
Genes Associated with Foliar Resistance to Septoria Nodorum Blotch of Hexaploid Wheat (Triticum aestivum L.)

Abstract
:1. Introduction
2. Results
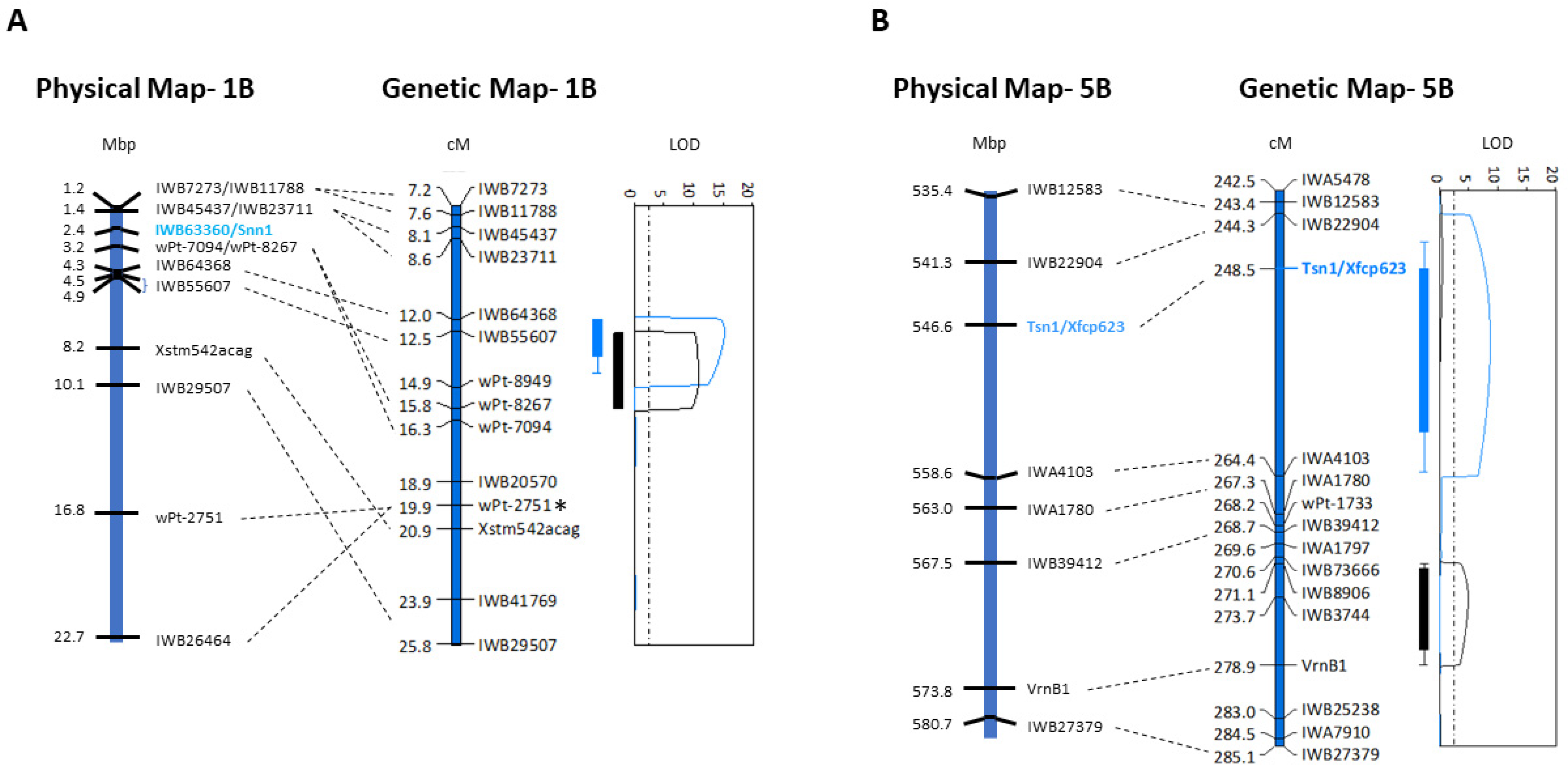
2.1. Alignment of Genetic and Physical Maps and Gene Annotation on Chromosomes 1BS and 5BL
2.2. Genes on Chromosome 1BS Associated with Foliar SNB Resistance
2.3. Genes on Chromosome 5BL Associated with Foliar SNB Resistance
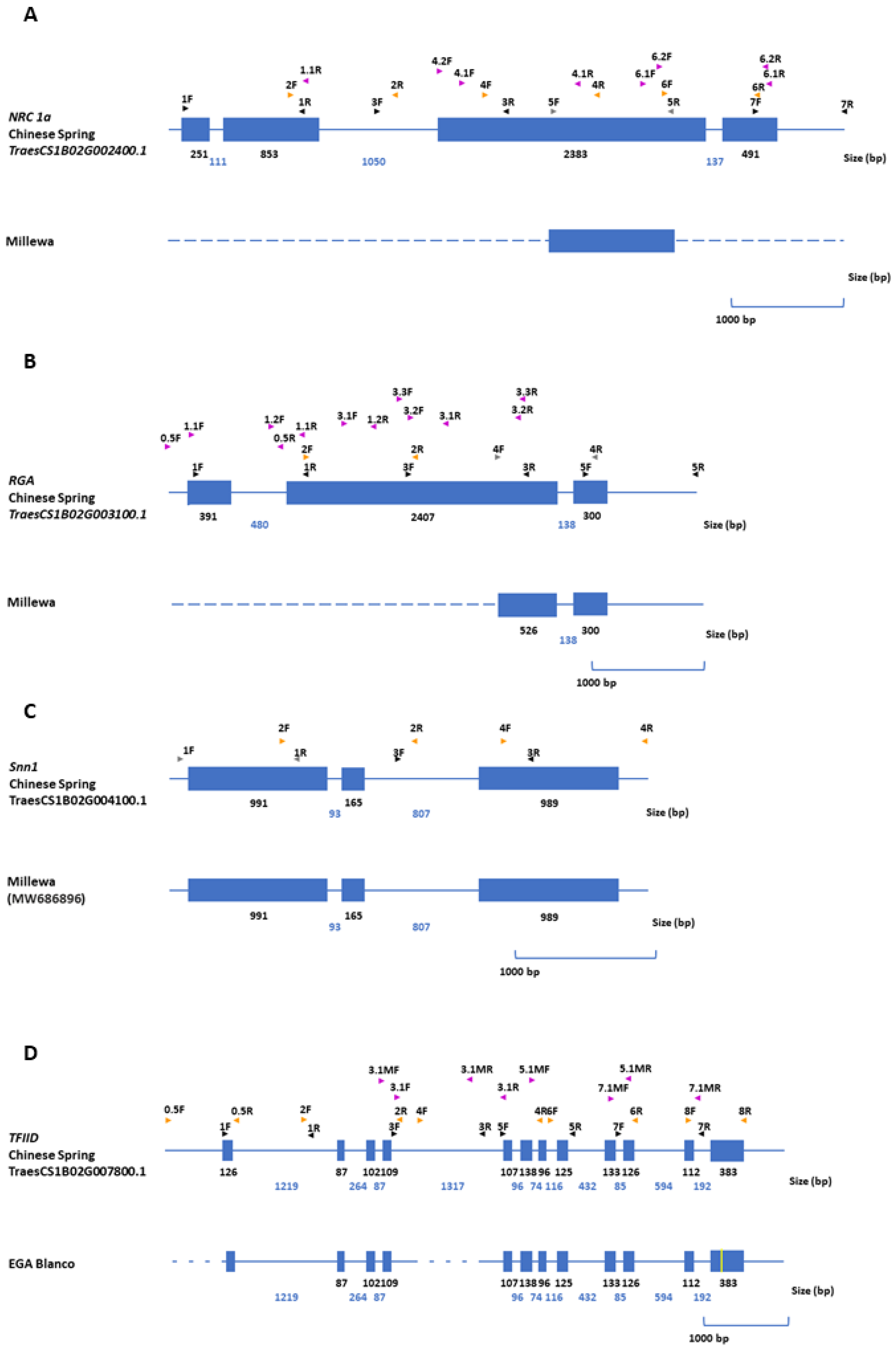
2.4. Gene Structure Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Material and Evaluation for SNB Response
4.2. Aligning Genetic and Physical Maps and Annotation of Gene Content
4.3. Primer Design and Tiling Paths
4.4. PCR Amplification and Sequencing of Genomic Tiling Paths
4.5. Gene Marker Development, Genotyping and QTL Analysis
4.6. Gene Structure Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murray, G.M.; Brennan, J.P. Estimating disease losses to the Australian wheat industry. Australas. Plant Pathol. 2009, 38, 558–570. [Google Scholar] [CrossRef]
- Mullaney, E.J.; Scharen, A.L.; Bryan, M.D. Resistance to Septoria nodorum in a durum wheat cultivar as determined by stage of host development. Can. J. Bot. 1983, 61, 2248–2250. [Google Scholar] [CrossRef]
- Spadafora, V.J.; Cole, H.; Frank, J.A. Effects of leaf and glume blotch caused by Leptosphaeria nodorum on yield and yield components of soft red winter wheat in Pennsylvania. Phytopathology 1987, 77, 1326–1329. [Google Scholar] [CrossRef] [Green Version]
- Wainshilbaum, S.J.; Lipps, P.E. Effect of temperature and growth stage of wheat on development of leaf and glume blotch caused by Septoria tritici and S. nodorum. Plant Dis. 1991, 75, 993–998. [Google Scholar] [CrossRef]
- Shankar, M.; Walker, E.; Golzar, H.; Loughman, R.; Wilson, R.E.; Francki, M.G. Quantitative Trait Loci for Seedling and Adult Plant Resistance to Stagonospora nodorum in Wheat. Phytopathology 2008, 98, 886–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francki, M.G.; Shankar, M.; Walker, E.; Loughman, R.; Golzar, H.; Ohm, H. New Quantitative Trait Loci in Wheat for Flag Leaf Resistance to Stagonospora nodorum Blotch. Phytopathology 2011, 101, 1278–1284. [Google Scholar] [CrossRef]
- Francki, M.G.; Walker, E.; McMullan, C.J.; Morris, W.G. Multi-Location Evaluation of Global Wheat Lines Reveal Multiple QTL for Adult Plant Resistance to Septoria Nodorum Blotch (SNB) Detected in Specific Environments and in Response to Different Isolates. Front. Plant Sci. 2020, 11, 771. [Google Scholar] [CrossRef]
