
Therapies Targeted at Non-Coding RNAs in Prevention and Limitation of Myocardial Infarction and Subsequent Cardiac Remodeling—Current Experience and Perspectives

 ,
,
Abstract
:1. Introduction
2. Agents Targeted at ncRNAs
3. The ncRNA as Potential Targets in Therapy of Atherosclerosis: Experience from Preclinical Studies
3.1. Targeting ncRNA in Prevention of Myocardial Infarction—Atherosclerotic Plaque Progression
3.2. Targeting ncRNA Regulating the Process of Myocardial Infarction
3.2.1. Targeting the Process of Cardiomyocytes Cell Death during Acute Phase of Myocardial Infarction
3.2.2. Targeting ncRNA in Prevention of Unfavorable Cardiac Remodeling after Myocardial Infarction
3.2.3. Preclinical Studies Examples
4. Challenges in Therapies Aimed to ncRNA
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Makałowski, W. The human genome structure and organization. Acta Biochim. Pol. 2001, 48, 587–598. [Google Scholar] [CrossRef]
- Gonzaga-Jauregui, C.; Lupski, J.R.; Gibbs, R.A. Human Genome Sequencing in Health and Disease. Annu. Rev. Med. 2012, 63, 35–61. [Google Scholar] [CrossRef] [Green Version]
- Cobb, J.; Büsst, C.; Petrou, S.; Harrap, S.; Ellis, J. Searching for Functional Genetic Variants in Non-Coding DNA. Clin. Exp. Pharmacol. Physiol. 2008, 35, 372–375. [Google Scholar] [CrossRef]
- Shabalina, S.A.; Spiridonov, N.A. The mammalian transcriptome and the function of non-coding DNA sequences. Genome Biol. 2004, 5, 105. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, Y.; Furuno, M.; Kasukawa, T.; Adachi, J.; Bono, H.; Kondo, S.; Nikaido, I.; Osato, N.; Saito, R.; Suzuki, H.; et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 2002, 420, 563–573. [Google Scholar]
- Hombach, S.; Kretz, M. Non-coding RNAs: Classification, Biology and Functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17. [Google Scholar] [CrossRef]
- Fabbri, M.; Girnita, L.; Varani, G.; Calin, G.A. Decrypting noncoding RNA interactions, structures, and functional networks. Genome Res. 2019, 29, 1377–1388. [Google Scholar] [CrossRef] [Green Version]
- Akhade, V.S.; Pal, D.; Kanduri, C. Long Noncoding RNA: Genome Organization and Mechanism of Action. Adv. Exp. Med. Biol. 2017, 1008, 47–74. [Google Scholar] [CrossRef]
- Alessio, E.; Bonadio, R.S.; Buson, L.; Chemello, F.; Cagnin, S. A Single Cell but Many Different Transcripts: A Journey into the World of Long Non-Coding RNAs. Int. J. Mol. Sci. 2020, 21, 302. [Google Scholar] [CrossRef] [Green Version]
- Czech, B.; Munafo, M.; Ciabrelli, F.; Eastwood, E.L.; Fabry, M.H.; Kneuss, E.; Hannon, G.J. piRNA-Guided Genome Defense: From Biogenesis to Silencing. Annu. Rev. Genet. 2018, 52, 131–157. [Google Scholar] [CrossRef]
- Li, X.; Yang, L.; Chen, L.-L. The Biogenesis, Functions, and Challenges of Circular RNAs. Mol. Cell 2018, 71, 428–442. [Google Scholar] [CrossRef] [Green Version]
- Poller, W.; Dimmeler, S.; Heymans, S.; Zeller, T.; Haas, J.; Karakas, M.; Leistner, D.; Jakob, P.; Nakagawa, S.; Blankenberg, S.; et al. Non-coding RNAs in cardiovascular diseases: Diagnostic and therapeutic perspectives. Eur. Heart J. 2018, 39, 2704–2716. [Google Scholar] [CrossRef] [Green Version]
- Broderick, J.A.; Zamore, P.D. MicroRNA therapeutics. Gene Ther. 2011, 18, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Lennox, K.A.; Behlke, M.A. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011, 18, 1111–1120. [Google Scholar] [CrossRef] [Green Version]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, T.; Bonauer, A.; Dimmeler, S. RNA Therapeutics in Cardiovascular Disease. Circ. Res. 2018, 123, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA–guided activation of endogenous human genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Long, B.; Zhou, L.-Y.; Liu, F.; Zhou, Q.-Y.; Liu, C.-Y.; Fan, Y.-Y.; Li, P.-F. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat. Commun. 2014, 5, 3596. [Google Scholar] [CrossRef] [Green Version]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Kobayashi, N.; Todo, Y.; Watari, H. Long Non-coding RNA NEAT1: A Novel Target for Diagnosis and Therapy in Human Tumors. Front. Genet. 2018, 9, 471. [Google Scholar] [CrossRef] [Green Version]
- Vegter, E.L.; Van Der Meer, P.; De Windt, L.J.; Pinto, Y.M.; Voors, A.A. MicroRNAs in heart failure: From biomarker to target for therapy. Eur. J. Heart Fail. 2016, 18, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Skuratovskaia, D.A.; Vulf, M.A.; Komar, A.; Kirienkova, E.; Litvinova, L.S. Epigenetic regulation as a promising tool for treatment of atherosclerosis. Front. Biosci. 2020, 12, 173–199. [Google Scholar] [CrossRef]
- Leopoulou, M.; Mistakidi, V.C.; Oikonomou, E.; Latsios, G.; Papaioannou, S.; Deftereos, S.; Siasos, G.; Antonopoulos, A.; Charalambous, G.; Tousoulis, D. Acute Coronary Syndrome with Non-ruptured Plaques (NONRUPLA): Novel Ideas and Perspectives. Curr. Atheroscler. Rep. 2020, 22, 21. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; de Winther, M.P.; Weber, C.; Daemen, M.J.; Lutgens, E.; Soehnlein, O. Atherosclerotic plaque destabilization: Mechanisms, models, and therapeutic strategies. Circ. Res. 2014, 114, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Basatemur, G.L.; Jørgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef]
- Kavurma, M.M.; Rayner, K.J.; Karunakaran, D. The walking dead: Macrophage inflammation and death in atherosclerosis. Curr. Opin. Lipidol. 2017, 28, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef]
- Bryan, M.T.; Duckles, H.; Feng, S.; Hsiao, S.T.; Kim, H.R.; Serbanovic-Canic, J.; Evans, P.C. Mechanoresponsive Networks Controlling Vascular Inflammation. Arter. Thromb. Vasc. Biol. 2014, 34, 2199–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfenniger, A.; Wong, C.; Sutter, E.; Cuhlmann, S.; Dunoyer-Geindre, S.; Mach, F.; Horrevoets, A.J.; Evans, P.C.; Krams, R.; Kwak, B.R. Shear stress modulates the expression of the atheroprotective protein Cx37 in endothelial cells. J. Mol. Cell. Cardiol. 2012, 53, 299–309. [Google Scholar] [CrossRef]
