
E3 Ubiquitin Ligase SPL2 Is a Lanthanide-Binding Protein


Abstract
:1. Introduction
2. Results
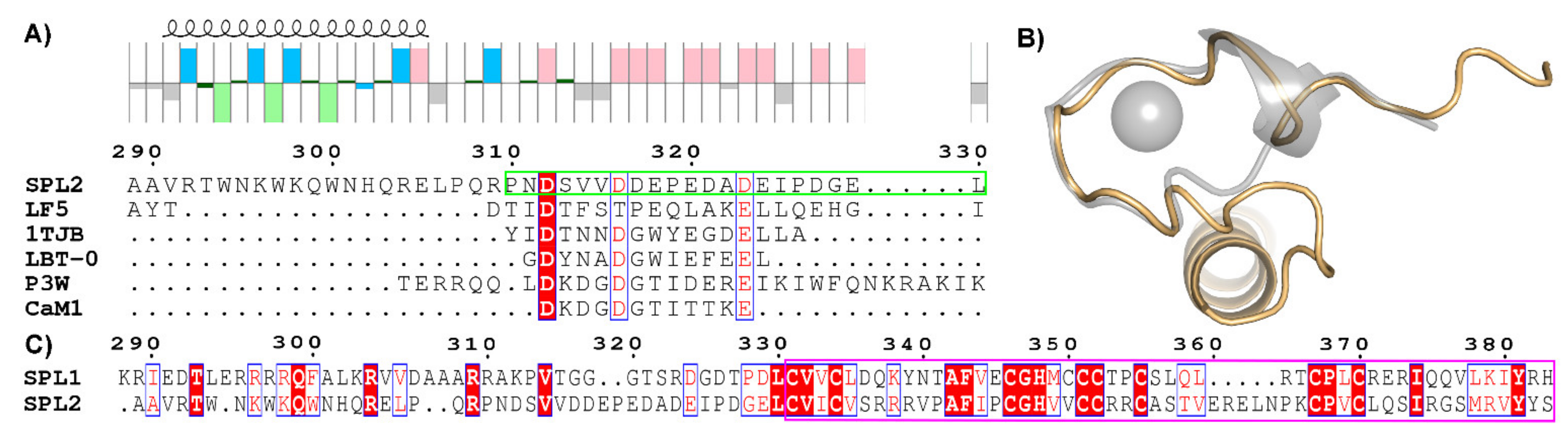
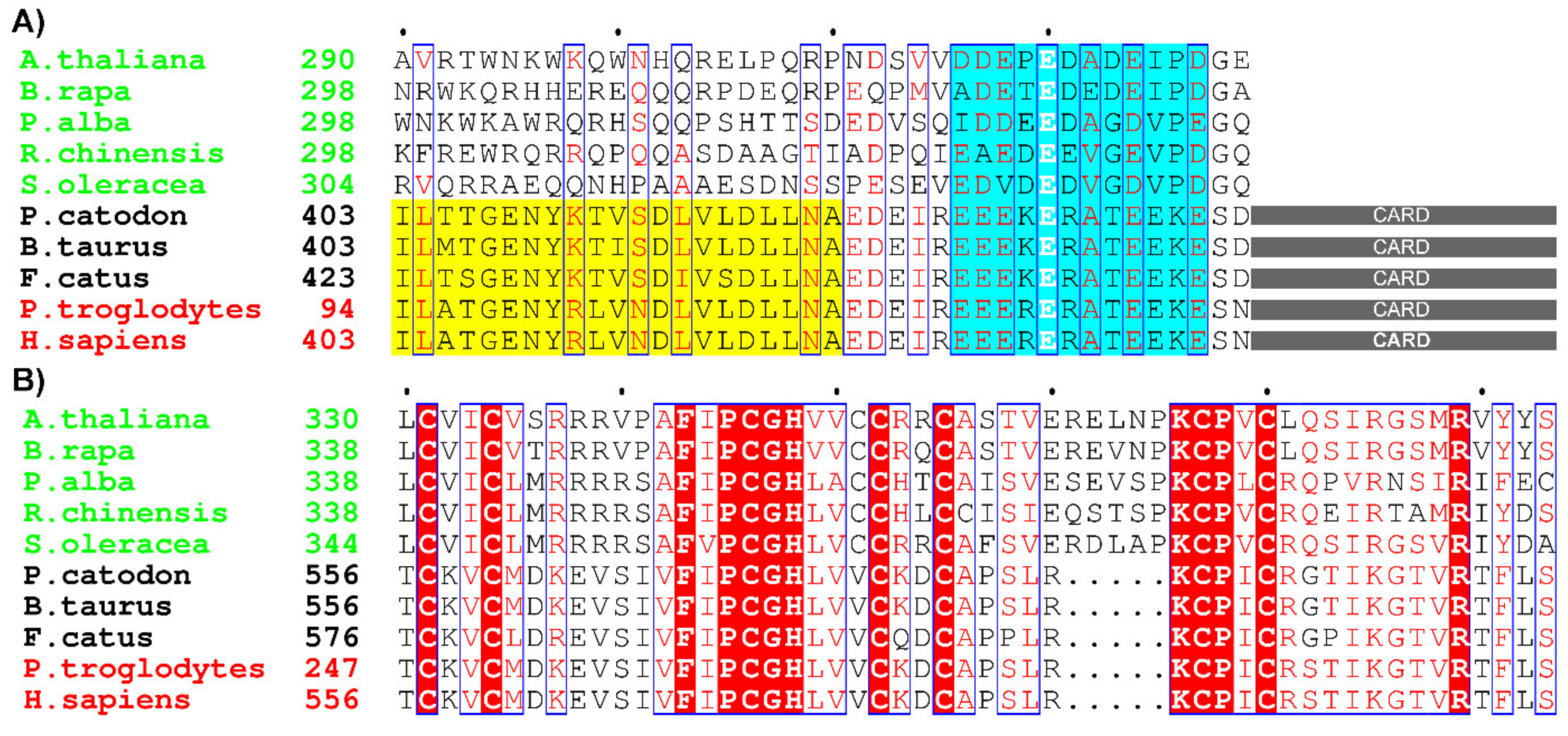
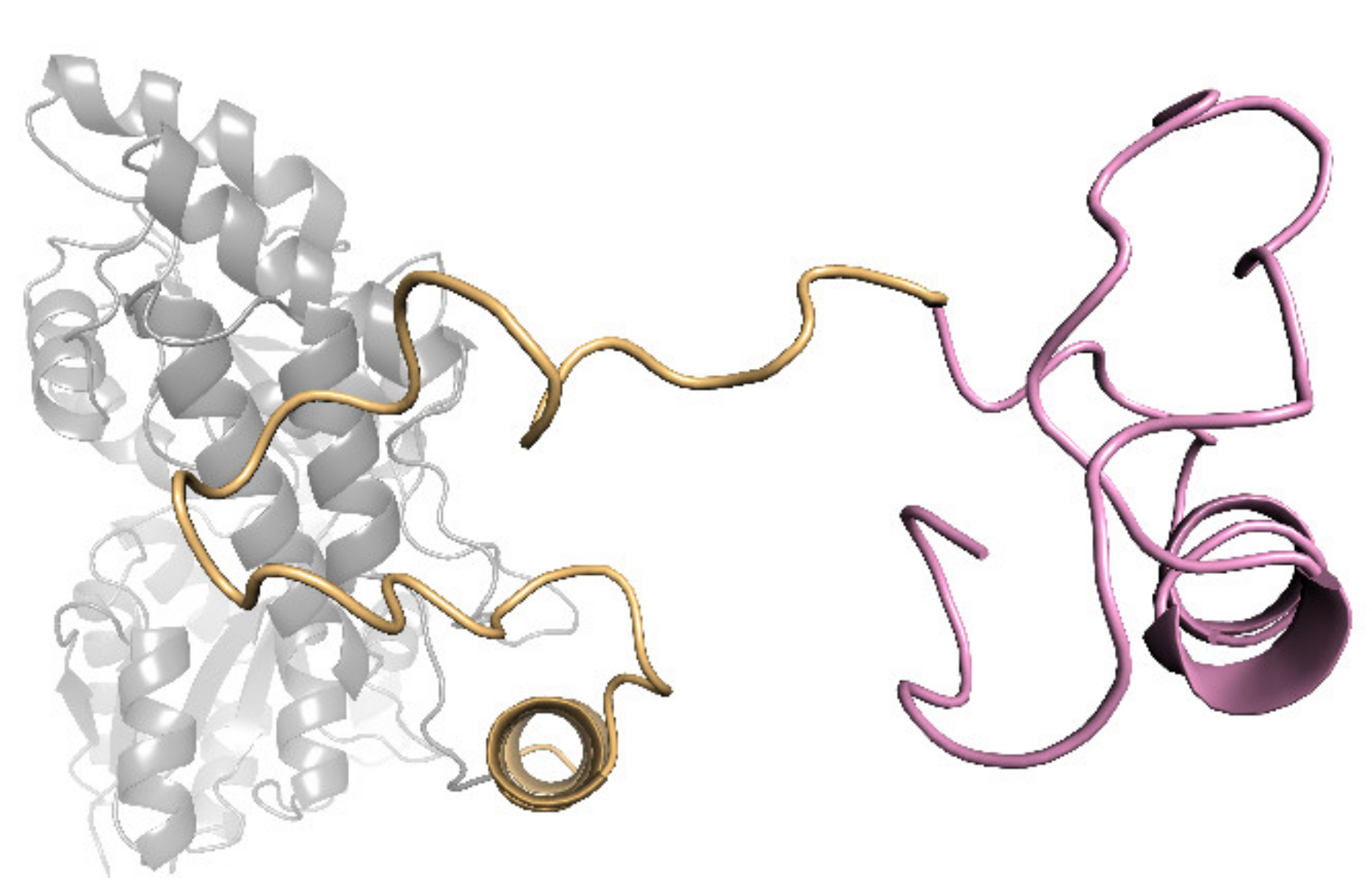
2.1. Sequence Analysis and Prediction of Lanthanide-Binding Motif

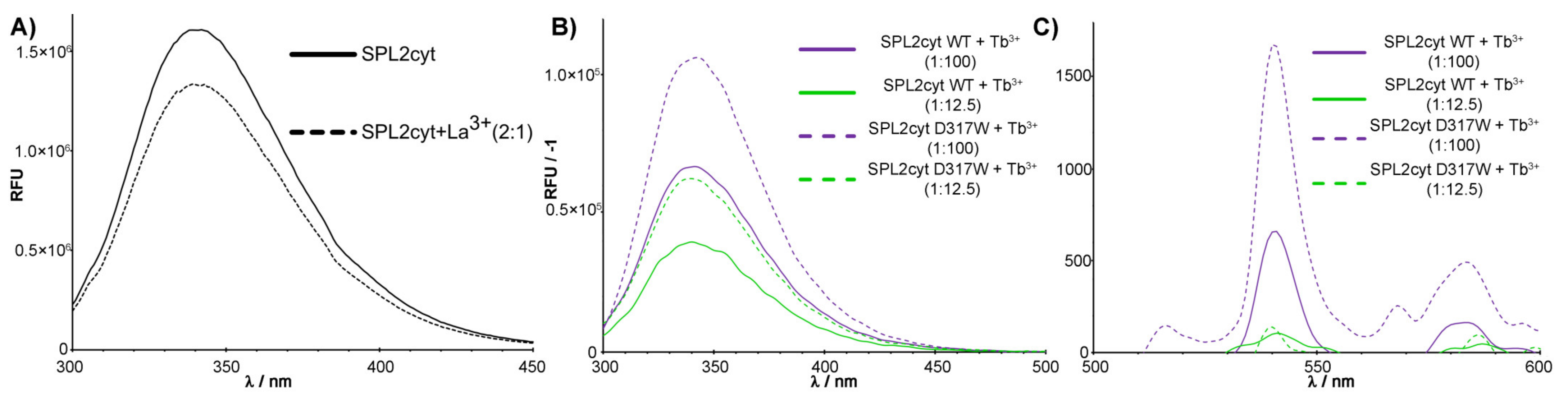
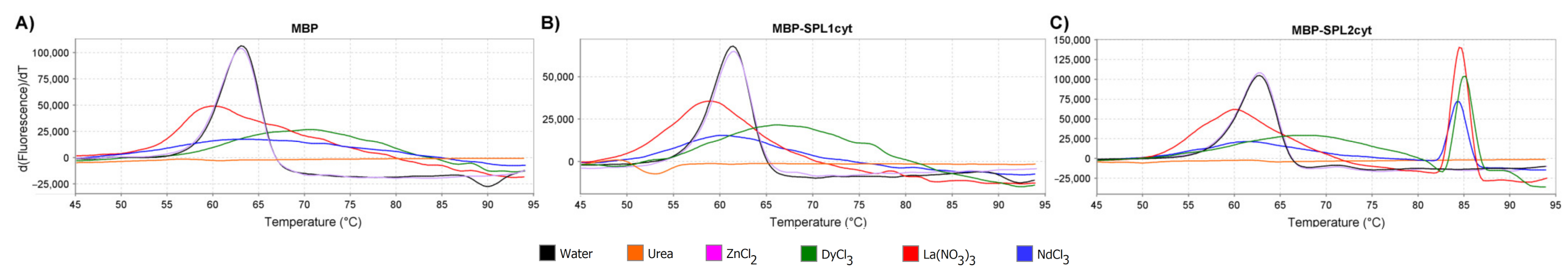
2.2. Emission Spectra of SPL2cyt and Protein–Tb3+ Complexes
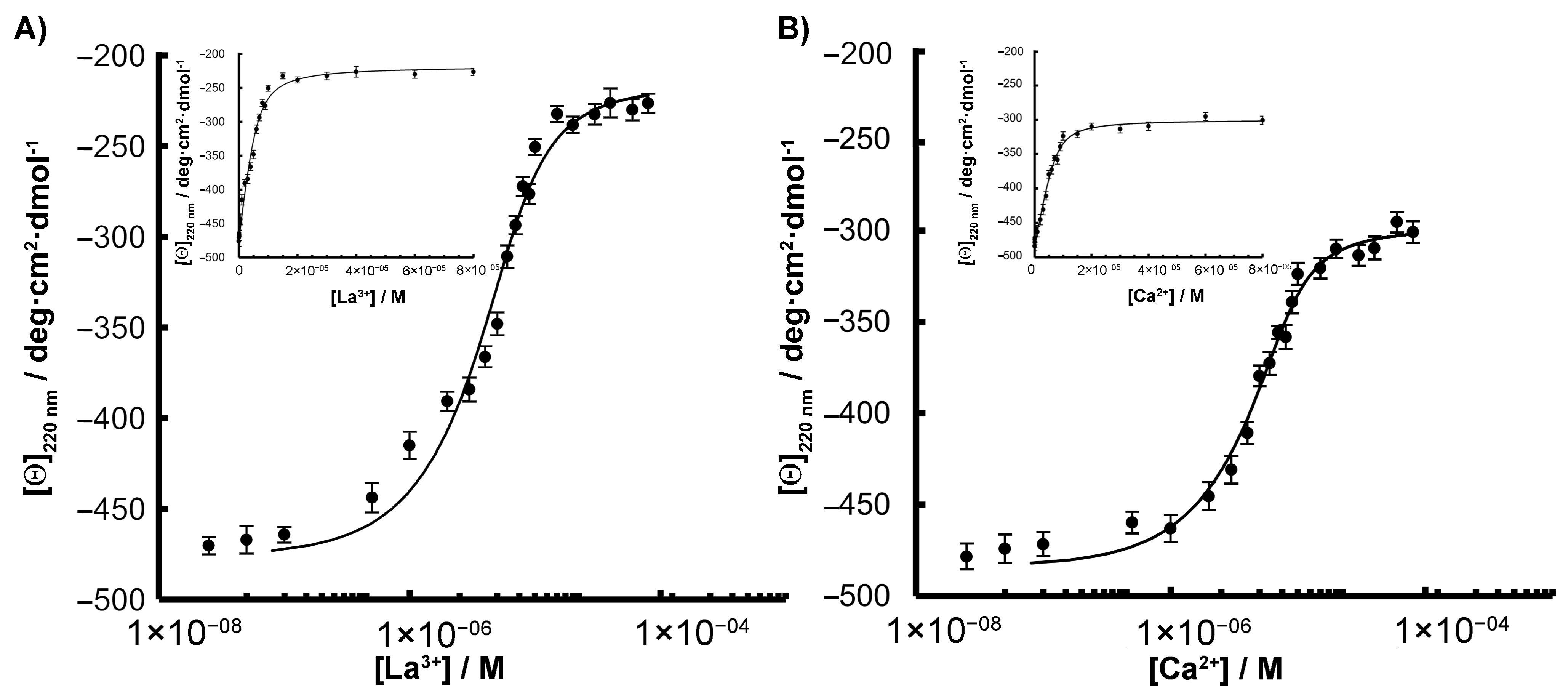
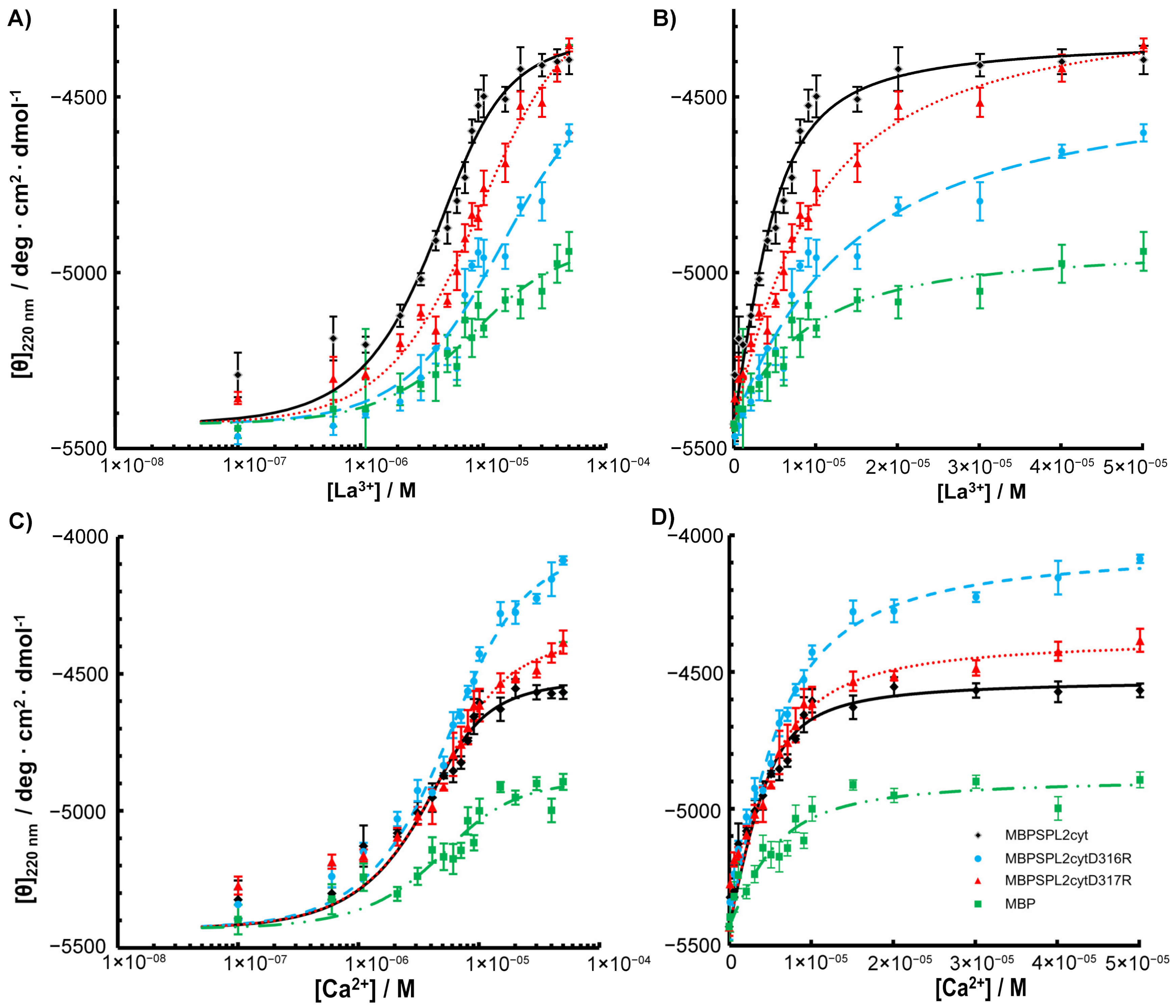
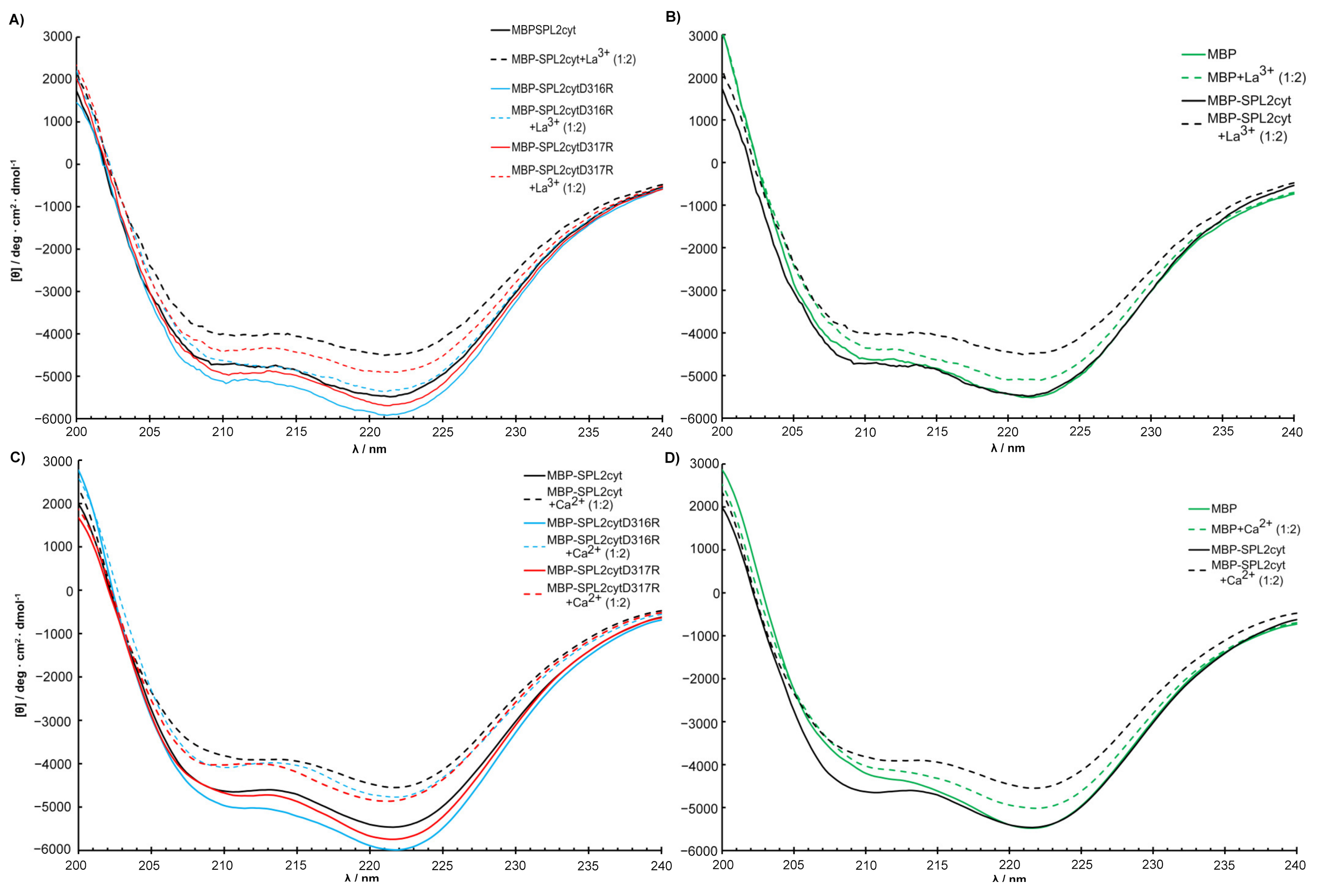
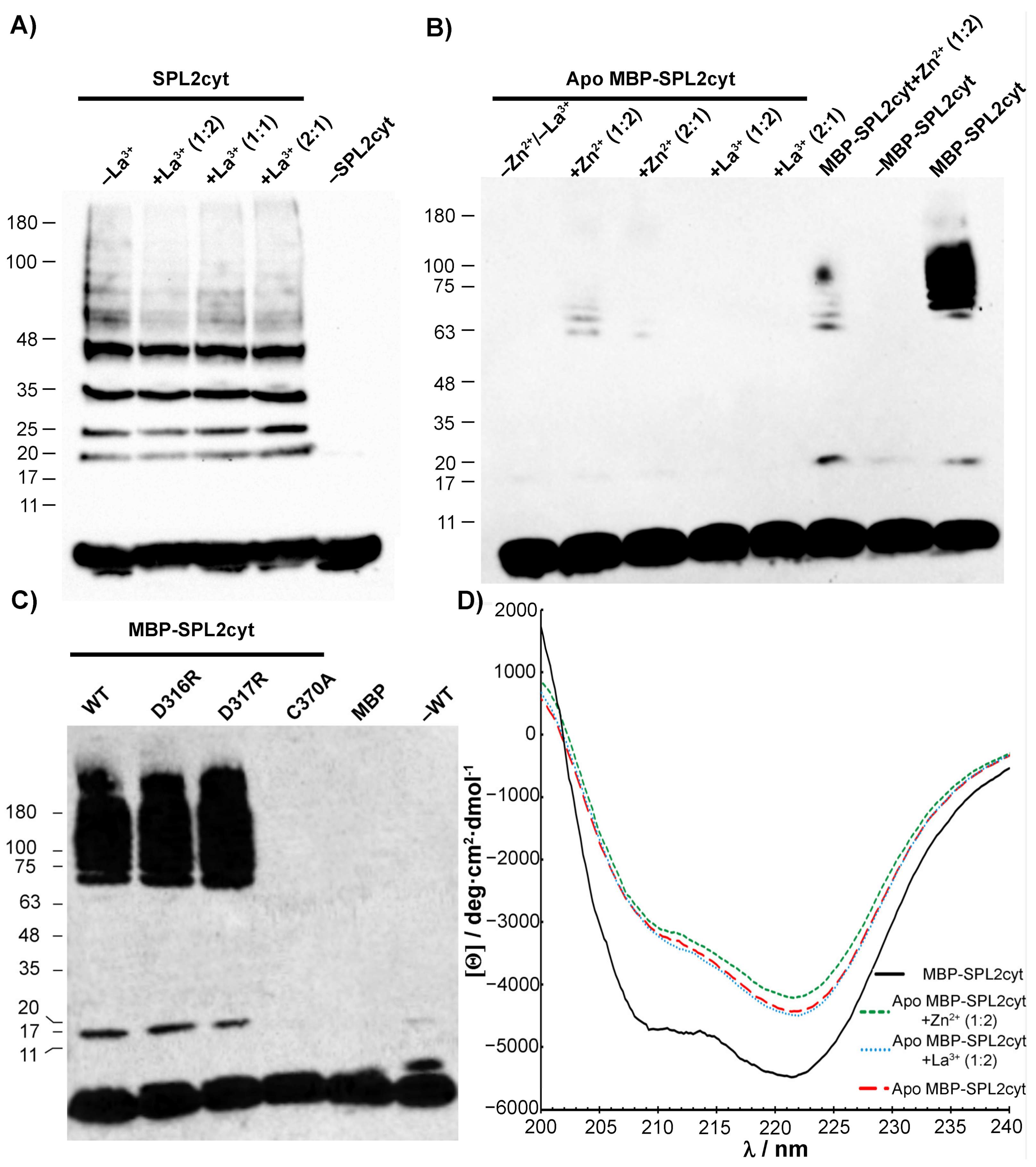
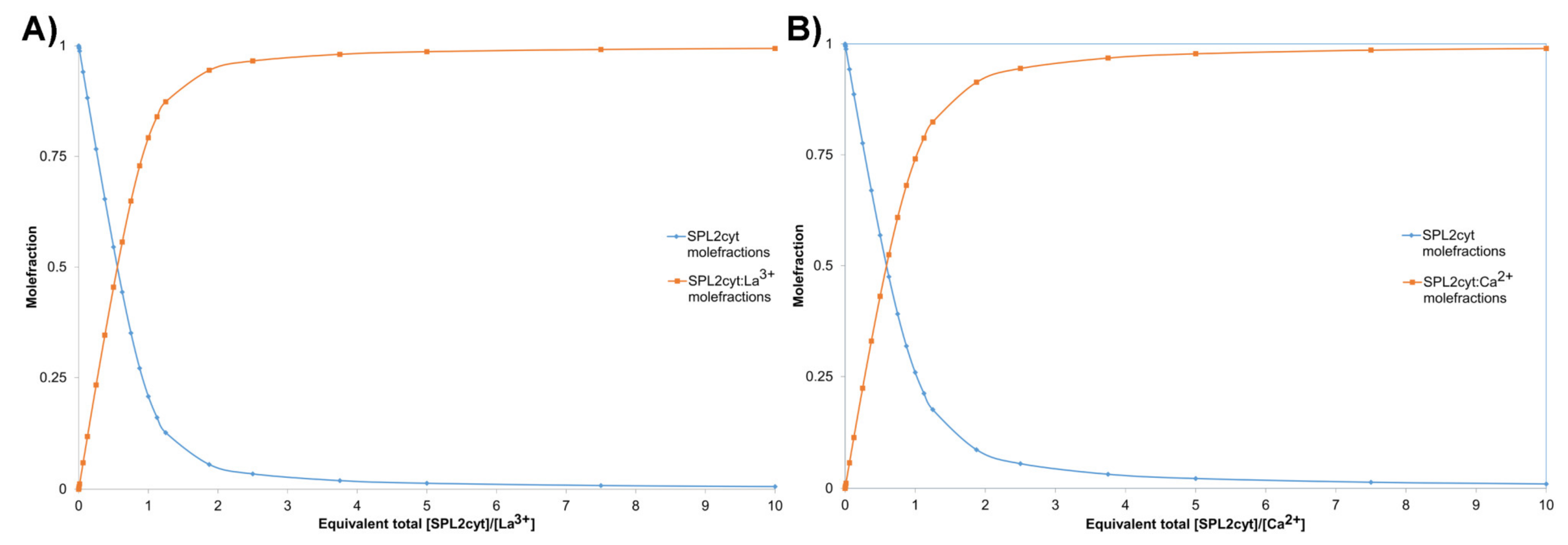
2.3. CD Measurements and Metal Binding of SPL2cyt and Its Mutants
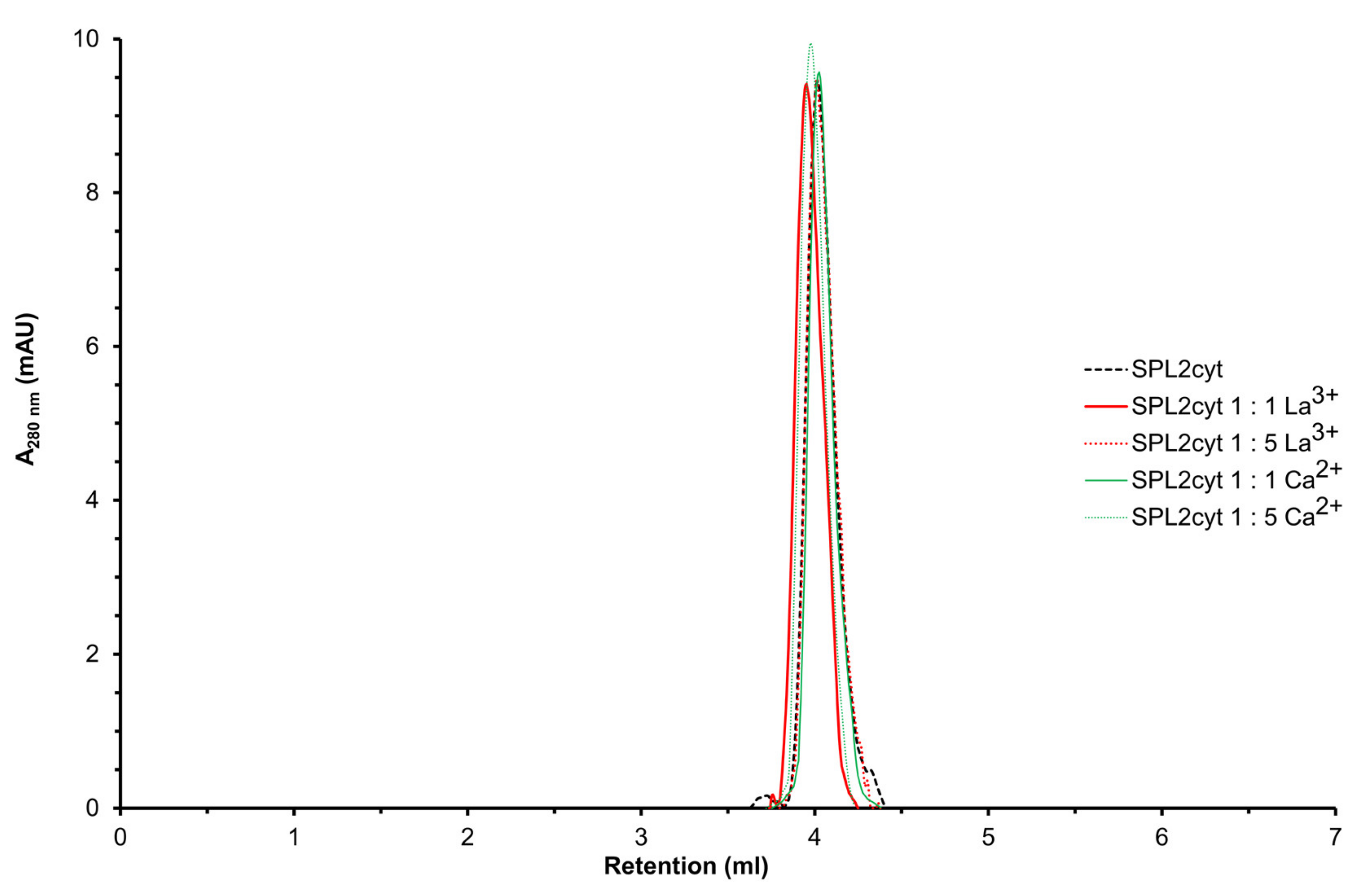
2.4. Interactions with Ca2+ and La3+ Induce Structural Transitions in SPL2cyt
2.5. SPL2 Auto-Ubiquitination in the Presence of Lanthanide Ions
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Gel Electrophoresis, In-Gel Luminescence, and Immunoblotting
4.3. In Vitro Ubiquitylation Assay
4.4. Fluorescence Measurements
4.5. Circular Dichroism (CD) Spectroscopy
4.6. Analytical Size-Exclusion Chromatography
4.7. Dynamic Light Scattering
4.8. Multiple Sequence Alignments and Structural Prediction
4.9. Thermal Shift Assay
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A




Appendix B
Appendix B.1. Materials
Appendix B.2. Cloning and Protein Expression
Appendix B.3. Protein Purification, MBP-Tag Removal, Zinc-Free Protein Preparation, and Purity Assessment
References