- Lu, Q.; Lillemo, M. Molecular mapping of adult plant resistance to Parastogonospora nodorum leaf blotch in bread wheat lines ‘Shanghai-3/Catbird’ and ‘Naxos’. Theor. Appl. Genet. 2014, 127, 2635–2644. [Google Scholar] [CrossRef] [Green Version]
- Ruud, A.K.; Dieseth, J.A.; Ficke, A.; Furuki, E.; Phan, H.T.T.; Oliver, R.P.; Tan, K.; Lillemo, M. Genome-Wide Association Mapping of Resistance to Septoria Nodorum Leaf Blotch in a Nordic Spring Wheat Collection. Plant Genome 2019, 12, 180105. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Corsi, B.; Ficke, A.; Tan, K.-C.; Cockram, J.; Lillemo, M. Genetic mapping using a wheat multi-founder population reveals a locus on chromosome 2A controlling resistance to both leaf and glume blotch caused by the necrotrophic fungal pathogen Parastagonospora nodorum. Theor. Appl. Genet. 2020, 133, 785–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, M.C.; Solomon, P.S. Just the surface. Advances in the discovery and characterization of necrotrophic wheat effectors. Curr. Opin. Microbiol. 2018, 46, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Friesen, T.L.; Ling, H.; Meinhardt, S.W.; Oliver, R.P.; Rasmussen, J.B.; Faris, J.D. The Tsn1–ToxA interaction in the wheat–Stagonospora nodorum pathosystem parallels that of the wheat–tan spot system. Genome 2006, 49, 1265–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesen, T.L.; Chu, C.-G.; Liu, Z.H.; Xu, S.S.; Halley, S.; Faris, J.D. Host-selective toxins produced by Stagonospora nodorum confer disease susceptibility in adult wheat plants under field conditions. Theor. Appl. Genet. 2009, 118, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.T.T.; Ryback, K.; Furuki, E.; Breen, S.; Solomon, P.S.; Oliver, R.P.; Tan, K.C. Differential effector expression un-derpins epistasis in a plant fungal disease. Plant J. 2016, 87, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Zhang, Z.; Friesen, T.L.; Raats, D.; Fahima, T.; Brueggeman, R.S.; Lu, S.; Trick, H.N.; Liu, Z.; Chao, W.; et al. The hijacking of a receptor kinase-driven pathway by a wheat fungal pathogen leads to disease. Sci. Adv. 2016, 2, e1600822. [Google Scholar] [CrossRef] [Green Version]
- Ruud, A.K.; Wingju, S.; Belova, T.; Friesen, T.L.; Lillemo, M. Mapping of SnTox3-Snn3 as a major determinant of field susceptibility to Septoria nodorum leaf blotch in the SHA3/CBRD × Naxos population. Theor. Appl. Genet. 2017, 130, 1361–1374. [Google Scholar] [CrossRef]
