
S1P Lyase Regulates Intestinal Stem Cell Quiescence via Ki-67 and FOXO3


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
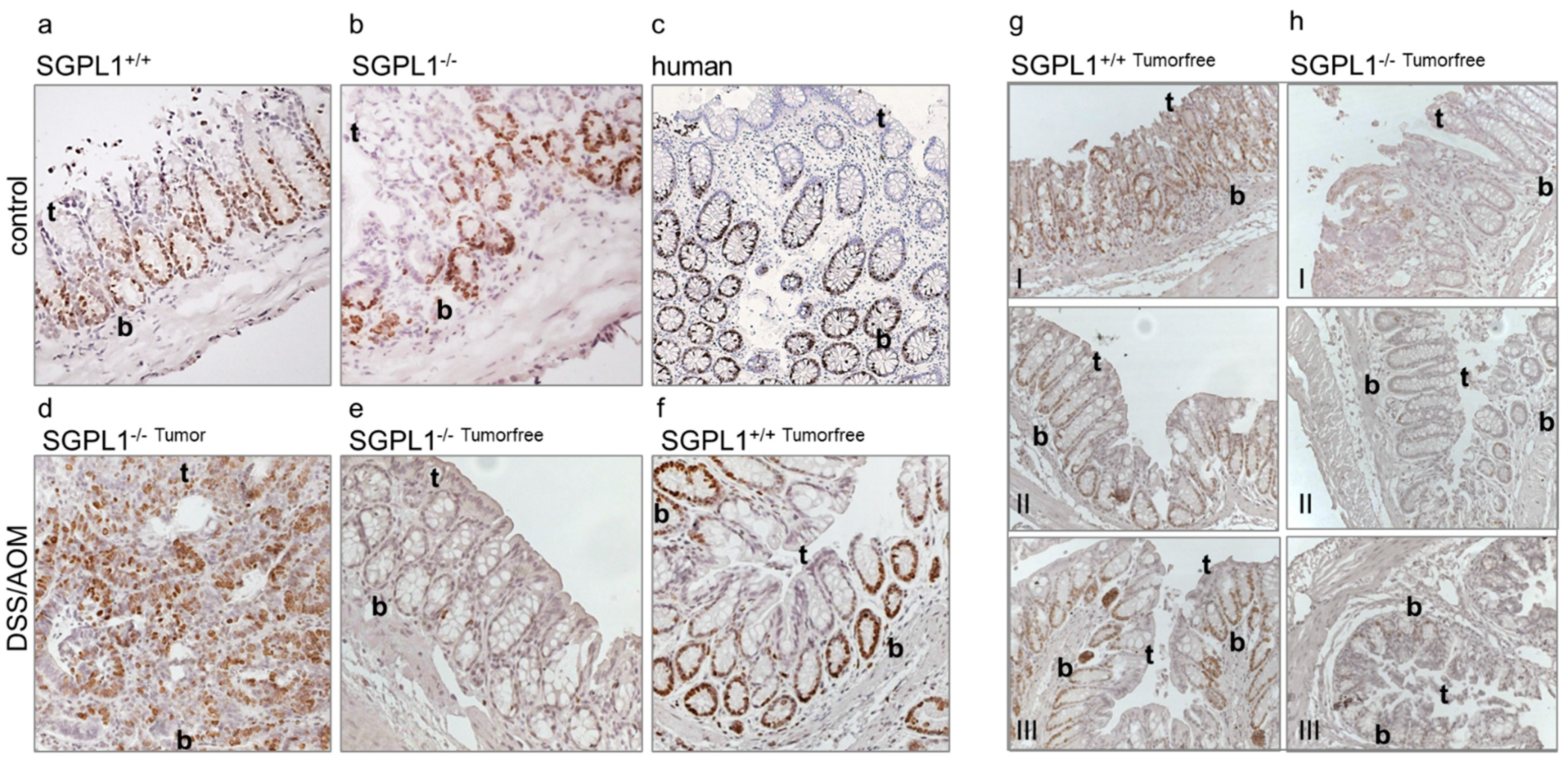
2.1. SGPL1 Knockout Leads to a Shift of Cell Configuration in Colonic Crypts
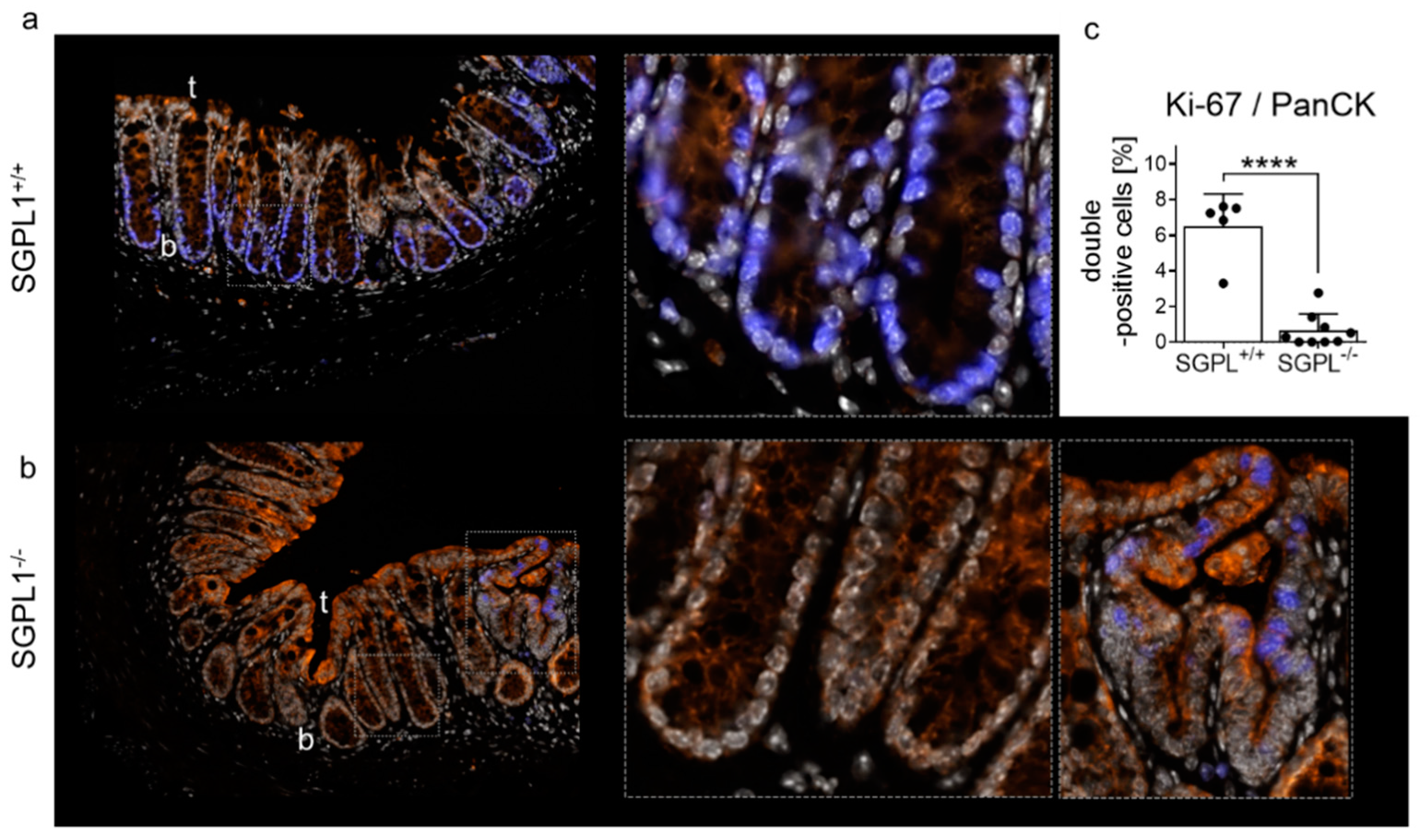
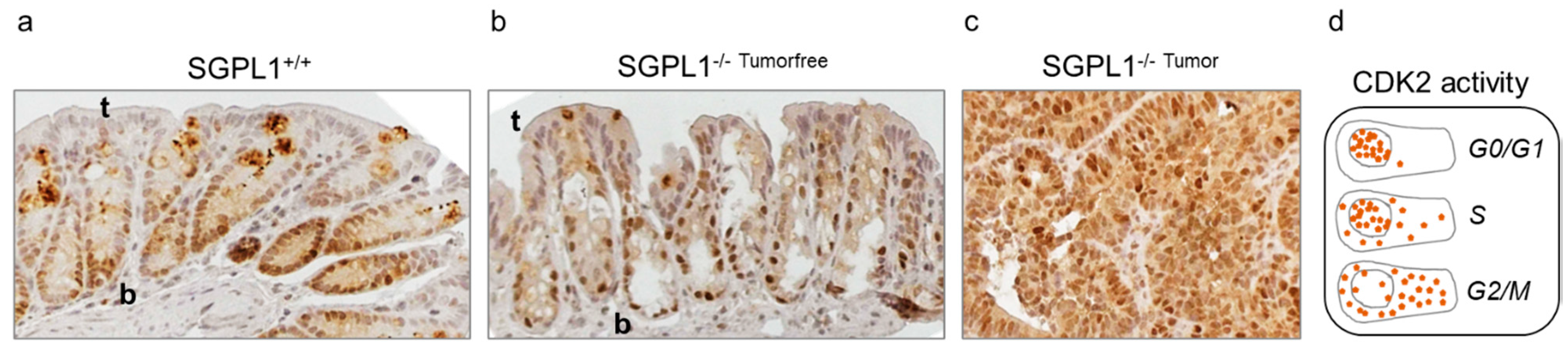
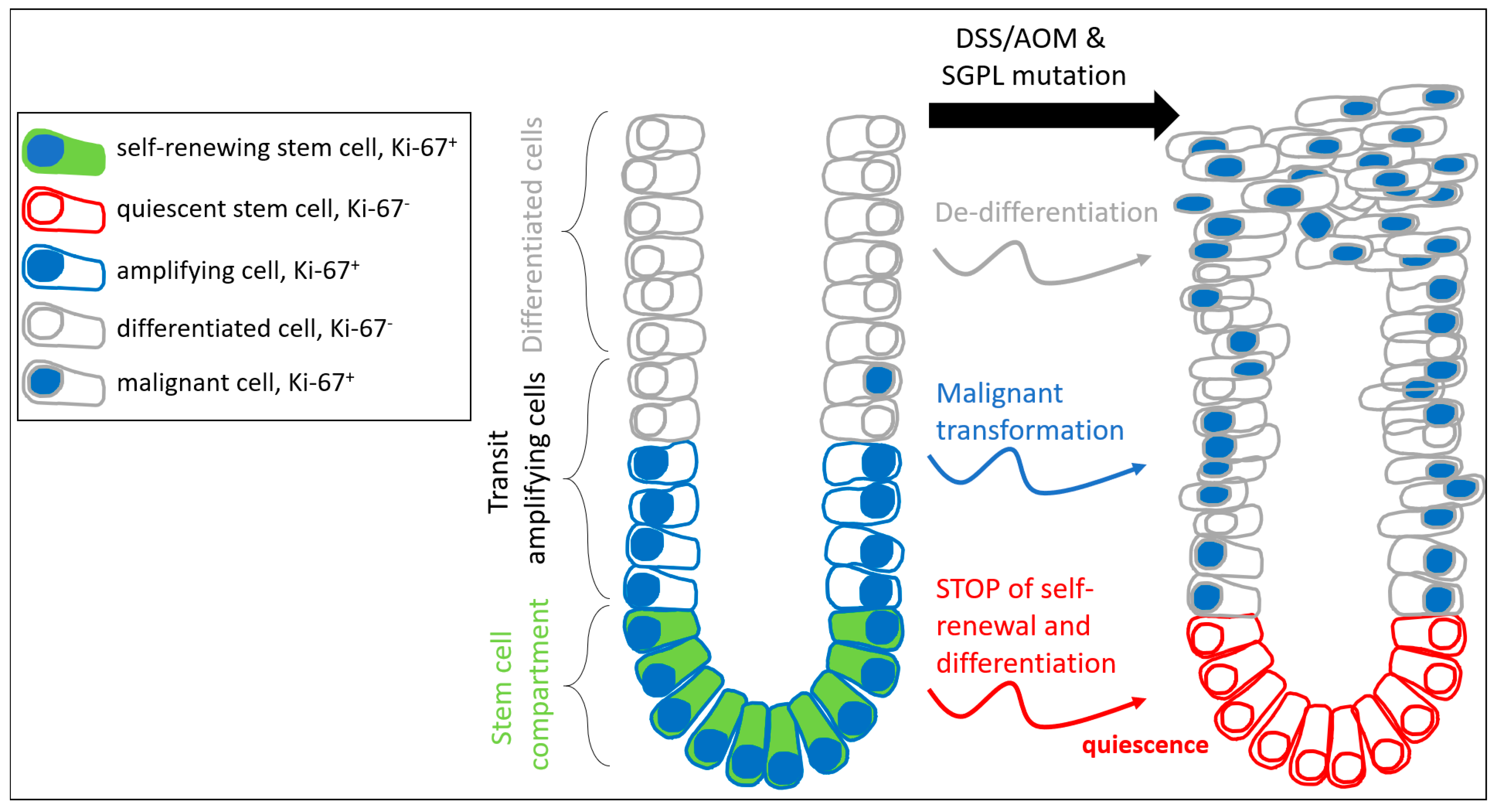
2.2. SGPL1 Knockout Tumors Are Strongly Ki-67 Positive, but the Intestinal Stem Cell Compartment Is Completely Ki-67 Negative
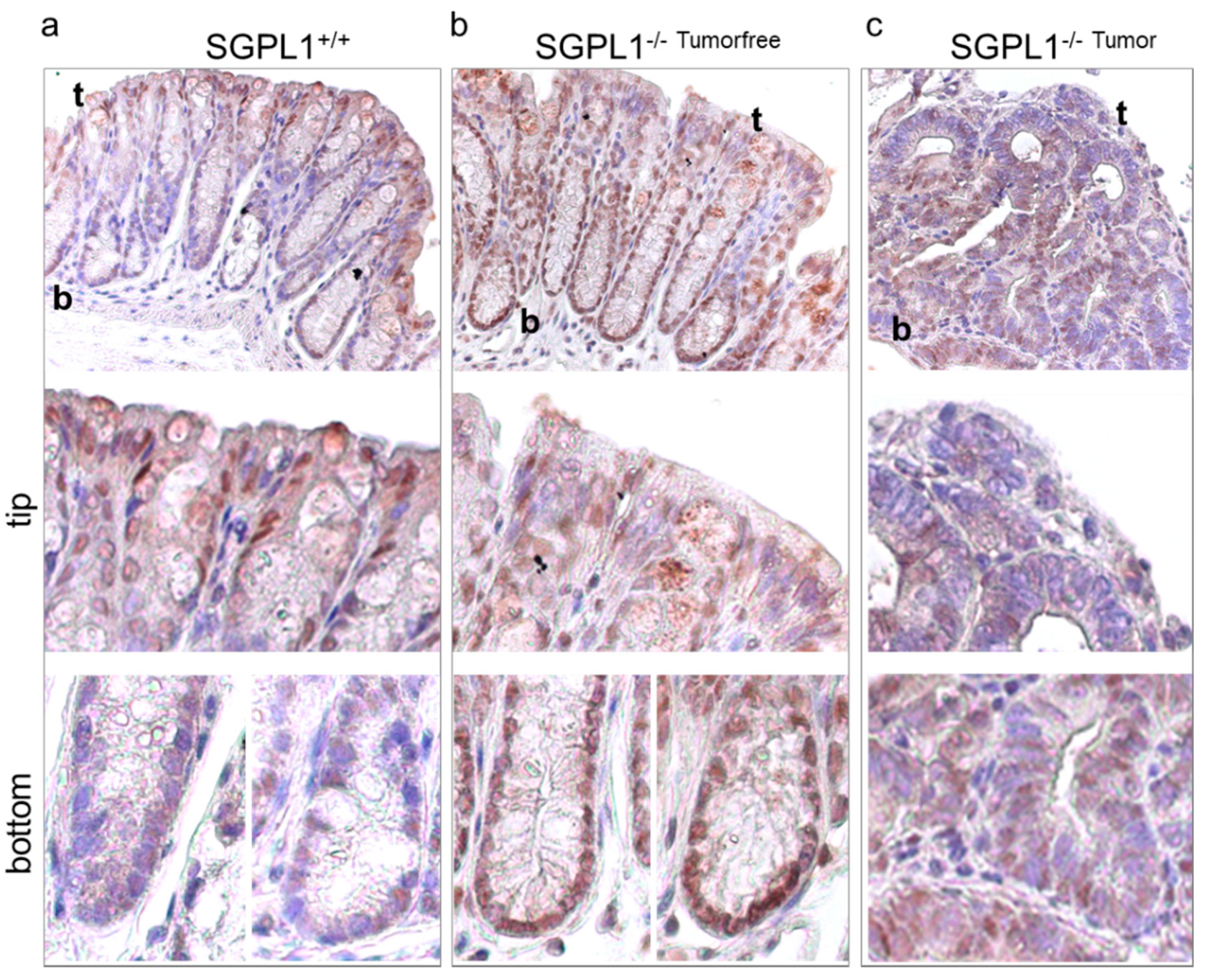
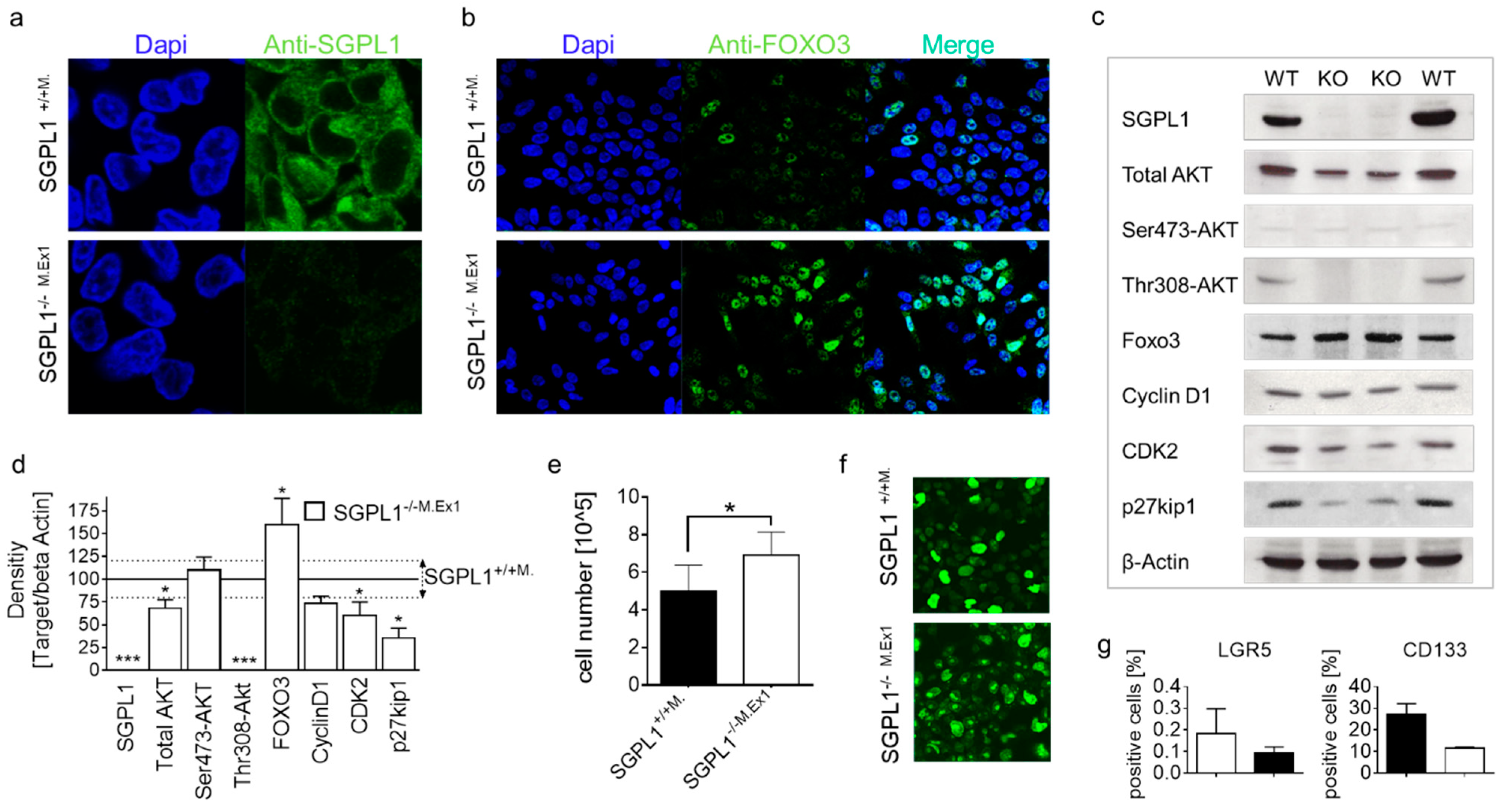
2.3. FOXO3 Expression Is Strongly Enhanced in SGPL1 Knockout Colon Tissue
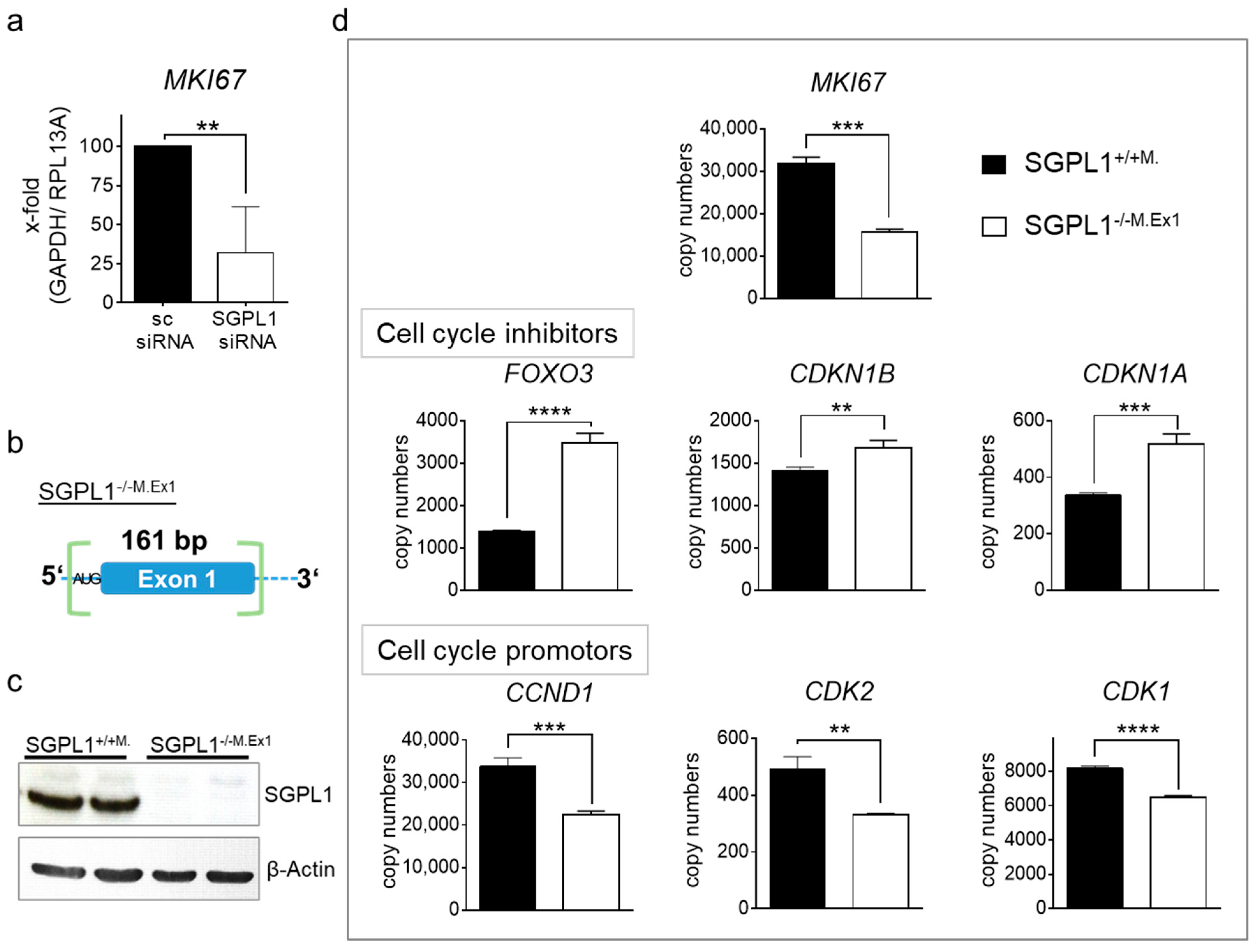
2.4. Transcriptome Analysis of Human SGPL1 Knockout Colorectal Cancer Cells
2.5. SGPL1 Knockout Dampens Established PDK1/AKT Signaling in Human Colorectal Cancer Cells but Does Not Induce Cell Cycle Arrest
3. Discussion
4. Materials and Methods
4.1. Experimental Model of Colitis-Associated Colon Cancer (CAC)
4.2. Immunohistochemistry
4.3. Cultivation of DLD-1
4.4. Transfection with siRNA
4.5. CRISPR/Cas9
4.6. RNA-Sequencing
4.7. Flow Cytometry
4.8. Liquid Chromatography Tandem Mass Spectrometry
4.9. Western Blot
4.10. Immunofluorescence Staining
4.11. RNA Isolation and Real-Time PCR