- Selvin, P.R. Principles and Biophysical Applications of Lanthanide-Based Probes. Annu. Rev. Biophys. Biomol. Struct. 2002, 31, 275–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibi, Y.; Asai, K.; Arafuka, H.; Hamajima, M.; Iwama, T.; Kawai, K. Molecular Structure of La3+-Induced Methanol Dehydrogenase-like Protein in MethylobacteriumRadiotolerans. J. Biosci. Bioeng. 2011, 111, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Deblonde, G.J.P.; Mattocks, J.A.; Park, D.M.; Reed, D.W.; Cotruvo, J.A.; Jiao, Y. Selective and Efficient Biomacromolecular Extraction of Rare-Earth Elements Using Lanmodulin. Inorg. Chem. 2020, 59, 11855–11867. [Google Scholar] [CrossRef] [PubMed]
- Tyler, G. Rare Earth Elements in Soil and Plant Systems—A Review. Plant Soil. 2004, 267, 191–206. [Google Scholar] [CrossRef]
- Pang, X.; Li, D.; Peng, A. Application of Rare-Earth Elements in the Agriculture of China and Its Environmental Behavior in Soil. J. Soils Sedim. 2001, 1, 124–129. [Google Scholar] [CrossRef]
- Tracz, M.; Bialek, W. Beyond K48 and K63: Non-Canonical Protein Ubiquitination. Cell. Mol. Biol. Lett. 2021, 26, 1–17. [Google Scholar] [CrossRef]
- Ling, Q.; Huang, W.; Baldwin, A.; Jarvis, P. Chloroplast Biogenesis Is Regulated by Direct Action of the Ubiquitin-Proteasome System. Science 2012, 338, 655–659. [Google Scholar] [CrossRef]
- Ling, Q.; Broad, W.; Trösch, R.; Töpel, M.; Sert, T.D.; Lymperopoulos, P.; Baldwin, A.; Jarvis, R.P. Ubiquitin-Dependent Chloroplast-Associated Protein Degradation in Plants. Science 2019, 363, eaav4467. [Google Scholar] [CrossRef]
- Campbell, A.; Williams, R.; Brown, C.; Meng, J.; Uversky, V.; Dunker, A. TOP-IDP-Scale: A New Amino Acid Scale Measuring Propensity for Intrinsic Disorder. Protein Pept. Lett. 2008, 15, 956–963. [Google Scholar]