- Feinberg, M.W.; Moore, K.J. MicroRNA Regulation of Atherosclerosis. Circ. Res. 2016, 118, 703–720. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Thavarajah, T.; Gu, W.; Cai, J.; Xu, Q. Impact of miRNA in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2018, 38, e159–e170. [Google Scholar] [CrossRef] [Green Version]
- Fasolo, F.; Di Gregoli, K.; Maegdefessel, L.; Johnson, J.L. Non-coding RNAs in cardiovascular cell biology and atherosclerosis. Cardiovasc. Res. 2019, 115, 1732–1756. [Google Scholar] [CrossRef]
- Pierce, J.B.; Feinberg, M.W. Long Noncoding RNAs in Atherosclerosis and Vascular Injury: Pathobiology, Biomarkers, and Targets for Therapy. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2002–2017. [Google Scholar] [CrossRef]
- Soh, J.; Iqbal, J.; Queiroz, J.; Fernandez-Hernando, C.; Hussain, M.M. MicroRNA-30c reduces hyperlipidemia and atherosclerosis in mice by decreasing lipid synthesis and lipoprotein secretion. Nat. Med. 2013, 19, 892–900. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-Y.; Yang, T.-L.; Huang, Y.-H.; Lee, C.-I.; Chen, L.-J.; Shih, Y.-T.; Wei, S.-Y.; Wang, W.-L.; Wu, C.-C.; Chiu, J.-J. Induction of microRNA-10a using retinoic acid receptor-α and retinoid x receptor-α agonists inhibits atherosclerotic lesion formation. Atherosclerosis 2018, 271, 36–44. [Google Scholar] [CrossRef]
- Loyer, X.; Potteaux, S.; Vion, A.-C.; Guérin, C.L.; Boulkroun, S.; Rautou, P.-E.; Ramkhelawon, B.; Esposito, B.; Dalloz, M.; Paul, J.-L.; et al. Inhibition of MicroRNA-92a Prevents Endothelial Dysfunction and Atherosclerosis in Mice. Circ. Res. 2014, 114, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Su, G.; Sun, G.; Liu, H.; Shu, L.; Liang, Z. Downregulation of miR-34a promotes endothelial cell growth and suppresses apoptosis in atherosclerosis by regulating Bcl-2. Heart Vessel. 2018, 33, 1185–1194. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef] [Green Version]
- Badi, I.; Mancinelli, L.; Polizzotto, A.; Ferri, D.; Zeni, F.; Burba, I.; Milano, G.; Brambilla, F.; Saccu, C.; Bianchi, M.E.; et al. miR-34a Promotes Vascular Smooth Muscle Cell Calcification by Downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase). Arter. Thromb. Vasc. Biol. 2018, 38, 2079–2090. [Google Scholar] [CrossRef]
- Schober, A.; Nazari-Jahantigh, M.; Wei, Y.; Bidzhekov, K.; Gremse, F.; Grommes, J.; Megens, R.T.A.; Heyll, K.; Noels, H.; Hristov, M.; et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat. Med. 2014, 20, 368–376. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.-C.; Tang, Y.-Y.; Peng, J.; Zhao, G.-J.; Yang, J.; Yao, F.; Ouyang, X.-P.; He, P.-P.; Xie, W.; Tan, Y.-L.; et al. MicroRNA-19b promotes macrophage cholesterol accumulation and aortic atherosclerosis by targeting ATP-binding cassette transporter A1. Atherosclerosis 2014, 236, 215–226. [Google Scholar] [CrossRef]
- Distel, E.; Barrett, T.J.; Chung, K.; Girgis, N.M.; Parathath, S.; Essau, C.C.; Murphy, A.J.; Moore, K.J.; Fisher, E.A. miR33 Inhibition Overcomes Deleterious Effects of Diabetes Mellitus on Atherosclerosis Plaque Regression in Mice. Circ. Res. 2014, 115, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouimet, M.; Ediriweera, H.N.; Gundra, U.M.; Sheedy, F.; Ramkhelawon, B.; Hutchison, S.B.; Rinehold, K.; Van Solingen, C.; Fullerton, M.D.; Cecchini, K.; et al. MicroRNA-33–dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J. Clin. Investig. 2015, 125, 4334–4348. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Wu, X.; Dai, D.; Li, J.; Mehta, J.L. MicroRNA-98 regulates foam cell formation and lipid accumulation through repression of LOX-1. Redox Biol. 2018, 16, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Yu, F.; Wang, Y.; Hui, Y.; Carnevale, K.; Fu, M.; Lu, H.; Fan, D. MicroRNA-155 Deficiency Results in Decreased Macrophage Inflammation and Attenuated Atherogenesis in Apolipoprotein E–Deficient Mice. Arter. Thromb. Vasc. Biol. 2014, 34, 759–767. [Google Scholar] [CrossRef]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J.; et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J. Clin. Investig. 2012, 122, 4190–4202. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, R.M.; Chaudhuri, A.A.; Rao, D.S.; Baltimore, D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. USA 2009, 106, 7113–7118. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.-P.; Gong, D.; Zhao, Z.-W.; He, P.-P.; Yu, X.-H.; Ye, Q.; Huang, C.; Zhang, X.; Chen, L.-Y.; Xie, W.; et al. MicroRNA-182 Promotes Lipoprotein Lipase Expression and Atherogenesisby Targeting Histone Deacetylase 9 in Apolipoprotein E-Knockout Mice. Circ. J. 2018, 82, 28–38. [Google Scholar] [CrossRef] [Green Version]
- He, P.-P.; Ouyang, X.-P.; Li, Y.; Lv, Y.-C.; Wang, Z.-B.; Yao, F.; Xie, W.; Tan, Y.-L.; Li, L.; Zhang, M.; et al. MicroRNA-590 Inhibits Lipoprotein Lipase Expression and Prevents Atherosclerosis in apoE Knockout Mice. PLoS ONE 2015, 10, e0138788. [Google Scholar] [CrossRef]
- Chen, W.; Yu, F.; Di, M.; Li, M.; Chen, Y.; Zhang, Y.; Liu, X.; Huang, X.; Zhang, M. MicroRNA-124-3p inhibits collagen synthesis in atherosclerotic plaques by targeting prolyl 4-hydroxylase subunit alpha-1 (P4HA1) in vascular smooth muscle cells. Atherosclerosis 2018, 277, 98–107. [Google Scholar] [CrossRef]
- Canfrán-Duque, A.; Rotllan, N.; Zhang, X.; Fernández-Fuertes, M.; Ramírez-Hidalgo, C.; Araldi, E.; Daimiel, L.; Busto, R.; Fernández-Hernando, C.; Suárez, Y. Macrophage deficiency of miR-21 promotes apoptosis, plaque necrosis, and vascular inflammation during atherogenesis. EMBO Mol. Med. 2017, 9, 1244–1262. [Google Scholar] [CrossRef]
- Sun, P.; Tang, L.-N.; Li, G.-Z.; Xu, Z.-L.; Xu, Q.-H.; Wang, M.; Li, L. Effects of MiR-21 on the proliferation and migration of vascular smooth muscle cells in rats with atherosclerosis via the Akt/ERK signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2216–2222. [Google Scholar] [PubMed]