- Downie, R.C.; Bouvet, L.; Furuki, E.; Gosman, N.; Gardner, K.A.; Mackay, I.J.; Mantello, C.C.; Mellers, G.; Phan, H.T.T.; Rose, G.A.; et al. Assessing European Wheat Sensitivities to Parastagonospora nodorum Necrotrophic Effectors and Fine-Mapping the Snn3-B1 Locus Conferring Sensitivity to the Effector SnTox3. Front. Plant Sci. 2018, 9, 881. [Google Scholar] [CrossRef] [PubMed]
- Abeysekara, N.S.; Friesen, T.L.; Keller, B.; Faris, J.D. Identification and characterization of a novel host–toxin interaction in the wheat–Stagonospora nodorum pathosystem. Theor. Appl. Genet. 2009, 120, 117–126. [Google Scholar] [CrossRef]
- Friesen, T.L.; Chu, C.; Xu, S.S.; Faris, J.D. SnTox5-Snn5: A novel Stagonospora nodorum effector- wheat gene interaction and its relationship with SnToxA-Tsn1 and SnTox3-Snn3-B1 interactions. Mol. Plant Path. 2012, 13, 1101–1109. [Google Scholar] [CrossRef]
- Gao, Y.; Faris, J.D.; Liu, Z.; Kim, Y.M.; Syme, R.A.; Oliver, R.P.; Xu, S.S.; Friesen, T.L. Identification and Characterization of the SnTox6-Snn6 Interaction in the Parastagonospora nodorum–Wheat Pathosystem. Mol. Plant-Microbe Interact. 2015, 28, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Friesen, T.L.; Saini, J.; Xu, S.S.; Rasmussen, J.B.; Faris, J.D. The wheat Snn7 gene confers susceptibility on recog-nition of the Parastagonospora nodorum necrotrophic effector SnTox7. Plant Genome 2015, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Gonzalelz-Hernandez, J.L.; Singh, P.K.; Mergoum, M.; Adhikari, T.B.; Kianian, S.F.; Simsek, S.; Elias, E.M. A quantitative trait locus on chromosome 5B controls resistance of Triticum turgidum (L.) var. diccocoides to Stagnospora nodorum blotch. Euphytica 2009, 166, 199–206. [Google Scholar] [CrossRef]
- Cowger, C.; Ward, B.; Brown-Guedira, G.; Brown, J.K.M. Role of Effector-Sensitivity Gene Interactions and Durability of Quantitative Resistance to Septoria Nodorum Blotch in Eastern U.S. Wheat. Front. Plant Sci. 2020, 11, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faris, J.D.; Zhang, Z.; Lu, H.; Lu, S.; Reddy, L.; Cloutier, S.; Fellers, J.P.; Meinhardt, S.W.; Rasmussen, J.B.; Xu, S.S.; et al. A unique wheat disease resistance-like gene governs effector-triggered susceptibility to necrotrophic pathogens. Proc. Natl. Acad. Sci. USA 2010, 107, 13544–13549. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loreng, J. Necrotrophic exploitation abs subversion of plant defense: A lifestyle or just a phase, and implications in breeding. Phytopathology 2019, 109, 332–346. [Google Scholar] [CrossRef]
- Bellincampi, D.; Cervone, F.; Lionetti, V. Plant cell wall dynamics and wall related susceptibility in plant-pathogen interactions. Front. Plant Sci. 2014, 5, 228. [Google Scholar] [CrossRef] [Green Version]
