven by primary breast cancer prevents metastasis-initiating cell colonization. Nat. Cell Biol. 2018, 20, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Karin, M.; Sun, B. Targeting cancer-promoting inflammation—Have anti-inflammatory therapies come of age? Nat. Rev. Clin. Oncol. 2021, 18, 261–279. [Google Scholar] [CrossRef]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.G.; Passegue, E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, eaan4673. [Google Scholar] [CrossRef]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Corces-Zimmerman, M.R.; Hong, W.J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Heffernan, T.P.; Gera, S.; Kovacs, J.J.; et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 2018, 23, 700–713 e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095 e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, R.L.; Busque, L.; Levine, R.L. Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell 2018, 22, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFalpha facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.H.; Jiang, Y.; Meydan, C.; Shank, K.; Pandey, S.; Barreyro, L.; Antony-Debre, I.; Viale, A.; Socci, N.; Sun, Y.; et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 2015, 27, 502–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristinsson, S.Y.; Bjorkholm, M.; Hultcrantz, M.; Derolf, A.R.; Landgren, O.; Goldin, L.R. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 2897–2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristinsson, S.Y.; Landgren, O.; Samuelsson, J.; Bjorkholm, M.; Goldin, L.R. Autoimmunity and the risk of myeloproliferative neoplasms. Haematologica 2010, 95, 1216–1220. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Choudhary, G.S.; Pellagatti, A.; Choi, K.; Bolanos, L.C.; Bhagat, T.D.; Gordon-Mitchell, S.; Von Ahrens, D.; Pradhan, K.; Steeples, V.; et al. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nat. Cell Biol. 2019, 21, 640–650. [Google Scholar] [CrossRef]
- Pellagatti, A.; Cazzola, M.; Giagounidis, A.; Perry, J.; Malcovati, L.; Della Porta, M.G.; Jadersten, M.; Killick, S.; Verma, A.; Norbury, C.J.; et al. Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia 2010, 24, 756–764. [Google Scholar] [CrossRef] [Green Version]
- Pollyea, D.A.; Harris, C.; Rabe, J.L.; Hedin, B.R.; De Arras, L.; Katz, S.; Wheeler, E.; Bejar, R.; Walter, M.J.; Jordan, C.T.; et al. Myelodysplastic syndrome-associated spliceosome gene mutations enhance innate immune signaling. Haematologica 2019, 104, e388–e392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018, 23, 833–849 e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisel, M.; Hinterleitner, R.; Pacis, A.; Chen, L.; Earley, Z.M.; Mayassi, T.; Pierre, J.F.; Ernest, J.D.; Galipeau, H.J.; Thuille, N.; et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018, 557, 580–584. [Google Scholar] [CrossRef]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Muto, T.; Walker, C.S.; Choi, K.; Hueneman, K.; Smith, M.A.; Gul, Z.; Garcia-Manero, G.; Ma, A.; Zheng, Y.; Starczynowski, D.T. Adaptive response to inflammation contributes to sustained myelopoiesis and confers a competitive advantage in myelodysplastic syndrome HSCs. Nat. Immunol. 2020, 21, 535–545. [Google Scholar] [CrossRef]