- Nitz, M.; Sherawat, M.; Franz, K.J.; Peisach, E.; Allen, K.N.; Imperiali, B. Structural Origin of the High Affinity of a Chemically Evolved Lanthanide-Binding Peptide. Angew. Chem. Int. Ed. 2004, 43, 3682–3685. [Google Scholar] [CrossRef]
- Caravan, P.; Greenwood, J.M.; Welch, T.; Franklin, S.J. Gadolinium-Binding Helix–Turn–Helix Peptides: DNA-Dependent MRI Contrast Agents. Chem. Commun. 2003, 20, 2574–2575. [Google Scholar] [CrossRef]
- MacManus, J.P.; Hogue, C.W.; Marsden, B.J.; Sikorska, M.; Szabo, A.G. Terbium Luminescence in Synthetic Peptide Loops from Calcium-Binding Proteins with Different Energy Donors. J. Biol. Chem. 1990, 265, 10358–10366. [Google Scholar] [CrossRef]
- Schiffer, M.; Chang, C.H.; Stevens, F.J. The Functions of Tryptophan Residues in Membrane Proteins. Protein Eng. Des. Sel. 1992, 5, 213–214. [Google Scholar] [CrossRef]
- De Planque, M.R.R.; Kruijtzer, J.A.W.; Liskamp, R.M.J.; Marsh, D.; Greathouse, D.V.; Koeppe, R.E.; De Kruijff, B.; Killian, J.A. Different Membrane Anchoring Positions of Tryptophan and Lysine in Synthetic Transmembrane α-Helical Peptides. J. Biol. Chem. 1999, 274, 20839–20846. [Google Scholar] [CrossRef] [Green Version]
- Killian, J.A.; Von Heijne, G. How Proteins Adapt to a Membrane-Water Interface. Trends Biochem. Sci. 2000, 25, 429–434. [Google Scholar] [CrossRef]
- Franz, K.J.; Nitz, M.; Imperiali, B. Lanthanide-Binding Tags as Versatile Protein Coexpression Probes. ChemBioChem 2003, 4, 265–271. [Google Scholar] [CrossRef]