- The International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 6194. [Google Scholar]
- The International Wheat Genome Sequencing Consortium. Shifting the limits of wheat research and breeding using a fully annotated reference genome. Science 2018, 361, 6430. [Google Scholar]
- Clavijo, B.; Venturini, L.; Schudoma, C.; Garcia, A.G.; Kaithakottil, G.; Wright, J.; Borrill, P.; Kettleborough, G.; Heavens, D.; Chapman, H.; et al. An improved assembly and annotation of the allohexaplod wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 2017, 27, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Montenegro, J.D.; Golicz, A.A.; Bayer, P.E.; Hurgobin, B.; Lee, H.; Chan, C.-K.K.; Visendi, P.; Lai, K.; Doležel, J.; Batley, J.; et al. The pangenome of hexaploidy bread wheat. Plant J. 2017, 90, 1007–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkowiak, S.; Gao, L.; Monat, C.; Haberer, G.; Kassa, M.T.; Brinton, J.; Ramirez-Gonzalez, R.H.; Kolodziej, M.C.; Delorean, E.; Thambugala, D.; et al. Faculty Opinions recommendation of Multiple wheat genomes reveal global variation in modern breeding. Nature 2020, 588, 277. [Google Scholar] [CrossRef] [PubMed]
- Francki, M.G.; Walker, E.; Li, D.A.; Forrest, K. High-density SNP mapping reveals closely linked QTL for resistance to Stagonospora nodorum blotch (SNB) in flag leaf and glume of hexaploid wheat. Genome 2018, 61, 145–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badaeva, E.D.; Dedkova, O.S.; Gay, G.; Pukhalskyi, V.A.; Zelenin, A.V.; Bernard, S.; Bernard, M. Chromosomal rear-rangements in wheat: Their types and distribution. Genome 2007, 50, 907–926. [Google Scholar] [CrossRef]
- Przewieslik-Allen, A.M.; Wilkinson, P.A.; Burridge, A.J.; Winfield, M.O.; Dai, X.; Beaumont, M.; King, J.; Yang, C.-Y.; Griffiths, S.; Wingen, L.U.; et al. The role of gene flow and chromosomal instability in shaping the bread wheat genome. Nat. Plants 2021, 7, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhao, X.; Mace, E.; Henry, R.; Jordan, D. Exploring and Exploiting Pan-genomics for Crop Improvement. Mol. Plant 2019, 12, 156–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Ravet, K.; Pearce, S. Extensive structural variation in the Bowman-Birk inhibitor family in common wheat (Triticum aestivum L.). BMC Genom. 2021, 22, 1–21. [Google Scholar] [CrossRef]
- Liu, M.; Stiller, J.; Holušová, K.; Vrána, J.; Liu, D.; Doležel, J.; Liu, C. Chromosome-specific sequencing reveals an extensive dispensable genome component in wheat. Sci. Rep. 2016, 6, 36398. [Google Scholar] [CrossRef]