- Barreyro, L.; Chlon, T.M.; Starczynowski, D.T. Chronic immune response dysregulation in MDS pathogenesis. Blood 2018, 132, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Raaijmakers, M.H.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.L.; Ma, C.; O’Connell, R.M.; Mehta, A.; DiLoreto, R.; Heath, J.R.; Baltimore, D. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell 2014, 14, 445–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.G.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.J.; Adisty, M.N.; Van Strien, P.M.H.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, H.K.; Bryder, D.; Park, C.Y. Molecular mechanisms underlying lineage bias in aging hematopoiesis. Semin. Hematol. 2017, 54, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Craver, B.M.; El Alaoui, K.; Scherber, R.M.; Fleischman, A.G. The Critical Role of Inflammation in the Pathogenesis and Progression of Myeloid Malignancies. Cancers 2018, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A.; List, A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Rhyasen, G.; Bolanos, L.; Rasch, C.; Varney, M.; Wunderlich, M.; Goyama, S.; Jansen, G.; Cloos, J.; Rigolino, C.; et al. Cytotoxic effects of bortezomib in myelodysplastic syndrome/acute myeloid leukemia depend on autophagy-mediated lysosomal degradation of TRAF6 and repression of PSMA1. Blood 2012, 120, 858–867. [Google Scholar] [CrossRef]
- Daher, M.; Hidalgo Lopez, J.E.; Randhawa, J.K.; Jabbar, K.J.; Wei, Y.; Pemmaraju, N.; Borthakur, G.; Kadia, T.; Konopleva, M.; Kantarjian, H.M.; et al. An exploratory clinical trial of bortezomib in patients with lower risk myelodysplastic syndromes. Am. J. Hematol. 2017, 92, 674–682. [Google Scholar] [CrossRef] [Green Version]
- Eksioglu, E.A.; Chen, X.; Heider, K.H.; Rueter, B.; McGraw, K.L.; Basiorka, A.A.; Wei, M.; Burnette, A.; Cheng, P.; Lancet, J.; et al. Novel therapeutic approach to improve hematopoiesis in low risk MDS by targeting MDSCs with the Fc-engineered CD33 antibody BI 836858. Leukemia 2017, 31, 2172–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Shichita, T.; Okada, M.; Komine, R.; Noguchi, Y.; Yoshimura, A.; Morita, R. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 2015, 6, 7360. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [Green Version]


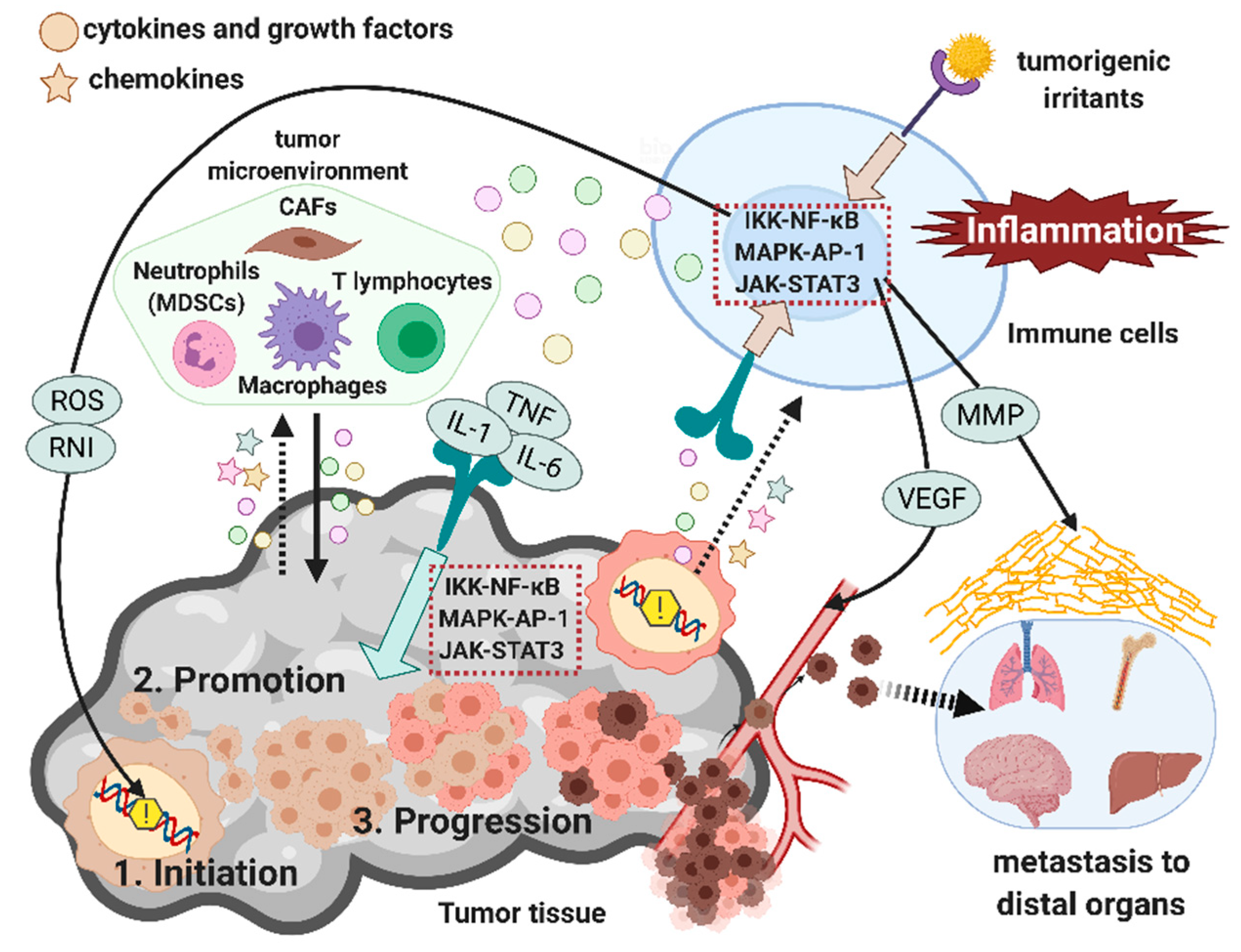{kind=link}
| Types of Cancers | Features | References |
|---|---|---|
| Esophageal cancer | IL-6 and IL-8 are produced by ESCC cells and they are implicated in immune evasion via STAT3 activation. | [110] |
| LIF produced by ESCC cells is necessary for tumor cell proliferation and migration/invasion. | [103] | |
| IL-6 induces STAT3 and ERK1/2 signaling and IL-6 knockout attenuates ESCC cell proliferation. | [191] | |
| IL-6 secreted by CAFs induces EMT and therapy resistance in esophageal adenocarcinoma. | [192] | |
| miR-204-5p functions as a tumor suppressor by directly inhibiting IL-11 expression. | [193] | |
| Gastric cancer | IL-8 and IL-17 are induced by the activation of NF-κB pathway and initiate gastric neoplasia. | [122] |
| CD8+ T cell infiltration is enhanced in EBV+ GC. | [129] | |
| CAFs that secret IL-6 enhances cancer cell migration and EMT by activating STAT3. | [132] | |
| IL-11-producing CAFs facilitate the chemotherapeutic drugs resistance of GC cells. | [133] | |
| IL-22 enhances the viability of GC cells through upregulating the JNK signaling pathway. | [134] | |
| IL-22 stimulation promotes the migration and invasion of GC cells by regulating the AKT/MMP-9 signaling axis. | [135] | |
| IL-22-expressing CAFs promote GC cell invasion via STAT3 and ERK signaling. | [136] | |