- Di Gregoli, K.; Jenkins, N.; Salter, R.; White, S.; Newby, A.C.; Johnson, J.L. MicroRNA-24 regulates macrophage behavior and retards atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1990–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, K.; Zhu, X.; Zheng, Z.; Mo, Z.-C.; Peng, X.-S.; Zeng, Y.-Z.; Ou, H.-X.; Zhang, Q.-H.; Qi, H.-Z.; Zhao, G.-J.; et al. MicroRNA-24 aggravates atherosclerosis by inhibiting selective lipid uptake from HDL cholesterol via the post-transcriptional repression of scavenger receptor class B type I. Atherosclerosis 2018, 270, 57–67. [Google Scholar] [CrossRef]
- Shan, Z.; Qin, S.; Li, W.; Wu, W.; Yang, J.; Chu, M.; Li, X.; Huo, Y.; Schaer, G.L.; Wang, S.; et al. An Endocrine Genetic Signal Between Blood Cells and Vascular Smooth Muscle Cells: Role of MicroRNA-223 in Smooth Muscle Function and Atherogenesis. J. Am. Coll. Cardiol. 2015, 65, 2526–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Fang, Z.; Jiang, T.; Zhang, Q.; Liu, C.; Zhang, C.; Xiang, Y. Serum microRNAs profile from genome-wide serves as a fingerprint for diagnosis of acute myocardial infarction and angina pectoris. BMC Med. Genom. 2013, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; De Ronde, M.W.J.; Kok, M.G.M.; Beijk, M.A.; De Winter, R.J.; Van Der Wal, A.C.; Sondermeijer, B.M.; Meijers, J.C.M.; Creemers, E.E.; Pinto-Sietsma, S.-J. MiR-223-3p and miR-122-5p as circulating biomarkers for plaque instability. Open Heart 2020, 7, e001223. [Google Scholar] [CrossRef] [PubMed]
- Lovren, F.; Pan, Y.; Quan, A.; Singh, K.K.; Shukla, P.C.; Gupta, N.; Steer, B.M.; Ingram, A.J.; Gupta, M.; Al-Omran, M.; et al. MicroRNA-145 Targeted Therapy Reduces Atherosclerosis. Circulation 2012, 126, S81–S90. [Google Scholar] [CrossRef] [Green Version]
- Sala, F.; Aranda, J.F.; Rotllan, N.; Ramirez, C.M.; Aryal, B.; Elia, L.; Condorelli, G.; Catapano, A.L.; Fernandez-Hernando, C.; Norata, G.D. MiR-143/145 deficiency attenuates the progression of atherosclerosis in Ldlr-/-mice. Thromb. Haemost. 2014, 112, 796–802. [Google Scholar] [CrossRef] [Green Version]
- Di Gregoli, K.; Anuar, N.N.M.; Bianco, R.; White, S.J.; Newby, A.C.; George, S.J.; Johnson, J.L. MicroRNA-181b Controls Atherosclerosis and Aneurysms Through Regulation of TIMP-3 and Elastin. Circ. Res. 2017, 120, 49–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan-Peng, X.; Sheng-Cai, C.; Xia, Y.-P.; Chen, S.-C.; Baral, S.; Mao, L.; Jin, H.-J.; Ling-Qiang, Z.; Wang, M.-D.; Chen, J.-G.; et al. MiR-181b Antagonizes Atherosclerotic Plaque Vulnerability Through Modulating Macrophage Polarization by Directly Targeting Notch1. Mol. Neurobiol. 2017, 54, 6329–6341. [Google Scholar] [CrossRef]
- Holdt, L.M.; Stahringer, A.; Sass, K.; Pichler, G.; Kulak, N.A.; Wilfert, W.; Kohlmaier, A.; Herbst, A.; Northoff, B.; Nicolaou, A.; et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat. Commun. 2016, 7, 12429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haemmig, S.; Yang, D.; Sun, X.; Das, D.; Ghaffari, S.; Molinaro, R.; Chen, L.; Deng, Y.; Freeman, D.; Moullan, N.; et al. Long noncoding RNA SNHG12 integrates a DNA-PK–mediated DNA damage response and vascular senescence. Sci. Transl. Med. 2020, 12, eaaw1868. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Meng, K.; Jiang, L.; Zhong, Y.; Yang, Y.; Lan, Y.; Zeng, Q.; Cheng, L. Thymic stromal lymphopoietin-induced HOTAIR activation promotes endothelial cell proliferation and migration in atherosclerosis. Biosci. Rep. 2017, 37, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lyu, Q.; Xu, S.; Lyu, Y.; Choi, M.; Christie, C.K.; Slivano, O.J.; Rahman, A.; Jin, Z.-G.; Long, X.; Xu, Y.; et al. SENCR stabilizes vascular endothelial cell adherens junctions through interaction with CKAP4. Proc. Natl. Acad. Sci. USA 2019, 116, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.-W.; Guo, F.-X.; Xu, Y.-J.; Li, P.; Lu, Z.-F.; McVey, D.G.; Zheng, L.; Wang, Q.; Ye, J.H.; Kang, C.-M.; et al. Long noncoding RNA NEXN-AS1 mitigates atherosclerosis by regulating the actin-binding protein NEXN. J. Clin. Investig. 2019, 129, 1115–1128. [Google Scholar] [CrossRef]
- Ye, Z.-M.; Yang, S.; Xia, Y.-P.; Hu, R.-T.; Chen, S.; Li, B.-W.; Chen, S.-L.; Luo, X.-Y.; Mao, L.; Li, Y.; et al. LncRNA MIAT sponges miR-149-5p to inhibit efferocytosis in advanced atherosclerosis through CD47 upregulation. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Sallam, T.; Jones, M.; Thomas, B.J.; Wu, X.; Gilliland, T.; Qian, K.; Eskin, A.; Casero, D.; Zhang, Z.; Sandhu, J.; et al. Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nat. Med. 2018, 24, 304–312. [Google Scholar] [CrossRef]
- Ahmed, A.S.I.; Dong, K.; Liu, J.; Wen, T.; Yu, L.; Xu, F.; Kang, X.; Osman, I.; Hu, G.; Bunting, K.M.; et al. Long noncoding RNA NEAT1 (nuclear paraspeckle assembly transcript 1) is critical for phenotypic switching of vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA 2018, 115, E8660–E8667. [Google Scholar] [CrossRef] [Green Version]
- Song, T.-F.; Huang, L.-W.; Yuan, Y.; Wang, H.-Q.; He, H.-P.; Ma, W.-J.; Huo, L.-H.; Zhou, H.; Wang, N.; Zhang, T.-C. LncRNA MALAT1 regulates smooth muscle cell phenotype switch via activation of autophagy. Oncotarget 2017, 9, 4411–4426. [Google Scholar] [CrossRef] [Green Version]
- Cremer, S.; Michalik, K.M.; Fischer, A.; Pfisterer, L.; Jaé, N.; Winter, C.; Boon, R.A.; Muhly-Reinholz, M.; John, D.; Uchida, S.; et al. Hematopoietic Deficiency of the Long Noncoding RNA MALAT1 Promotes Atherosclerosis and Plaque Inflammation. Circulation 2019, 139, 1320–1334. [Google Scholar] [CrossRef]
- Cho, H.; Shen, G.-Q.; Wang, X.; Wang, F.; Archacki, S.; Li, Y.; Yu, G.; Chakrabarti, S.; Chen, Q.; Wang, Q.K. Long noncoding RNA ANRIL regulates endothelial cell activities associated with coronary artery disease by up-regulating CLIP1, EZR, and LYVE1 genes. J. Biol. Chem. 2019, 294, 3881–3898. [Google Scholar] [CrossRef]
- Holdt, L.M.; Beutner, F.; Scholz, M.; Gielen, S.; Gabel, G.; Bergert, H.; Schuler, G.; Thiery, J.; Teupser, D. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 620–627. [Google Scholar] [CrossRef] [Green Version]