- Della, C.R.; Qiu, Y.; Ou, S.; Hufford, M.B.; Hirsch, C.N. How the pan-genome is changing crop genomics and improvement. Genome Biol. 2021, 22, 3. [Google Scholar] [CrossRef]
- Czembor, P.C.; Arseniuk, E.; Radecka-Janusik, M.; Piechota, U.; Słowacki, P. Quantitative trait loci analysis of adult plant resistance to Parastagonospora nodorum blotch in winter wheat cv. Liwilla (Triticum aestivum L.). Eur. J. Plant Pathol. 2019, 155, 1001–1016. [Google Scholar] [CrossRef] [Green Version]
- Andersen, E.J.; Nepal, M.P.; Purintun, J.M.; Nelson, D.; Mermigka, G.; Sarris, P.F. Wheat Disease Resistance Genes and Their Diversification Through Integrated Domain Fusions. Front. Genet. 2020, 11, 898. [Google Scholar] [CrossRef] [PubMed]
- Sekhwar, M.K.; Li, P.; Lam, I.; Wang, X.; Cloutier, S.; You, F.M. Disease resistance gene analogues (RGAs) in plants. Int. J. Mol. Sci. 2015, 16, 19248–19290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baggs, E.; Dagdas, G.; Krasileva, K. NLR diversity, helpers and integrated domains: Making sense of the NLR IDentity. Curr. Opin. Plant Biol. 2017, 38, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Leibman-Markus, M.; Pizarro, L.; Bar, M.; Coaker, G.; Avni, A. NRC proteins—A critical node for pattern and effector mediated signaling. Plant Signal. Behav. 2018, 13, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Belhaj, K.; Bozkurt, T.O.; Birk, M.S.; Kamoun, S. Helper NLR proteins NRC 2a/b and NRC 3 but not NRC 1 are required for Pto-mediated cell death and resistance in Nicotiana benthamiana. New Phytol. 2016, 209, 1344–1352. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-H.; Abd-El-Haliem, A.; Bozkurt, T.O.; Belhaj, K.; Terauchi, R.; Vossen, J.H.; Kamoun, S. NLR network mediates immunity to diverse plant pathogens. Proc. Natl. Acad. Sci. USA 2017, 114, 8113–8118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hȕckellhoven, R. Cell wall-associated mechanisms of disease resistance and susceptibility. Ann. Rev. Phytopathol. 2007, 45, 101–127. [Google Scholar] [CrossRef] [PubMed]
- Deeks, M.J.; Calcutt, J.R.; Ingle, E.K.; Hawkins, T.J.; Chapman, S.; Richardson, A.C.; Mentlak, D.A.; Dixon, M.R.; Cartwright, F.; Smertenko, A.P.; et al. A Superfamily of Actin-Binding Proteins at the Actin-Membrane Nexus of Higher Plants. Curr. Biol. 2012, 22, 1595–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Hawes, C.; Hussey, P.J. Plant Endoplasmic Reticulum–Plasma Membrane Contact Sites. Trends Plant Sci. 2017, 22, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Aoyagi, N.; Wassarman, D.A. Genes encoding Drosophila melanogaster RNA polymerase II general transcription factors: Diversity in TFIIA and TFIID components contributes to gene-specific regulation. J. Cell Biol. 2000, 150, F45–F49. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.B.; Greber, B.J.; Nogales, E. Recent insights into the structure of TFIID, its assembly and its binding to core promoter. Curr. Opin. Struct. Biol. 2020, 61, 17–24. [Google Scholar] [CrossRef]
- Stephens, A.E.; Gardiner, D.M.; White, R.G.; Munn, A.L.; Manners, J.M. Phases of Infection and Gene Expression of Fusarium graminearum During Crown Rot Disease of Wheat. Mol. Plant-Microbe Interact. 2008, 21, 1571–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yundaeng, C.; Somta, P.; Chen, J.; Yuan, X.; Chankaew, S.; Chen, X. Fine mapping of QTL conferring Cercospora leaf spot disease resistance in mungbean revealed TAF5 as candidate gene for the resistance. Theor. Appl. Genet. 2021, 134, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, E. Nullisomic-tetrasomic combinations in hexaploid wheat. In Chromosome Manipulations and Plant Genetics; Lewis, D.R., Ed.; Oliver and Boyd: London, UK, 1966; pp. 29–47. [Google Scholar]
- Francki, M.G.; Walker, E.; Crawford, A.C.; Broughton, S.; Ohm, H.W.; Barclay, I.; Wilson, R.E.; McLean, R. Comparison of genetic and cytogenetic maps of hexaploid wheat (Triticum aestivum L.) using SSR and DArT markers. Mol. Genet. Genom. 2008, 281, 181–191. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop. J. 2015, 3, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Voorrips, R.E. MapChart: Software for the Graphical Presentation of Linkage Maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | IWGSC ID | Polymorphism a | Physical Map Position (bp) b | Genetic Map Position (cM) |
|---|---|---|---|---|
| 1BR_NRC_5A | TraesCS1B02G002400.1 | PAV (EGA Blanco) | 1,782,029 | 7.7 |
| 1B_NRC_A1 | TraesCS1B02G002800.1 | PAV (EGA Blanco) | 1,846,624 | 14.4 |
| 1BR_RPP13_1B | TraesCS1B02G002900.1 | PAV (EGA Blanco) | 1,943,148 | 14.4 |
| 1BR_RPP13_2C | TraesCS1B02G003000.1 | PAV (EGA Blanco) | 1,971,915 | 14.4 |
| 1BR_RGA_4B | TraesCS1B02G003100.1 | PAV (EGA Blanco) | 2,004,576 | 14.4 |
| 1BR_RPP13_11D | TraesCS1B02G003200.1 | PAV (EGA Blanco) | 2,057,816 | 10.4 |
| 1BR_RPP13_13E | TraesCS1B02G003300.1 | PAV (EGA Blanco) | 2,078,761 | 10.9 |
| 1BR_RPP13_7F | TraesCS1B02G003400.1 | PAV (EGA Blanco) | 2,105,792 | 10.4 |
| 1B_RGA_E1 | TraesCS1B02G004100.1 | PAV (EGA Blanco) | 2,357,713 | 14.9 |
| 1B_NRC_C1 | TraesCS1B02G004600.1 | PAV (EGA Blanco) | 2,756,985 | 14.9 |
| 1BR_RR_3A | TraesCS1B02G004700.1 | PAV (EGA Blanco) | 2,919,258 | 14.4 |