- Berwick, M.R.; Lewis, D.J.; Jones, A.W.; Parslow, R.A.; Dafforn, T.R.; Cooper, H.J.; Wilkie, J.; Pikramenou, Z.; Britton, M.M.; Peacock, A.F.A. De Novo Design of Ln(III) Coiled Coils for Imaging Applications. J. Am. Chem. Soc. 2014, 136, 1166–1169. [Google Scholar] [CrossRef]
- Cotruvo, J.A.; Featherston, E.R.; Mattocks, J.A.; Ho, J.V.; Laremore, T.N. Lanmodulin: A Highly Selective Lanthanide-Binding Protein from a Lanthanide-Utilizing Bacterium. J. Am. Chem. Soc. 2018, 140, 15056–15061. [Google Scholar] [CrossRef]
- Evans, C.H. Biochemistry of the Lanthanides; Springer: Boston, MA, USA, 1990. [Google Scholar]
- Brittain, H.G.; Richardson, F.S.; Martin, R.B. Terbium(III) Emission as a Probe of Calcium(II) Binding Sites in Proteins. J. Am. Chem. Soc. 1976, 98, 8255–8260. [Google Scholar] [CrossRef]
- Eberhard, M.; Erne, P. Calcium and Magnesium Binding to Rat Parvalbumin. Eur. J. Biochem. 1994, 222, 21–26. [Google Scholar] [CrossRef]
- Kruk, J.; Burda, K.; Jemioła-Rzemińska, M.; Strzałka, K. The 33 KDa Protein of Photosystem II Is a Low-Affinity Calcium- and Lanthanide-Binding Protein. Biochemistry 2003, 42, 14862–14867. [Google Scholar] [CrossRef]
- Murphy, P.; Xu, Y.; Rouse, S.L.; Jaffray, E.G.; Plechanovová, A.; Matthews, S.J.; Carlos Penedo, J.; Hay, R.T. Functional 3D Architecture in an Intrinsically Disordered E3 Ligase Domain Facilitates Ubiquitin Transfer. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Burda, K.; Strzałka, K.S.; Schmid, G.H. Europium- and Dysprosium-Ions as Probes for the Study of Calcium Binding Sites in Photosystem II. Z. Naturforsch. Sect. C J. Biosci. 1995, 50, 220–230. [Google Scholar] [CrossRef]