- Horie, T.; Baba, O.; Kuwabara, Y.; Chujo, Y.; Watanabe, S.; Kinoshita, M.; Horiguchi, M.; Nakamura, T.; Chonabayashi, K.; Hishizawa, M.; et al. MicroRNA-33 deficiency reduces the progression of atherosclerotic plaque in ApoE-/- mice. J. Am. Heart Assoc. 2012, 1, e003376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouimet, M.; Ediriweera, H.; Afonso, M.S.; Ramkhelawon, B.; Singaravelu, R.; Liao, X.; Bandler, R.C.; Rahman, K.; Fisher, E.A.; Rayner, K.J.; et al. microRNA-33 Regulates Macrophage Autophagy in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2017, 37, 1058–1067. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wang, M.; He, Q.; Li, Z.; Zhao, Y.; Wang, W.; Ma, J.; Li, Y.; Chang, G. MicroRNA-98 rescues proliferation and alleviates ox-LDL-induced apoptosis in HUVECs by targeting LOX-1. Exp. Ther. Med. 2017, 13, 1702–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santovito, D.; Mandolini, C.; Marcantonio, P.; De Nardis, V.; Bucci, M.; Paganelli, C.; Magnacca, F.; Ucchino, S.; Mastroiacovo, D.; Desideri, G.; et al. Overexpression of microRNA-145 in atherosclerotic plaques from hypertensive patients. Expert Opin. Ther. Targets 2013, 17, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Vengrenyuk, Y.; Nishi, H.; Long, X.; Ouimet, M.; Savji, N.; Martinez, F.O.; Cassella, C.P.; Moore, K.J.; Ramsey, S.A.; Miano, J.M.; et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, J.E.; Ercan, B.; Morin, K.M.; Liu, C.T.; Tamer, L.; Ayaz, L.; Kanadasi, M.; Cicek, D.; Seyhan, A.I.; Akilli, R.E.; et al. The distribution of circulating microRNA and their relation to coronary disease. F1000Res 2012, 1, 50. [Google Scholar] [CrossRef]
- Rayner, K.J.; Sheedy, F.; Esau, C.C.; Hussain, F.N.; Temel, R.E.; Parathath, S.; Van Gils, J.M.; Rayner, A.J.; Chang, A.N.; Suarez, Y.; et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Investig. 2011, 121, 2921–2931. [Google Scholar] [CrossRef] [Green Version]
- Marquart, T.J.; Wu, J.; Lusis, A.J.; Baldan, A. Anti-miR-33 therapy does not alter the progression of atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 455–458. [Google Scholar] [CrossRef] [Green Version]
- Rotllan, N.; Ramirez, C.M.; Aryal, B.; Esau, C.C.; Fernandez-Hernando, C. Therapeutic silencing of microRNA-33 inhibits the progression of atherosclerosis in Ldlr-/- mice--brief report. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1973–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Wu, N.; Leong, M.C.; Zhang, W.; Ye, Z.; Li, R.; Huang, J.; Zhang, Z.; Li, L.; Yao, X.; et al. miR-145 improves metabolic inflammatory disease through multiple pathways. J. Mol. Cell Biol. 2020, 12, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Wezel, A.; Welten, S.M.J.; Razawy, W.; Lagraauw, H.M.; de Vries, M.R.; Goossens, E.A.C.; Boonstra, M.C.; Hamming, J.F.; Kandimalla, E.R.; Kuiper, J.; et al. Inhibition of MicroRNA-494 Reduces Carotid Artery Atherosclerotic Lesion Development and Increases Plaque Stability. Ann. Surg. 2015, 262, 841–848. [Google Scholar] [CrossRef] [PubMed]
- van Ingen, E.; Foks, A.C.; Kröner, M.J.; Kuiper, J.; Quax, P.H.; Bot, I.; Nossent, A.Y. Antisense Oligonucleotide Inhibition of MicroRNA-494 Halts Atherosclerotic Plaque Progression and Promotes Plaque Stabilization. Mol. Ther. Nucleic Acids 2019, 18, 638–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welten, S.M.; De Jong, R.C.; Wezel, A.; De Vries, M.R.; Boonstra, M.C.; Parma, L.; Jukema, J.W.; Van Der Sluis, T.C.; Arens, R.; Bot, I.; et al. Inhibition of 14q32 microRNA miR-495 reduces lesion formation, intimal hyperplasia and plasma cholesterol levels in experimental restenosis. Atherosclerosis 2017, 261, 26–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieu, H.D.; Withycombe, S.K.; Walker, Q.; Rong, J.X.; Walzem, R.L.; Wong, J.S.; Hamilton, R.L.; Fisher, E.A.; Young, S.G. Eliminating Atherogenesis in Mice by Switching Off Hepatic Lipoprotein Secretion. Circulation 2003, 107, 1315–1321. [Google Scholar] [CrossRef] [Green Version]
- Parathath, S.; Grauer, L.; Huang, L.-S.; Sanson, M.; Distel, E.; Goldberg, I.J.; Fisher, E.A. Diabetes Adversely Affects Macrophages During Atherosclerotic Plaque Regression in Mice. Diabetes 2011, 60, 1759–1769. [Google Scholar] [CrossRef] [Green Version]
- Mansoor, M.; Melendez, A.J. Advances in Antisense Oligonucleotide Development for Target Identification, Validation, and as Novel Therapeutics. Gene Regul. Syst. Biol. 2008, 2, 275–295. [Google Scholar] [CrossRef]
- Davalos, A.; Goedeke, L.; Smibert, P.; Ramirez, C.M.; Warrier, N.P.; Andreo, U.; Salinas, D.C.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.-M.; Chen, K.-C.; Hsu, P.-Y.; Lin, H.-F.; Wang, Y.-S.; Chen, C.-Y.; Liao, Y.-C.; Juo, S.-H.H. microRNA let-7g suppresses PDGF-induced conversion of vascular smooth muscle cell into the synthetic phenotype. J. Cell. Mol. Med. 2017, 21, 3592–3601. [Google Scholar] [CrossRef]
- Wang, J.; Hu, X.; Hu, X.; Gao, F.; Li, M.; Cui, Y.; Wei, X.; Qin, Y.; Zhang, C.; Zhao, Y.; et al. MicroRNA-520c-3p targeting of RelA/p65 suppresses atherosclerotic plaque formation. Int. J. Biochem. Cell Biol. 2021, 131, 105873. [Google Scholar] [CrossRef]
- Sun, D.; Ma, T.; Zhang, Y.; Zhang, F.; Cui, B. Overexpressed miR-335-5p reduces atherosclerotic vulnerable plaque formation in acute coronary syndrome. J. Clin. Lab. Anal. 2021, 35, e23608. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tao, G.; Liu, Q.; Liu, K.; Yang, X. MicroRNA let-7g alleviates atherosclerosis via the targeting of LOX-1 in vitro and in vivo. Int. J. Mol. Med. 2017, 40, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Ye, Z.-M.; Chen, S.; Luo, X.-Y.; Chen, S.-L.; Mao, L.; Li, Y.; Jin, H.; Yu, C.; Xiang, F.-X.; et al. MicroRNA-23a-5p promotes atherosclerotic plaque progression and vulnerability by repressing ATP-binding cassette transporter A1/G1 in macrophages. J. Mol. Cell. Cardiol. 