| Colorectal cancer | APC loss activates IL-23-synthesizing myeloid cells and expands tumor-resident IL-17-producing T lymphocytes. | [36] |
| Th1, Th2, CD8+ T cells, macrophages, and B cells are recruited in MSI-H tumors compared to microsatellite stable (MSS) cancers. | [151] | |
| IL-6 promotes early CAC tumorigenesis via STAT3 activation. | [194] | |
| Inhibition of IL-11 signaling attenuates colitis-promoted carcinogenesis. | [195,196] | |
| TGF-β signaling in CD4+ T cells promotes the emergence of IL-22-producing Th17 cells and thereby colorectal tumorigenesis. | [197] | |
| Excessive IL-22 in the colon cancer microenvironment leads to tumor growth, inhibition of apoptosis, and promotion of metastasis via STAT3 activation. | [198] | |
| Liver cancer | Th2-like cytokines such as IL-4, IL-5, IL-8, and IL-10 are associated with a more aggressive and metastatic HCC phenotype. | [158] |
| IL-6 secreted by immune cells such as macrophages and Kupffer cells activates inflammatory signaling pathways in hepatocytes via JAK–STAT3 and NF-κB and promotes cell proliferation. | [157] | |
| IL-22 controls the activity of a wide variety of HCC cell survival and proliferation genes. | [165] | |
| HCCs promote immunologic tolerance through the secretion of IL-10 and TGF-β. | [173] | |
| Inhibition of IL-6 signaling turns macrophages into M1-type and reduces HCC tumor formation. | [199] | |
| Direct suppression of IKKβ by miR-451 inhibits HCC cell proliferation. | [200] | |
| Pancreatic cancer | Chronic inflammation leads to production of pro-inflammatory cytokines such as TNF and IL-6. IL-6 activates JAK–STAT3 signaling and promotes pancreatic cancer cell growth and progression. | [185] |
| LIF produced by pancreatic stellate cells promote pancreatic tumorigenesis. | [187] | |
| TNF produced by myeloid cells and pancreatic cancer cells stimulates the production of other cytokines which enhances primary tumor growth. | [188] | |
| Persistent STAT3 activation mediated by loss of p53 promotes pancreatic tumor growth. | [201] | |
| Tumor-induced IL-6 results in the change of metabolic response and thus impairs anti-tumor immunity. | [202] | |
| IL-6 stimulates pancreatic cancer cell proliferation and survival. | [203] | |
| LIF expression is induced by oncogenic KRAS in PDAC and LIF depletion prevents engraftment in pancreatic xenograft models. | [204] | |
| ILC3s promote the proliferation, migration, and invasion of pancreatic cancer cells by secreting IL-22 to activate AKT signaling. | [205] | |
| IL-22 promotes acinar to ductal metaplasia, stem cell features, and increased expression of EMT markers. | [206] |
| Types of Cancers | Features | References |
|---|---|---|
| Lung cancer | Stage I lung adenocarcinoma lesions already harbor significantly altered T cell and NK cell compartments. | [211] |
| CAFs and matched normal fibroblasts show 46 differentially expressed genes, encoding significantly enriched extracellular proteins regulated by the TGF-β signaling pathway. | [214] | |
| CAFs support T cell suppression within the tumor microenvironment by a mechanism dependent on immune checkpoint activation. | [218] | |
| CAFs constitute a supporting niche for cancer stemness through IGF-2/IGF-1R signaling and this blockade inhibits Nanog expression. | [216] | |
| Stabilized HIF-1α protein expression inhibits the TGF-β-stimulated appearance of EMT phenotypes across cell types and species. | [219] | |
| Co-occurring genomic alterations, particularly in TP53 and LKB1, have emerged as core determinants of oncogene-driven lung cancer subgroup. | [224] | |