- Klusener, B.; Boheim, G.; Lift, H.; Engelberthl, J.; Weiler, E.W. Gadolinium-Sensitive, Voltage-Dependent Calcium Release Channels in the Endoplasmic Reticulum of a Higher Plant Mechanoreceptor Organ. EMBO J. 1995, 14, 2708–2714. [Google Scholar] [CrossRef]
- Amann, B.T.; Mulqueen, P.; Horrocks, W.D. A Continuous Spectrophotometric Assay for the Activation of Plant NAD Kinase by Calmodulin, Calcium (II), and Europium (III) Ions. J. Biochem. Biophys. Methods 1992, 25, 207–217. [Google Scholar] [CrossRef]
- Dressler, L.; Golbik, R.; Ulbrich-Hofmann, R. Lanthanides as Substitutes for Calcium Ions in the Activation of Plant α-Type Phospholipase D. Biol. Chem. 2014, 395, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gu, Y.; Zhao, G.; Tao, Y.; Luo, J.; Hu, T. Effects of Rare Earth Ions on Activity of RuBPcase in Tobacco. Plant Sci. 2000, 152, 145–151. [Google Scholar] [CrossRef]
- Chen, W.J.; Tao, Y.; Gu, Y.H.; Zhao, G.W. Effect of Lanthanide Chloride on Photosynthesis and Dry Matter Accumulation in Tobacco Seedlings. Biol. Trace Elem. Res. 2001, 79, 169–176. [Google Scholar] [CrossRef]
- Mu, F.; Gonza, J.M.; Aguilar-Hernandez, V.; Guzman, P. Repertoire of Plant RING E3 Ubiquitin Ligases Revisited: New Groups Counting Gene Families and Single Genes. PLoS ONE 2018, 13, e0203442. [Google Scholar]
- Ling, Q.; Jarvis, P. Regulation of Chloroplast Protein Import by the Ubiquitin E3 Ligase SP1 Is Important for Stress Tolerance in Plants. Curr. Biol. 2015, 25, 2527–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witcombe, J.R.; Hollington, P.A.; Howarth, C.J.; Reader, S.; Steele, K.A. Breeding for Abiotic Stresses for Sustainable Agriculture. Phil. Trans. R. Soc. B 2008, 363, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.