4.12. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duan, R.D.; Nilsson, A. Metabolism of sphingolipids in the gut and its relation to inflammation and cancer development. Prog. Lipid Res. 2009, 48, 62–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiebs, A.; Friesen, O.; Katzy, E.; Ferreiros, N.; Pfeilschifter, J.M.; Radeke, H.H. Activation-Induced cell death of dendritic cells is dependent on sphingosine kinase 1. Front. Pharmacol. 2016, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Arlt, O.; Schwiebs, A.; Japtok, L.; Ruger, K.; Katzy, E.; Kleuser, B.; Radeke, H.H. Sphingosine-1-phosphate modulates dendritic cell function: Focus on non-migratory effects in vitro and in vivo. Cell Physiol. Biochem. 2014, 34, 27–44. [Google Scholar] [CrossRef]
- Kawamori, T.; Kaneshiro, T.; Okumura, M.; Maalouf, S.; Uflacker, A.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2009, 23, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.Q.; Su, Y.J.; Qin, M.B.; Mao, Y.B.; Huang, J.A.; Tang, G.D. Sphingosine kinase 1 promotes tumor progression and confers malignancy phenotypes of colon cancer by regulating the focal adhesion kinase pathway and adhesion molecules. Int. J. Oncol. 2013, 42, 617–626. [Google Scholar] [CrossRef] [Green Version]