2018, 123, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Liu, Y.; Wu, M.; Meng, Y.; Lu, M.; Guo, J.; Zhou, Y. Downregulation of microRNA-135b promotes atherosclerotic plaque stabilization in atherosclerotic mice by upregulating erythropoietin receptor. IUBMB Life 2020, 72, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Eken, S.M.; Jin, H.; Chernogubova, E.; Li, Y.; Simon, N.; Sun, C.; Korzunowicz, G.; Busch, A.; Bäcklund, A.; Österholm, C.; et al. MicroRNA-210 Enhances Fibrous Cap Stability in Advanced Atherosclerotic Lesions. Circ. Res. 2017, 120, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, V.; Rotllan, N.; Araldi, E.; Luciano, A.; Skroblin, P.; Abonnenc, M.; Perrotta, P.; Yin, X.; Bauer, A.; Leslie, K.L.; et al. Chronic miR-29 antagonism promotes favorable plaque remodeling in atherosclerotic mice. EMBO Mol. Med. 2016, 8, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Cai, J.; Han, Y.; Chen, J.; Huang, Z.-P.; Chen, C.; Cai, Y.; Huang, H.; Yang, Y.; Liu, Y.; et al. LincRNA-p21 Regulates Neointima Formation, Vascular Smooth Muscle Cell Proliferation, Apoptosis, and Atherosclerosis by Enhancing p53 Activity. Circulation 2014, 130, 1452–1465. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Fu, Y.; Gu, X.; Xi, X.; Peng, X.; Wang, C.; Sun, Q.; Wang, X.; Qian, F.; Qin, Z.; et al. Macrophage-Enriched lncRNA RAPIA: A Novel Therapeutic Target for Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1464–1478. [Google Scholar] [CrossRef]
- Basalay, M.V.; Yellon, D.M.; Davidson, S.M. Targeting myocardial ischaemic injury in the absence of reperfusion. Basic Res. Cardiol. 2020, 115, 1–16. [Google Scholar] [CrossRef]
- Azevedo-Gaiolla, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.R.; Zornoff, L.A.M. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Braz. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef]
- Konstam, M.A.; Kramer, D.G.; Patel, A.R.; Maron, M.S.; Udelson, J.E. Left Ventricular Remodeling in Heart Failure: Current Concepts in Clinical Significance and Assessment. JACC Cardiovasc. Imaging 2011, 4, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Samidurai, A.; Salloum, F.N. Deciphering Non-coding RNAs in Cardiovascular Health and Disease. Front. Cardiovasc. Med. 2018, 5, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Liu, C.; Zhao, Y.; Ponnusamy, M.; Li, P.; Wang, K. Role of noncoding RNAs in regulation of cardiac cell death and cardiovascular diseases. Cell. Mol. Life Sci. 2017, 75, 291–300. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Liang, X.-L.; Zhang, C.-L.; Pang, Y.-H.; Lu, Y.-X. LncRNA KLF3-AS1 in human mesenchymal stem cell-derived exosomes ameliorates pyroptosis of cardiomyocytes and myocardial infarction through miR-138-5p/Sirt1 axis. Stem Cell Res. Ther. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, A.-Y.; Yang, Q.; Yang, K. miR-133a mediates the hypoxia-induced apoptosis by inhibiting TAGLN2 expression in cardiac myocytes. Mol. Cell. Biochem. 2014, 400, 173–181. [Google Scholar] [CrossRef]
- Zhang, X.-G.; Wang, L.-Q.; Guan, H.-L. Investigating the expression of miRNA-133 in animal models of myocardial infarction and its effect on cardiac function. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5934–5940. [Google Scholar]
- Gao, F.; Kataoka, M.; Liu, N.; Liang, T.; Huang, Z.-P.; Gu, F.; Ding, J.; Liu, J.; Zhang, F.; Ma, Q.; et al. Therapeutic role of miR-19a/19b in cardiac regeneration and protection from myocardial infarction. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, X.; Ren, X.-P.; Chen, J.; Liu, H.; Yang, J.; Medvedovic, M.; Hu, Z.; Fan, G.-C. MicroRNA-494 Targeting Both Proapoptotic and Antiapoptotic Proteins Protects Against Ischemia/Reperfusion-Induced Cardiac Injury. Circulation 2010, 122, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- Han, F.; Chen, Q.; Su, J.; Zheng, A.; Chen, K.; Sun, S.; Wu, H.; Jiang, L.; Xu, X.; Yang, M.; et al. MicroRNA-124 regulates cardiomyocyte apoptosis and myocardial infarction through targeting Dhcr24. J. Mol. Cell. Cardiol. 2019, 132, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Y.; Wang, B.J.; Ma, M.; Yu, K.; Zhang, Q.; Zhang, X.W. MicroRNA-325-3p protects the heart after myocardial infarction by inhibiting RIPK3 and programmed necrosis in mice. BMC Mol. Biol. 2019, 20, 17. [Google Scholar]
- Abbate, A.; Biondi-Zoccai, G.G.; Baldi, A. Pathophysiologic role of myocardial apoptosis in post-infarction left ventricular remodeling. J. Cell. Physiol. 2002, 193, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Hullinger, T.G.; Montgomery, R.L.; Seto, A.G.; Dickinson, B.A.; Semus, H.M.; Lynch, J.M.; Dalby, C.M.; Robinson, K.; Stack, C.; Latimer, P.A.; et al. Inhibition of miR-15 Protects Against Cardiac Ischemic Injury. Circ. Res. 2012, 110, 71–81. [Google Scholar] [CrossRef]
- Zhong, Z.; Wu, H.; Zhong, W.; Zhang, Q.; Yu, Z. Expression profiling and bioinformatics analysis of circulating microRNAs in patients with acute myocardial infarction. J. Clin. Lab. Anal. 2019, 34, e23099. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Gao, X.-M.; Winbanks, C.E.; Boey, E.J.H.; Tham, Y.K.; Kiriazis, H.; Gregorevic, P.; Obad, S.; Kauppinen, S.; Du, X.-J.; et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. USA 2012, 109, 17615–17620. [Google Scholar] [CrossRef] [Green Version]
- Qian, L.; Van Laake, L.W.; Huang, Y.; Liu, S.; Wendland, M.F.; Srivastava, D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J. Exp. Med. 2011, 208, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Moghaddam, A.S.; Afshari, J.T.; Esmaeili, S.-A.; Saburi, E.; Joneidi, Z.; Momtazi-Borojeni, A.A. Cardioprotective microRNAs: Lessons from stem cell-derived exosomal microRNAs to treat cardiovascular disease. Atherosclerosis 2019, 285, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuoka, M.; Fujita, H.; Numao, K.; Nakamura, Y.; Shimizu, H.; Sekiguchi, M.; Hohjoh, H. MiR-199-3p enhances muscle regeneration and ameliorates aged muscle and muscular dystrophy. Commun. Biol. 2021, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Wang, X.-G.; Cheng, J.-D.; You, S.-Z.; Ma, S.-H.; Zhong, X.; Quan, L.; Luo, B. The up-regulation of endothelin-1 and down-regulation of miRNA-125a-5p, -155, and -199a/b-3p in human atherosclerotic coronary artery. Cardiovasc. Pathol. 2014, 23, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.W.; Zhang, Y.N.; Hu, H.J.; Zhang, P.Q.; Cui, W. Downregulation of microRNA199a5p attenuates hypoxia/reoxygenationinduced cytotoxicity in cardiomyocytes by targeting the HIF1alphaGSK3betamPTP axis. Mol. Med. Rep. 2019, 19, 5335–5344. [Google Scholar] [PubMed] [Green Version]