| 1BR_LRR_3C | TraesCS1B02G004900.1 | PAV (EGA Blanco) | 3,145,388 | 14.0 |
| 1BR_RR_2C | TraesCS1B02G005600.1 | PAV (Millewa) | 3,656,683 | 7.2 |
| 1B_Lr21_B3 | TraesCS1B02G006300.1 | PAV (Millewa) | 3,925,448 | 7.2 |
| 1BR_LRR_1D | TraesCS1B02G006400.1 | PAV (Millewa) | 3,935,573 | 4.0 * |
| 1BR_LRR_4E | TraesCS1B02G006800.1 | PAV (Millewa) | 3,966,850 | 7.7 |
| 1B_RPM1_A1 | TraesCS1B02G006900.1 | PAV (Millewa) | 4,049,415 | 6.8 |
| 1BR_LRR_2F | TraesCS1B02G007200.1 | PAV (Millewa) | 4,534,576 | 7.7 |
| 1BR_LRR_4G | TraesCS1B02G007300.1 | PAV (Millewa) | 4,146,127 | 0 * |
| IWB64368 | TraesCS1B02G007800.1 | SNP | 4,320,651 | 9.8 |
| 1BR_LRR_1H | TraesCS1B02G008500.1 | PAV (Millewa) | 4,483,578 | 7.7 |
| 1BR_LRR_1I | TraesCS1B02G008800.1 | PAV (Millewa) | 4,534,869 | 7.7 |
| 1BR_LRR_4J | TraesCS1B02G008900.1 | PAV (Millewa) | 4,541,238 | 7.7 |
| 1B_Pm3_F1 | TraesCS1B02G051200.1 | PAV (Millewa) | 6,272,172 | 24.8 |
| 1B_RPP13_C1 | TraesCS1B02G014500.1 | PAV (EGA Blanco) | 7,040,210 | 28.1 |
| 1B_Pm3_D1 | TraesCS1B02G014800.1 | PAV (EGA Blanco) | 7,257,001 | 25.2 |
| 1B_Pm3_D6 | TraesCS1B02G014900.1 | PAV (Millewa) | 7,253,228 | 26.6 |
| 1B_Pm3_I1 | TraesCS1B02G042500.1 | PAV (Millewa) | 22,155,020 | 42.5 |
| QTL | Marker (Gene) | Polymorphism a | Physical Position (bp) b | Genetic Position (cM) c | LOD d | PVE e | Additive f |
|---|---|---|---|---|---|---|---|
| QSnl07.daw-1B | 1BR_RR_2C | PAV (Millewa) | 3,656,683 | 7.2 | 0.1 | 0.1 | 0.4 |
| 1BR_NRC_5A | PAV (EGA Blanco) | 1,782,029 | 7.7 | 13.6 | 20.3 | −5.9 | |
| IWB64368 | SNP | 4,320,856 | 9.8 | 18.6 | 25.8 | −6.7 | |
| 1BR_RPP13_11D | PAV (EGA Blanco) | 2,057,816 | 10.4 | 0.0 | 0.0 | 0.0 | |
| 1BR_RPP13_13E | PAV (EGA Blanco) | 2,078,761 | 10.9 | 0.0 | 0.0 | 0.0 | |
| QSnl08.daw-1B | 1BR_LRR_3C | PAV (EGA Blanco) | 3,145,388 | 14.0 | 0.0 | 0.0 | 0.1 |
| 1BR_RGA_4B | PAV (EGA Blanco) | 2,004,576 | 14.4 | 10.0 | 16.9 | −3.9 | |
| X3476283 * (Snn1) | PAV (EGA Blanco) | 2,357,713 | 14.9 | 10.1 | 17.1 | −3.9 | |
| IWB7273 | SNP | 1,203,706 | 16.3 | 0.0 | 0.0 | −0.1 | |
| IWB11788 | SNP | 1,203,968 | 16.8 | 0.0 | 0.0 | −0.1 | |
| QSnl07.daw-5B | IWB12583 | SNP | 539,072,791 | 229.8 | 0.1 | 0.2 | 0.5 |
| IWB22904 | SNP | 541,343,146 | 230.7 | 0.1 | 0.1 | 0.4 | |
| Xfcp623 (Tsn1) | PAV | ND | 234.9 | 7.5 | 10.1 | −4.2 | |
| Xfcp620 (WAK35) | PAV | 546,568,033 | 234.9 | 7.5 | 10.1 | −4.2 | |
| IWA4103 | SNP | 558,625,387 | 250.8 | 0.6 | 0.7 | −1.1 | |
| 5B_RGA_SNP_3A1 | SNP | 562,793,027 | 253.6 | 0.4 | 0.4 | −0.9 | |
| QSnl08.daw-5B | Xfcp623 (Tsn1) | PAV | ND | 234.9 | 0.3 | 0.5 | −0.6 |