- Park, S.B.; Choi, B.I.; Lee, B.J.; Kim, N.J.; Jeong, Y.A.; Joo, M.K.; Kim, H.J.; Park, J.J.; Kim, J.S.; Noh, Y.S.; et al. Intestinal epithelial deletion of sphk1 prevents colitis-associated cancer development by inhibition of epithelial stat3 activation. Dig. Dis. Sci. 2020, 65, 2284–2293. [Google Scholar] [CrossRef] [PubMed]
- Chumanevich, A.A.; Poudyal, D.; Cui, X.; Davis, T.; Wood, P.A.; Smith, C.D.; Hofseth, L.J. Suppression of colitis-driven colon cancer in mice by a novel small molecule inhibitor of sphingosine kinase. Carcinogenesis 2010, 31, 1787–1793. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.Y.; Li, L.; Wang, X.H.; Wen, X.Z.; Ji, K.; Ye, L.; Cai, J.; Jiang, W.G.; Ji, J.F. Inhibition of sphingosine-1-phosphate phosphatase 1 promotes cancer cells migration in gastric cancer: Clinical implications. Oncol. Rep. 2015, 34, 1977–1987. [Google Scholar] [CrossRef]
- Huang, W.C.; Liang, J.; Nagahashi, M.; Avni, D.; Yamada, A.; Maceyka, M.; Wolen, A.R.; Kordula, T.; Milstien, S.; Takabe, K.; et al. Sphingosine-1-phosphate phosphatase 2 promotes disruption of mucosal integrity, and contributes to ulcerative colitis in mice and humans. FASEB J. 2016, 30, 2945–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faqar-Uz-Zaman, W.F.; Schmidt, K.G.; Thomas, D.; Pfeilschifter, J.M.; Radeke, H.H.; Schwiebs, A. S1P lyase siRNA dampens malignancy of DLD-1 colorectal cancer cells. Lipids 2021, 56, 155–166. [Google Scholar] [CrossRef]