- Wang, X.; Guo, Z.; Ding, Z.; Mehta, J.L. Inflammation, Autophagy, and Apoptosis after Myocardial Infarction. J. Am. Hear. Assoc. 2018, 7, e008024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Xie, J.; Li, R.; Shi, J.; Sun, J.; Gu, R.; Ding, L.; Wang, L.; Xu, B. Overexpression of microRNA-99a attenuates heart remodelling and improves cardiac performance after myocardial infarction. J. Cell. Mol. Med. 2014, 18, 919–928. [Google Scholar] [CrossRef]
- Gupta, S.K.; Foinquinos, A.; Thum, S.; Remke, J.; Zimmer, K.; Bauters, C.; de Groote, P.; Boon, R.A.; de Windt, L.J.; Preissl, S.; et al. Preclinical Development of a MicroRNA-Based Therapy for Elderly Patients With Myocardial Infarction. J. Am. Coll. Cardiol. 2016, 68, 1557–1571. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Qi, B.; Wu, X.; Peng, S.; Zhou, G.; Wei, Y.; Xu, J.; Chen, S.; Liu, S. Circular RNA Ttc3 regulates cardiac function after myocardial infarction by sponging miR-15b. J. Mol. Cell. Cardiol. 2019, 130, 10–22. [Google Scholar] [CrossRef]
- Garikipati, V.N.S.; Verma, S.K.; Cheng, Z.; Liang, D.; Truongcao, M.M.; Cimini, M.; Yue, Y.; Huang, G.; Wang, C.; Benedict, C.; et al. Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF-A axis. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Sun, J.; Wang, C. Long non-coding RNAs in cardiac hypertrophy. Hear. Fail. Rev. 2020, 25, 1037–1045. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling—Concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Sygitowicz, G.; Maciejak-Jastrzębska, A.; Sitkiewicz, D. MicroRNAs in the development of left ventricular remodeling and postmyocardial infarction heart failure. Pol. Arch. Intern. Med. 2020, 130, 59–65. [Google Scholar]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nat. Cell Biol. 2008, 456, 980–984. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, H.; Ge, D.; Xu, Y.; Xu, H.; Yang, Y.; Gu, M.; Zhou, Y.; Zhu, J.; Ge, T.; et al. Mir-21 Promotes Cardiac Fibrosis After Myocardial Infarction Via Targeting Smad7. Cell. Physiol. Biochem. 2017, 42, 2207–2219. [Google Scholar] [CrossRef]
- Yan, X.; Liu, Z.; Chen, Y. Regulation of TGF-beta signaling by Smad7. Acta Biochim. Biophys. Sin. 2009, 41, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, B.; Zhou, Q.; Wang, Y.; Liu, X.; Liu, Z.; Zhan, Z. MicroRNA-21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Bei, Y.; Kong, X.; Liu, X.; Lei, Z.; Xu, T.; Wang, H.; Xuan, Q.; Chen, P.; Xu, J.; et al. miR-17-3p Contributes to Exercise-Induced Cardiac Growth and Protects against Myocardial Ischemia-Reperfusion Injury. Theranostics 2017, 7, 664–676. [Google Scholar] [CrossRef]
- Yuan, X.; Pan, J.; Wen, L.; Gong, B.; Li, J.; Gao, H.; Tan, W.; Liang, S.; Zhang, H.; Wang, X. MiR-590-3p regulates proliferation, migration and collagen synthesis of cardiac fibroblast by targeting ZEB1. J. Cell. Mol. Med. 2020, 24, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Reboll, M.R.; Korf-Klingebiel, M.; Wollert, K.C. Angiogenesis after acute myocardial infarction. Cardiovasc. Res. 2021, 117, 1257–1273. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Tréguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nat. Cell Biol. 2013, 495, 107–110. [Google Scholar] [CrossRef]
- Icli, B.; Wara, A.; Moslehi, J.; Sun, X.; Plovie, E.; Cahill, M.; Marchini, J.F.; Schissler, A.; Padera, R.F.; Shi, J.; et al. MicroRNA-26a Regulates Pathological and Physiological Angiogenesis by Targeting BMP/SMAD1 Signaling. Circ. Res. 2013, 113, 1231–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Templin, C.; Volkmann, J.; Emmert, M.Y.; Mocharla, P.; Muller, M.; Kraenkel, N.; Ghadri, J.R.; Meyer, M.; Styp-Rekowska, B.; Briand, S.; et al. Increased Proangiogenic Activity of Mobilized CD34+ Progenitor Cells of Patients With Acute ST-Segment-Elevation Myocardial Infarction: Role of Differential MicroRNA-378 Expression. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 341–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, K.; Lei, W.; Wu, H.; Wu, J.; Yang, Z.; Yan, S.; Lu, X.A.; Li, J.; Xia, X.; Han, X.; et al. LncRNA-Safe contributes to cardiac fibrosis through Safe-Sfrp2-HuR complex in mouse myocardial infarction. Theranostics 2019, 9, 7282–7297. [Google Scholar] [CrossRef]
- Chen, G.; Huang, S.; Song, F.; Zhou, Y.; He, X. Lnc-Ang362 is a pro-fibrotic long non-coding RNA promoting cardiac fibrosis after myocardial infarction by suppressing Smad7. Arch. Biochem. Biophys. 2020, 685, 108354. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.-G.; Yuan, Y.-P.; Wu, H.-M.; Zhang, X.; Tang, Q.-Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramazani, Y.; Knops, N.; Elmonem, M.A.; Nguyen, T.Q.; Arcolino, F.O.; Heuvel, L.V.D.; Levtchenko, E.; Kuypers, D.; Goldschmeding, R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 2018, 68–69, 44–66. [Google Scholar] [CrossRef]
- Wang, X.; Yong, C.; Yu, K.; Yu, R.; Zhang, R.; Yu, L.; Li, S.; Cai, S. Long Noncoding RNA (lncRNA) n379519 Promotes Cardiac Fibrosis in Post-Infarct Myocardium by Targeting miR-30. Med. Sci. Monit. 2018, 24, 3958–3965. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, S.; Jia, Q.; Zhang, A.; Li, Y.; Zhu, Y.; Lv, S.; Zhang, J. The microRNA in ventricular remodeling: The miR-30 family. Biosci. Rep. 2019, 39, 20190788. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.H.; Chai, H.X.; Bai, M.; Zhang, Z. LncRNA-GAS5 regulates PDCD4 expression and mediates myocardial infarction-induced cardiomyocytes apoptosis via targeting MiR-21. Cell Cycle 2020, 19, 1363–1377. [Google Scholar] [CrossRef]