-Y.; Fang, M.-J.; Kuang, L.-Y.; Gelvin, S.B. Vectors for Multi-Color Bimolecular Fluorescence Complementation to Investigate Protein-Protein Interactions in Living Plant Cells. Plant Methods 2008, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Gookin, T.E.; Assmann, S.M. Significant Reduction of BiFC Non-Specific Assembly Facilitates in Planta Assessment of Heterotrimeric G-Protein Interactors. Plant J. 2014, 80, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, K.; Bernasconi, L.; Picard, D. Luminescence Resonance Energy Transfer between Genetically Encoded Donor and Acceptor for Protein-Protein Interaction Studies in the Molecular Chaperone HSP70/HSP90 Complexes. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Cho, U.; Chen, J.K. Lanthanide-Based Optical Probes of Biological Systems. Cell Chem. Biol. 2020, 27, 921–936. [Google Scholar] [CrossRef]
- Chen, T. Single-Chain Lanthanide Luminescence Biosensors for Cell-Based Imaging and Screening of Protein-Protein Interactions Single-Chain Lanthanide Luminescence. iScience 2020, 23, 101533. [Google Scholar] [CrossRef]
- Kaineder, K.; Mezricky, D. The Effect of Lanthanides on Photosynthesis, Growth, and Chlorophyll Profile of the Green Alga DesmodesmusQuadricauda. Photosynth. Res. 2016, 130, 335–346. [Google Scholar]
- Anan, Y.; Awaya, Y.; Ogihara, Y. Comparison in Accumulation of Lanthanide Elements among Three Brassicaceae Plant Sprouts. Bull. Environ. Contam. Toxicol. 2012, 89, 133–137. [Google Scholar] [CrossRef]