- Oskouian, B.; Sooriyakumaran, P.; Borowsky, A.D.; Crans, A.; Dillard-Telm, L.; Tam, Y.Y.; Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 17384–17389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Fujiya, M.; Konishi, H.; Murakami, Y.; Iwama, T.; Sasaki, T.; Kunogi, T.; Sakatani, A.; Ando, K.; Ueno, N.; et al. Heterogenous nuclear ribonucleoprotein H1 promotes colorectal cancer progression through the stabilization of mRNA of sphingosine-1-phosphate lyase 1. Int. J. Mol. Sci. 2020, 21, 4515. [Google Scholar] [CrossRef]
- Degagne, E.; Pandurangan, A.; Bandhuvula, P.; Kumar, A.; Eltanawy, A.; Zhang, M.; Yoshinaga, Y.; Nefedov, M.; de Jong, P.J.; Fong, L.G.; et al. Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J. Clin. Investig. 2014, 124, 5368–5384. [Google Scholar] [CrossRef] [Green Version]
- Schwiebs, A.; Herrero San Juan, M.; Schmidt, K.G.; Wiercinska, E.; Anlauf, M.; Ottenlinger, F.; Thomas, D.; Elwakeel, E.; Weigert, A.; Farin, H.F.; et al. Cancer-induced inflammation and inflammation-induced cancer in colon: A role for S1P lyase. Oncogene 2019, 38, 4788–4803. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.G.; Earnshaw, W.C. Ki-67 and the chromosome periphery compartment in mitosis. Trends Cell Biol. 2017, 27, 906–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, S.L.; Cappell, S.D.; Tsai, F.C.; Overton, K.W.; Wang, C.L.; Meyer, T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 2013, 155, 369–383. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Cuylen, S.; Blaukopf, C.; Politi, A.Z.; Muller-Reichert, T.; Neumann, B.; Poser, I.; Ellenberg, J.; Hyman, A.A.; Gerlich, D.W. Ki-67 acts as a biological surfactant to disperse mitotic chromosomes. Nature 2016, 535, 308–312. [Google Scholar] [CrossRef]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle regulation accounts for variability in Ki-67 expression levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef] [Green Version]
- Matheson, T.D.; Kaufman, P.D. The p150N domain of chromatin assembly factor-1 regulates Ki-67 accumulation on the mitotic perichromosomal layer. Mol. Biol. Cell 2017, 28, 21–29. [Google Scholar] [CrossRef]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef]
- Jeon, S.; Song, J.; Lee, D.; Kim, G.T.; Park, S.H.; Shin, D.Y.; Shin, K.O.; Park, K.; Shim, S.M.; Park, T.S. Inhibition of sphingosine 1-phosphate lyase activates human keratinocyte differentiation and attenuates psoriasis in mice. J. Lipid Res. 2020, 61, 20–32. [Google Scholar] [CrossRef]