| Tumor-promoting inflammation and immune modulation caused by KRAS mutation leads to immune escape in the TME. | [32] | |
| IL-6 is a potential druggable target for prevention and treatment of Kras-mutant lung tumors. | [223] | |
| NF-κB can be a potential companion drug target, together with EGFR, in EGFR-mutant lung cancers. | [225] | |
| Repetitive exposure to tobacco smoke promotes tumor development through Kras activation in lung epithelial cells. | [17] | |
| Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/TXA2. | [231] | |
| Prostate cancer | Exposure to environmental estrogens increases the risk of PCa. | [234] |
| IL-23 secreted by myeloid cells drives castration-resistant PCa. | [235] | |
| Inflammation and atrophy are involved in prostate carcinogenesis and the microbiome play an important role in establishing an inflammatory microenvironment. | [238] | |
| A HFD drives metastasis in a PTEN-null mouse model of PCa, and an SREBP signature was highly enriched in metastatic human CaP. | [247] | |
| A HFD fuels PCa progression by rewiring the metabolome and amplifying the MYC program. | [253] | |
| Senescence induced by PTEN deficiency or chemotherapy limits the progression of PCa and TIMP1 deletion allows senescence to promote metastasis. | [262] | |
| Adipocytes from periprostatic adipose tissue support the directed migration of PCa cells through CCR3/CCR7 axis promoted by obesity. | [250] | |
| Disruptions of CHD1 that define a subtype of ETS gene family fusion-negative PCa and ETS2 are also deregulated through mutation. | [236] | |
| Activation of PI3K–AKT–mTOR and MAPK signaling pathways in prostate tumors cooperate to upregulate c-Myc. | [252] | |
| In metastatic castration-resistant PCa patients, aberrations of AR, ETS genes, TP53, and PTEN are frequent, with TP53 and AR alterations enriched in mCRPC compared to primary PCa | [258] | |
| CAFs and M2-polarized macrophages synergize during prostate carcinoma progression. | [264] | |
| PTEN-null prostate tumors are infiltrated by TAMs expressing CXCR2, and activation of CXCR2–CXCL2 polarizes macrophages toward anti-inflammatory status. | [265] | |
| Breast cancer | eIF4GI reprograms the protein synthetic machinery for increased translation that promotes IBC tumor cell survival and formation of tumor emboli. | [273] |
| There is a crosstalk of immune and stromal cells in the local tumor microenvironment and IBC through IFNα signaling. | [275] | |
| The JAK2–STAT3 signaling pathway is required for growth of CD44+CD24- stem cell-like breast cancer cells in human tumors. | [279] | |
| Leptin signaling contributes to the metabolic features and shapes the tumor microenvironment. | [288] | |
| A fasting-mimicking diet promotes long-lasting tumor regression and reverts acquired resistance to drug treatment. | [292] | |
| JAK–STAT3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. | [295] | |
| BC patients with obesity harbored increased systemic concentrations of IL-6 and/or FGF-2 and their tumor vasculature was less sensitive to anti-VEGF-A treatment. | [297] | |
| Obesity induces an increase in tumor-infiltrating myeloid cells with an activated NLRC4 inflammasome in breast cancer. | [299] | |
| Inhibition of CCL2–CCR2 signaling blocks the recruitment of inflammatory monocytes and inhibits metastasis of breast tumors in a murine model. | [302] | |
| Inhibition of CCL2 and IL-6 markedly reduces metastases of breast cancer. | [303] | |