- Silke, J.; Kratina, T.; Chu, D.; Ekert, P.G.; Day, C.L.; Pakusch, M.; Huang, D.C.S.; Vaux, D.L. Determination of Cell Survival by RING-Mediated Regulation of Inhibitor of Apoptosis (IAP) Protein Abundance. Proc. Natl. Acad. Sci. USA 2005, 102, 16182–16187. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Fairmichael, C.; Longley, D.B.; Turkington, R.C. The Multiple Roles of the IAP Super-Family in Cancer. Pharmacol. Ther. 2020, 214, 107610. [Google Scholar] [CrossRef]
- Mace, P.D.; Linke, K.; Feltham, R.; Schumacher, F.R.; Smith, C.A.; Vaux, D.L.; Silke, J.; Day, C.L. Structures of the CIAP2 RING Domain Reveal Conformational Changes Associated with Ubiquitin-Conjugating Enzyme (E2) Recruitment. J. Biol. Chem. 2008, 283, 31633–31640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, M.D.; Moche, M.; Flodin, S.; Welin, M.; Trésaugues, L.; Johansson, I.; Nilsson, M.; Nordlund, P.; Nyman, T. Structures of BIR Domains from Human NAIP and CIAP2. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Kabaleeswaran, V.; Wang, Y.; Cheng, G.; Wu, H. Crystal Structures of the TRAF2: CIAP2 and the TRAF1: TRAF2: CIAP2 Complexes: Affinity, Specificity, and Regulation. Mol. Cell 2010, 38, 101–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamant, S.; Eliahu, N.; Rosenthal, D.; Goloubinoff, P. Chemical Chaperones Regulate Molecular Chaperones in Vitro and in Cells under Combined Salt and Heat Stresses. J. Biol. Chem. 2001, 276, 39586–39591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schägger, H. Tricine-SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Am Ende, C.W.; Meng, H.Y.; Ye, M.; Pandey, A.K.; Zondlo, N.J. Design of Lanthanide Fingers: Compact Lanthanide-Binding Metalloproteins. ChemBioChem 2010, 11, 1738–1747. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Prot. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Hibbert, D.B.; Thordarson, P. The death of the Job plot, transparency, open science and online tools, uncertainty estimation methods and other developments in supramolecular chemistry data analysis. Chem. Commun. 2016, 52, 12792–12805. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Dh (nm) | Kd (M) | ||
|---|---|---|---|---|
| −La3+ | +La3+ | +La3+ | +Ca2+ | |
| SPL2cyt | n.d. | n.d. | 9.45 × 10−7 | 1.21 × 10−6 |
| MBP-SPL2cyt | 7.72 ± 0.40 | 7.83 ± 0.27 | 1.72 × 10−6 | 1.13 × 10−6 |
| MBP-SPL2cyt-D316R | 8.99 ± 0.39 | 8.80 ± 0.22 | 1.13 × 10−5 | 3.03 × 10−6 |
| MBP-SPL2cyt-D317R | 9.19 ± 0.25 | 9.28 ± 0.20 | 6.80 × 10−6 | 2.09 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tracz, M.; Górniak, I.; Szczepaniak, A.; Białek, W. E3 Ubiquitin Ligase SPL2 Is a Lanthanide-Binding Protein. Int. J. Mol. Sci. 2021, 22, 5712. https://doi.org/10.3390/ijms22115712
Tracz M, Górniak I, Szczepaniak A, Białek W. E3 Ubiquitin Ligase SPL2 Is a Lanthanide-Binding Protein. International Journal of Molecular Sciences. 2021; 22(11):5712. https://doi.org/10.3390/ijms22115712
Chicago/Turabian StyleTracz, Michał, Ireneusz Górniak, Andrzej Szczepaniak, and Wojciech Białek. 2021. "E3 Ubiquitin Ligase SPL2 Is a Lanthanide-Binding Protein" International Journal of Molecular Sciences 22, no. 11: 5712. https://doi.org/10.3390/ijms22115712
APA StyleTracz, M., Górniak, I., Szczepaniak, A., & Białek, W. (2021). E3 Ubiquitin Ligase SPL2 Is a Lanthanide-Binding Protein. International Journal of Molecular Sciences, 22(11), 5712. https://doi.org/10.3390/ijms22115712