- Liu, F.; Levin, M.D.; Petrenko, N.B.; Lu, M.M.; Wang, T.; Yuan, L.J.; Stout, A.L.; Epstein, J.A.; Patel, V.V. Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy independent of angiotensin. J. Mol. Cell. Cardiol. 2008, 45, 715–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, C.E.; Nguyen, Q.; Lukowski, S.W.; Helfer, A.; Chiu, H.S.; Miklas, J.; Levy, S.; Suo, S.; Han, J.-D.J.; Osteil, P.; et al. Single-Cell Transcriptomic Analysis of Cardiac Differentiation from Human PSCs Reveals HOPX-Dependent Cardiomyocyte Maturation. Cell Stem Cell 2018, 23, 586–598.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Li, H.; Li, X.; Li, B.; Zhong, L.; Huang, S.; Zheng, H.; Li, M.; Jin, G.; Liao, W.; et al. Loss of long non-coding RNA CRRL promotes cardiomyocyte regeneration and improves cardiac repair by functioning as a competing endogenous RNA. J. Mol. Cell. Cardiol. 2018, 122, 152–164. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Liu, F.; Zhou, L.-Y.; Long, B.; Yuan, S.-M.; Wang, Y.; Liu, C.-Y.; Sun, T.; Zhang, X.-J.; Li, P.-F. The Long Noncoding RNA CHRF Regulates Cardiac Hypertrophy by Targeting miR-489. Circ. Res. 2014, 114, 1377–1388. [Google Scholar] [CrossRef] [Green Version]
- Seok, H.Y.; Chen, J.; Kataoka, M.; Huang, Z.-P.; Ding, J.; Yan, J.; Hu, X.; Wang, D.-Z. Loss of MicroRNA-155 Protects the Heart From Pathological Cardiac Hypertrophy. Circ. Res. 2014, 114, 1585–1595. [Google Scholar] [CrossRef]
- Du, W.W.; Yang, W.; Chen, Y.; Wu, Z.-K.; Foster, F.S.; Yang, Z.; Li, X.; Yang, B.B. Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur. Heart J. 2016, 38, 1402–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Long, B.; Liu, F.; Wang, J.-X.; Liu, C.-Y.; Zhao, B.; Zhou, L.-Y.; Sun, T.; Wang, M.; Cui-Yun, L.; et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur. Heart J. 2016, 37, 2602–2611. [Google Scholar] [CrossRef]
- Yan, M.; Chen, K.; Sun, R.; Lin, K.; Qian, X.; Yuan, M.; Wang, Y.; Ma, J.; Qing, Y.; Xu, J.; et al. Glucose impairs angiogenesis and promotes ventricular remodelling following myocardial infarction via upregulation of microRNA-17. Exp. Cell Res. 2019, 381, 191–200. [Google Scholar] [CrossRef]
- Huang, W.; Tian, S.-S.; Hang, P.-Z.; Sun, C.; Guo, J.; Du, Z.-M. Combination of microRNA-21 and microRNA-146a Attenuates Cardiac Dysfunction and Apoptosis During Acute Myocardial Infarction in Mice. Mol. Ther. Nucleic Acids 2016, 5, e296. [Google Scholar] [CrossRef]
- Li, J.; Rohailla, S.; Gelber, N.; Rutka, J.; Sabah, N.; Gladstone, R.A.; Wei, C.; Hu, P.; Kharbanda, R.K.; Redington, A.N. MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res. Cardiol. 2014, 109, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, S.X.; He, Q.; Zhang, H.; Friedberg, D.; Wang, F.; Redington, A.N. Intravenous miR-144 reduces left ventricular remodeling after myocardial infarction. Basic Res. Cardiol. 2018, 113, 36. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Huang, M.; Li, Z.; Jia, F.; Ghosh, Z.; Lijkwan, M.A.; Fasanaro, P.; Sun, N.; Wang, X.; Martelli, F.; et al. MicroRNA-210 as a Novel Therapy for Treatment of Ischemic Heart Disease. Circulation 2010, 122, S124–S131. [Google Scholar] [CrossRef] [Green Version]
- Martinez, E.C.; Lilyanna, S.; Wang, P.; Vardy, L.A.; Jiang, X.; Armugam, A.; Jeyaseelan, K.; Richards, A.M. MicroRNA-31 promotes adverse cardiac remodeling and dysfunction in ischemic heart disease. J. Mol. Cell. Cardiol. 2017, 112, 27–39. [Google Scholar] [CrossRef]
- Lesizza, P.; Prosdocimo, G.; Martinelli, V.; Sinagra, G.; Zacchigna, S.; Giacca, M. Single-Dose Intracardiac Injection of Pro-Regenerative MicroRNAs Improves Cardiac Function After Myocardial Infarction. Circ. Res. 2017, 120, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, J.; Geng, J.; Chen, F.; Wei, Z.; Liu, C.; Zhang, X.; Li, Q.; Zhang, J.; Gao, L.; et al. Long non-coding RNA MEG3 knockdown attenuates endoplasmic reticulum stress-mediated apoptosis by targeting p53 following myocardial infarction. J. Cell. Mol. Med. 2019, 23, 8369–8380. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; Van Der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latronico, M.V.; Condorelli, G. Therapeutic applications of noncoding RNAs. Curr. Opin. Cardiol. 2015, 30, 213–221. [Google Scholar] [CrossRef]
- Jiang, C.; Qi, Z.; He, W.; Li, Z.; Tang, Y.; Wang, Y.; Huang, Y.; Zang, H.; Yang, H.; Liu, J. Dynamically enhancing plaque targeting via a positive feedback loop using multifunctional biomimetic nanoparticles for plaque regression. J. Control. Release 2019, 308, 71–85. [Google Scholar] [CrossRef]
- Zhao, Y.; Gao, H.; He, J.; Jiang, C.; Lu, J.; Zhang, W.; Yang, H.; Liu, J. Co-delivery of LOX-1 siRNA and statin to endothelial cells and macrophages in the atherosclerotic lesions by a dual-targeting core-shell nanoplatform: A dual cell therapy to regress plaques. J. Control. Release 2018, 283, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Thum, T. RNA-based diagnostic and therapeutic strategies for cardiovascular disease. Nat. Rev. Cardiol. 2019, 16, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Golob, J.L.; Lugogo, N.; Lauring, A.S.; Lok, A.S. SARS-CoV-2 vaccines: A triumph of science and collaboration. JCI Insight 2021, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]



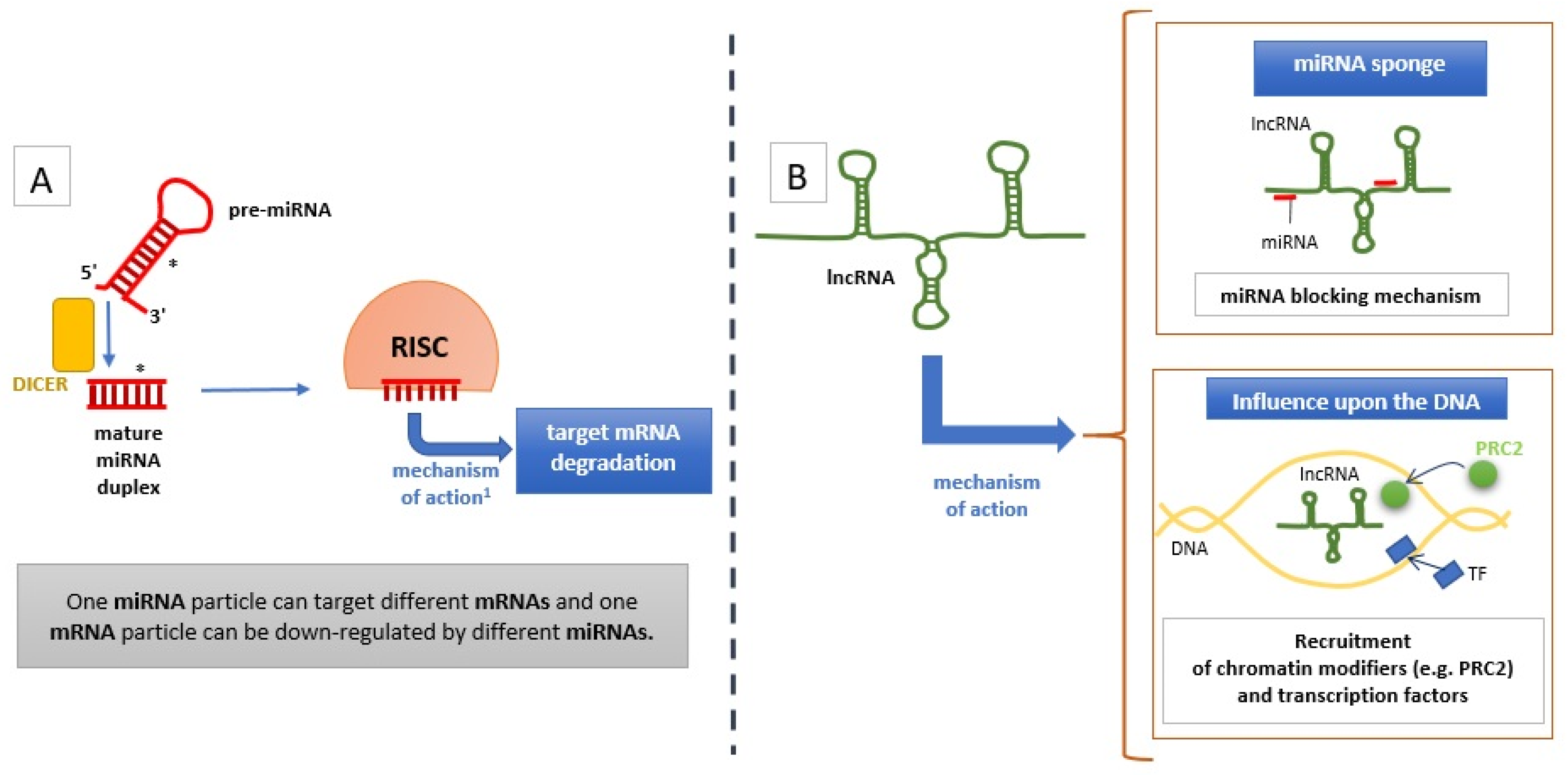{kind=link}
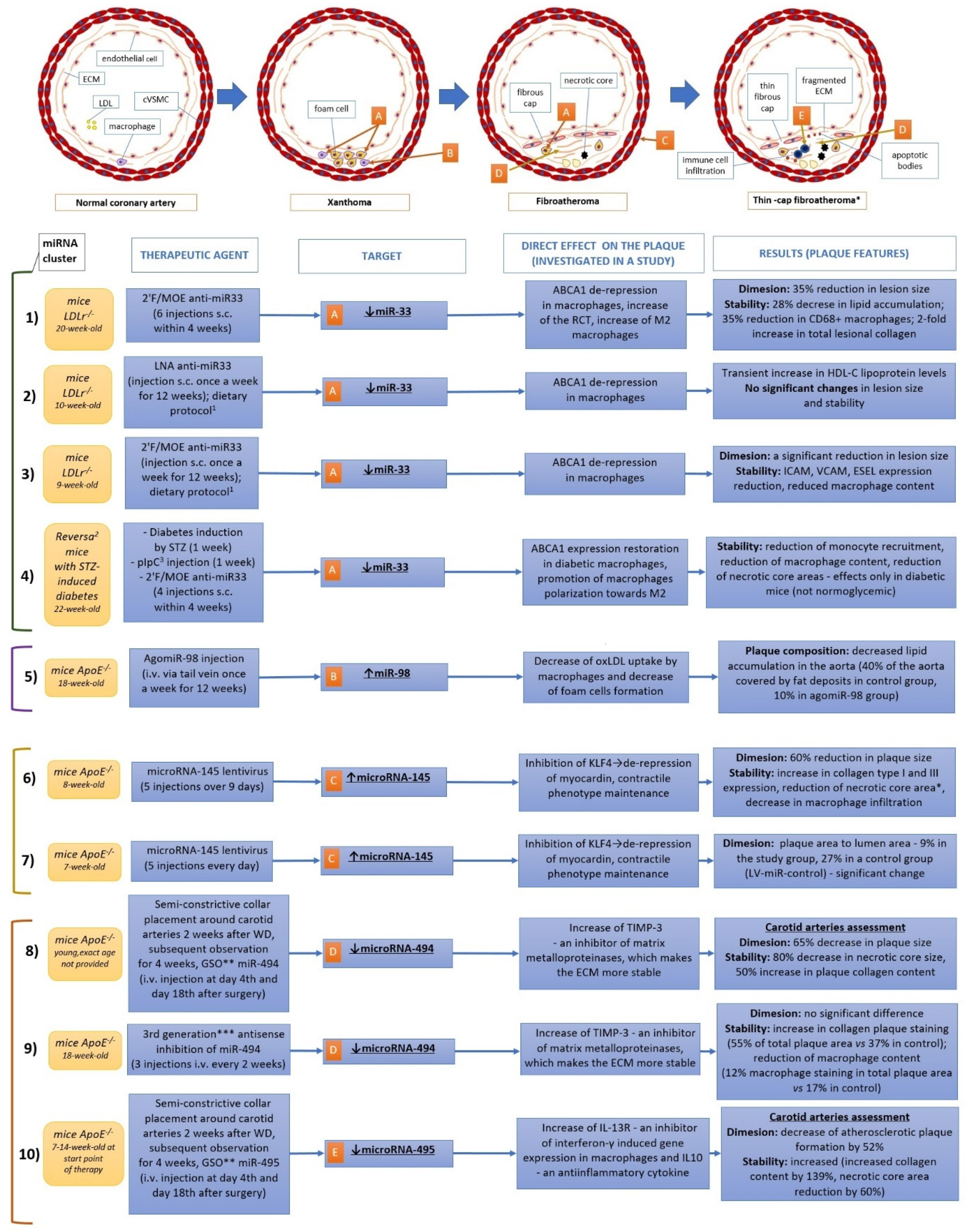{kind=link}
| Inhibition | Activation | |
|---|---|---|
| miRNA |
|
|
| lncRNA |
|
|
| The microRNAs in the Process of Atherosclerotic Plaque Progression (Categorized by Stages) | |||
|---|---|---|---|
| miRNA | Stage | Function | Mechanism, Target mRNA * and References |
| miR-30c | Initial—lipid metabolism | Atheroprotective | Down-regulation of MTTP *, an essential factor in lipidation of apoB [35] |
| miR-10a | Initial—EC | Atheroprotective | Down-regulation of GATA6 *, an inducer of adhesion molecule VCAM-1 in the EC [36] |
| miR-92a | Initial—EC | Atherogenic | Down-regulation of KLF2 * and KLF4 * expression [37] |
| miR-34a | Initial—EC | Atherogenic | Down-regulation of BCL2 * [38] (antiapoptotic mediator) and SIRT1 * (a class III histone deacetylase, participating in maintenance of cellular longevity) [39,40] |
| miR-126 | Initial—EC | Atheroprotective | Down-regulation of VCAM-1 * (cell adhesion molecule for leukocytes) and Dlk-1 * (a negative regulator of EC proliferation) [41] |