| Targeting breast cancer cell-initiated domino effect within the immune system (the γδ T cell/IL-17/neutrophil axis) inhibits metastasis. | [49] | |
| Blockade of WNT secretion in TP53-null cancer reverses macrophage production of IL-1β and subsequent neutrophilic inflammation, resulting in reduced metastasis. | [33] | |
| Ablation of the pro-inflammatory response or inhibition of the IL-1 receptor relieves the differentiation block of metastasis-initiating cancer cells into highly proliferative progeny, and results in metastatic colonization of breast cancer. | [304] | |
| Hematological malignancies | Germline genetic variation can shape somatic variation in hematopoietic stem cells, leading to CHIP. | [308] |
| Age-related clonal hematopoiesis is associated with increases in the risk of hematologic and cardiovascular disease. | [309] | |
| Preleukemic HSCs can survive induction chemotherapy, identifying these cells as a reservoir for the re-evolution of relapsed disease. | [311] | |
| Mutations in PPM1D, a DNA damage response regulator, drive clonal hematopoiesis in response to cytotoxic chemotherapy. | [312] | |
| JAK2V617F promotes clonal selection by conferring TNF resistance, while simultaneously generating a TNFα-rich environment. | [315] | |
| Chronic immune stimulation acts as a trigger for AML/MDS development. | [317] | |
| U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. | [319] | |
| NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. | [322] | |
| TET2-deficient mature myeloid cells and HSPCs increase in response to inflammation, resulting in production of inflammatory cytokines and resistance to apoptosis. | [323] | |
| Upon inflammation, MDS HSPCs switch from canonical to noncanonical NF-κB signaling, which is dependent on TLR–TRAF6-mediated activation of A20. | [327] | |
| Perturbation of specific mesenchymal stromal cells can disorder function and apoptosis of heterologous cells, and disrupt tissue homeostasis. | [329] | |
| Overproduction of niche factors such as CDH2, IGFBP2, VEGF-A, and LIF is associated with the ability of MSCs to enhance MDS expansion. | [330] | |
| Activation of p53-S100A8/9-TLR inflammatory signaling axis in the mesenchymal niche predicts leukemic evolution and progression in MDS. | [332] | |
| Leukemic stem cells isolated from de novo AML patients are uniquely reliant on amino acid metabolism for oxidative phosphorylation. | [344] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hibino, S.; Kawazoe, T.; Kasahara, H.; Itoh, S.; Ishimoto, T.; Sakata-Yanagimoto, M.; Taniguchi, K. Inflammation-Induced Tumorigenesis and Metastasis. Int. J. Mol. Sci. 2021, 22, 5421. https://doi.org/10.3390/ijms22115421
Hibino S, Kawazoe T, Kasahara H, Itoh S, Ishimoto T, Sakata-Yanagimoto M, Taniguchi K. Inflammation-Induced Tumorigenesis and Metastasis. International Journal of Molecular Sciences. 2021; 22(11):5421. https://doi.org/10.3390/ijms22115421
Chicago/Turabian StyleHibino, Sana, Tetsuro Kawazoe, Hidenori Kasahara, Shinji Itoh, Takatsugu Ishimoto, Mamiko Sakata-Yanagimoto, and Koji Taniguchi. 2021. "Inflammation-Induced Tumorigenesis and Metastasis" International Journal of Molecular Sciences 22, no. 11: 5421. https://doi.org/10.3390/ijms22115421
APA StyleHibino, S., Kawazoe, T., Kasahara, H., Itoh, S., Ishimoto, T., Sakata-Yanagimoto, M., & Taniguchi, K. (2021). Inflammation-Induced Tumorigenesis and Metastasis. International Journal of Molecular Sciences, 22(11), 5421. https://doi.org/10.3390/ijms22115421