- Cidado, J.; Wong, H.Y.; Rosen, D.M.; Cimino-Mathews, A.; Garay, J.P.; Fessler, A.G.; Rasheed, Z.A.; Hicks, J.; Cochran, R.L.; Croessmann, S.; et al. Ki-67 is required for maintenance of cancer stem cells but not cell proliferation. Oncotarget 2016, 7, 6281–6293. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. Targeting the non-canonical AKT-FOXO3a axis: A potential therapeutic strategy for oral squamous cell carcinoma. EBioMedicine 2019, 49, 6–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medema, R.H.; Kops, G.J.; Bos, J.L.; Burgering, B.M. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000, 404, 782–787. [Google Scholar] [CrossRef]
- Safarian, F.; Khallaghi, B.; Ahmadiani, A.; Dargahi, L. Activation of S1P(1) receptor regulates PI3K/Akt/FoxO3a pathway in response to oxidative stress in PC12 cells. J. Mol. Neurosci. 2015, 56, 177–187. [Google Scholar] [CrossRef]
- Ihlefeld, K.; Claas, R.F.; Koch, A.; Pfeilschifter, J.M.; Meyer Zu Heringdorf, D. Evidence for a link between histone deacetylation and Ca(2)+ homoeostasis in sphingosine-1-phosphate lyase-deficient fibroblasts. Biochem. J. 2012, 447, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical role of FOXO3a in carcinogenesis. Mol. Cancer 2018, 17, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penrose, H.M.; Cable, C.; Heller, S.; Ungerleider, N.; Nakhoul, H.; Baddoo, M.; Hartono, A.B.; Lee, S.B.; Burow, M.E.; Flemington, E.F.; et al. Loss of forkhead box O3 facilitates inflammatory colon cancer: Transcriptome profiling of the immune landscape and novel targets. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 391–408. [Google Scholar] [CrossRef] [Green Version]
- Huhn, M.; Juan, M.H.S.; Melcher, B.; Dreis, C.; Schmidt, K.G.; Schwiebs, A.; Collins, J.; Pfeilschifter, J.M.; Vieth, M.; Stein, J.; et al. Inflammation-Induced mucosal KYNU expression identifies human ileal crohn’s disease. J. Clin. Med. 2020, 9, 1360. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Shi, Y.; Li, R.; Gunther, S.; Ong, Y.T.; Potente, M.; Yuan, Z.; Liu, E.; Offermanns, S. YAP and TAZ protect against white adipocyte cell death during obesity. Nat. Commun. 2020, 11, 5455. [Google Scholar] [CrossRef] [PubMed]
- Dreis, C.; Ottenlinger, F.M.; Putyrski, M.; Ernst, A.; Huhn, M.; Schmidt, K.G.; Pfeilschifter, J.M.; Radeke, H.H. Tissue cytokine IL-33 modulates the cytotoxic CD8 T lymphocyte activity during nutrient deprivation by regulation of lineage-specific differentiation programs. Front. Immunol. 