| Xfcp620 (WAK35) | PAV | 546,568,033 | 234.9 | 0.3 | 0.5 | −0.6 | |
| IWA4103 | SNP | 558,625,387 | 250.8 | 2.8 | 4.6 | −2.0 | |
| 5B_RGA_SNP_3A1 | SNP | 562,793,027 | 253.6 | 4.3 | 6.9 | −2.4 | |
| 5B_RPP13_5B | PAV (Millewa) | 563,121,363 | 254.5 | 0.0 | 0.0 | 0.0 | |
| 5B_Dir_1C | PAV (Millewa) | 570,174,000 | 255.4 | 0.0 | 0.0 | 0.0 |
| Marker | IWGSC ID | Polymorphism a | Physical Map Position (bp) b | Genetic Map Position (cM) |
|---|---|---|---|---|
| Xfcp623 | ND c | PAV (EGA Blanco) | ND c | 234.9 |
| Xfcp620 | TraesCS5B02G368200.1 | SNP | 546,568,033 | 234.9 |
| IWA4103 | TraesCS5B02G380700.1 | SNP | 558,625,387 | 250.8 |
| 5B_RGA_SNP_3A1 | TraesCS5B02G383800.1 | SNP | 562,793,027 | 253.6 |
| 5B_RPP13_5B | TraesCS5B02G384300.1 | PAV (Millewa) | 563,121,363 | 254.5 |
| 5B_WRKY_2A | TraesCS5B02G384600.1 | PAV (EGA Blanco) | 563,332,765 | 255.9 |
| 5B_Dir_1C | TraesCS5B02G391000.1 | PAV (Millewa) | 570,174,000 | 255.4 |
| 5B_LRR_4D_SNP | TraesCS5B02G393400.1 | SNP | 571,609,010 | 258.3 |
| 5B_PiK2_SNP_11_A1 | TraesCS5B02G394100.1 | SNP | 571,998,218 | 260.1 |
| 5B_Pi_21_1A | TraesCS5B02G396100.1 | PAV (EGA Blanco) | 573,141,867 | 263 |
| 5B_RPK_2A | TraesCS5B02G402100.1 | PAV (Millewa) | 578,607,490 | 268 |
| 5B_SerThr_4C_SNP | TraesCS5B02G403300.1 | SNP | 580,085,277 | 266.2 |
| 5B_PerOX_2B | TraesCS5B02G404300.1 | PAV (EGA Blanco) | 580,431,388 | 264.4 |
| 5B_PerOX_SNP_1.1 | TraesCS5B02G404200.1 | SNP | 580,431,742 | 269.3 |
| 5B_Chi_1A_SNP_2 | TraesCS5B02G403700.1 | SNP | 589,164,993 | 270.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Walker, E.; Francki, M. Genes Associated with Foliar Resistance to Septoria Nodorum Blotch of Hexaploid Wheat (Triticum aestivum L.). Int. J. Mol. Sci. 2021, 22, 5580. https://doi.org/10.3390/ijms22115580
Li D, Walker E, Francki M. Genes Associated with Foliar Resistance to Septoria Nodorum Blotch of Hexaploid Wheat (Triticum aestivum L.). International Journal of Molecular Sciences. 2021; 22(11):5580. https://doi.org/10.3390/ijms22115580
Chicago/Turabian StyleLi, Dora, Esther Walker, and Michael Francki. 2021. "Genes Associated with Foliar Resistance to Septoria Nodorum Blotch of Hexaploid Wheat (Triticum aestivum L.)" International Journal of Molecular Sciences 22, no. 11: 5580. https://doi.org/10.3390/ijms22115580
APA StyleLi, D., Walker, E., & Francki, M. (2021). Genes Associated with Foliar Resistance to Septoria Nodorum Blotch of Hexaploid Wheat (Triticum aestivum L.). International Journal of Molecular Sciences, 22(11), 5580. https://doi.org/10.3390/ijms22115580