2019, 10, 1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiebs, A.; Thomas, D.; Kleuser, B.; Pfeilschifter, J.M.; Radeke, H.H. Nuclear translocation of SGPP-1 and decrease of SGPL-1 activity contribute to sphingolipid rheostat regulation of inflammatory dendritic cells. Mediators Inflamm. 2017, 2017, 5187368. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwiebs, A.; Faqar-Uz-Zaman, F.; Herrero San Juan, M.; Radeke, H.H. S1P Lyase Regulates Intestinal Stem Cell Quiescence via Ki-67 and FOXO3. Int. J. Mol. Sci. 2021, 22, 5682. https://doi.org/10.3390/ijms22115682
Schwiebs A, Faqar-Uz-Zaman F, Herrero San Juan M, Radeke HH. S1P Lyase Regulates Intestinal Stem Cell Quiescence via Ki-67 and FOXO3. International Journal of Molecular Sciences. 2021; 22(11):5682. https://doi.org/10.3390/ijms22115682
Chicago/Turabian StyleSchwiebs, Anja, Farha Faqar-Uz-Zaman, Martina Herrero San Juan, and Heinfried H. Radeke. 2021. "S1P Lyase Regulates Intestinal Stem Cell Quiescence via Ki-67 and FOXO3" International Journal of Molecular Sciences 22, no. 11: 5682. https://doi.org/10.3390/ijms22115682
APA StyleSchwiebs, A., Faqar-Uz-Zaman, F., Herrero San Juan, M., & Radeke, H. H. (2021). S1P Lyase Regulates Intestinal Stem Cell Quiescence via Ki-67 and FOXO3. International Journal of Molecular Sciences, 22(11), 5682. https://doi.org/10.3390/ijms22115682