| miR-19 | Progression—macrophages | Atherogenic | Down-regulation of ABCA1 * (a transporter essential for reverse cholesterol transport through HDL) [42] |
| miR-33 | Progression—macrophages | Atherogenic | Down-regulation of ABCA1 * (a transporter essential for reverse cholesterol transport through HDL) [43] and AMPK *, a kinase involved in FAO activation and macrophages polarization towards anti-inflammatory M2 phenotype [44] |
| miR-98 | Progression—macrophages | Atheroprotective | Down-regulation of LOX1 *, a receptor for oxLDL participating in foam cell formation [45] |
| miR-155 | Progression—macrophages | Atherogenic | Down-regulation of BCL6 *, an antiapoptotic protein and inhibitor of NF-kB signaling [46,47], and SHIP1 *, inositol phosphatase blocking PI3K pathways and modulating T-lymphocytes activity [46,48] |
| miR-182 | Progression—macrophages | Atherogenic | Down-regulation of HDAC9 *, which results in increase of lipoprotein lipase (LPL) expression in macrophages and increased uptake of lipoproteins [49] |
| miR-590 | Progression—macrophages | Atheroprotective | Down-regulation of LPL *, which results in decreased lipoprotein uptake by macrophages [50] |
| miR-124 | Advanced—VSMC, ECM | Atherogenic | Down-regulation of P4HA1 *, a key enzyme in collagen synthesis [51] |
| miR-21 | Advanced **—VSMC, ECM | Ambiguous | Atheroprotective: down-regulation MKK3 * (leading to ablation of MKK3-p38-CHOP pro-apoptotic pathways) [52] 1 Atherogenic: down-regulation of PTEN * (leading to Akt/ERK signaling pathways activation and VSMC proliferation) [53] 2 |
| miR-24 | Advanced **—VSMC, ECM | Ambiguous | Atheroprotective: down-regulation of MMP14 *, matrix metalloproteinase participating in extracellular matrix fragmentation [54] 3 Atherogenic: down-regulation of SR-BI * in liver, which causes inhibition of HDL uptake and reverse cholesterol transport [55] |
| miR-223 | Advanced **—VSMC, ECM | Ambiguous | Inhibition of IGF-1R * in VSMC, which results in atheroprotection on earlier stages [56], but might participate in destabilization on advanced stages (before rupture) [57,58] |
| miR-143/145 | Multi-stage | Ambiguous | Atheroprotective: down-regulation of KLF4 *, resulting in increased myocardin expression and VSMC contractile phenotype maintenance [59] Atherogenic: down-regulation of ABCA1 *, (a transporter essential for reverse cholesterol transport through HDL) [60] 4 |
| miR-181b | Multi-stage | Ambiguous | Atherogenic: down-regulation of TIMP3 *, an inhibitor of destabilizing metalloproteinases [61] Atheroprotective: down-regulation of NOTCH1 *, a transmembrane protein promoting macrophage pro-inflammatory polarization [62] |
| The lncRNA in the Process of Atherosclerotic Plaque Progression | |||
|---|---|---|---|
| lncRNA | Stage | Function | Mechanism and References |
| SNHG12 | Multi-stage | atheroprotective | Binding to DNA-PK, facilitating interaction with Ku70 and Ku80, resulting in appropriate response to DNA damage [64] |
| HOTAIR | Initial—EC | atheroprotective | Scaffold for PRC-2 and LDS-1 (histone modifications) [65], EC protection from senescence [66] |
| SENCR | Initial—EC | atheroprotective | Maintaining EC layer integrity by CKAP-4 binding in cytoplasm and prevention from VE-cadherin internalization induced by CKAP-4 [67] |
| NEXN-AS1 | Initial—EC | atheroprotective | Binding the DNA region of NEXN promotor and enhancing its expression (NEXN = inhibitor of TLR4 oligomerization and NF-kB activity) [68] |
| MIAT | Progression—macrophages | atherogenic | Sponging effect 1 on miR-149-5p and prevention from CD47 degradation, leading to impaired efferocytosis (CD47 = efferocytosis inhibitor, ‘don’t eat me’ signal, a target for miR-149-5p) [69] |
| MeXis | Progression—macrophages | atheroprotective | Indirect promotion of ABCA1 expression (by guiding transcription factor DDX-17 to a nearby ABCA1 locus) [70] |
| NEAT1 | Advanced—VSMC * | atherogenic | Inhibition of WDR-5 activity, a histone modifier that promotes contractile VSMC phenotype [71] |
| MALAT1 | Multi-stage | atheroprotective | Autophagy activation in VSMC (sponging 1 of miR-142-3p, a down-regulator of ATG7, an autophagy-related, beneficial protein) [72], decrease of hematopoietic cells in bone marrow, decrease leukocyte adhesion to EC (partially through miR-503 inhibition) [73] |
| ANRIL | Multi-stage | ambiguous | Atheroprotective 2: up-regulation of CLIP-1, EZR, and LYVE-1, which promotes EC physiological functions (e.g., nourishment through micropinocytosis) [74] Atherogenic: high-risk alleles of ANRIL (SNP in chromosome 9p21.3) promoting linear ANRIL isoform associated with increased atherosclerosis [75] |
| ncRNA of Interest | ncRNA Mechanism of Action | Intervention | Model | Outcome | References |
|---|---|---|---|---|---|
| miR-144 (miRNA) | mTOR down-regulation → promotion of autophagy | Enhancement MiR-144 i.v. administration (day 0, 1, 3, and later) 1 | C57BL/6 mice, MI by LAD ligation |
| [162,163] |
| miR-210 (miRNA) | Efna3 and Ptp1b (angiogenesis inhibitors) down-regulation | Enhancement Minicircle 2 DNA (containing miR-210 precursor) administration | Adult female FVB mice, MI by LAD ligation |
| [164] |
| miR-31 (miRNA) | Down-regulation of Tnnt2 (troponin T), Nr3c2, E2f6, and Timp4, factors responsible for cellular viability and ECM stability | Inhibition LNA-modified anti-miR injected s.c., after 2 and 16 days from MI | Male adult Wistar Rats, MI by LAD ligation |
| [165] |
| miR-199a-3p | Entothelin-1 (ET-1) down-regulation | Enhancement miR-199a-3p mimics (lipofectamine RNAiMAX) | Female CD1 mice, MI by LAD ligation |
| [166] |
| miR-590-3p | Collagen synthesis induction through ZEB1 down-regulation | Enhancement miR-590-3p mimics | Female CD1 mice, MI by LAD ligation |
| [166] |
| circFndc3b (circRNA) | Interaction with FUS RNA-binding protein → VEGF-A up-regulation | Enhancement AAV-9 mediated overexpression | C57BL/6 male mice, MI by LAD ligation |
| [129] |
| MEG3 (lncRNA) | Direct p53 binding and activation → promotion of ERS- and NF-kB mediated myocardial apoptosis | Inhibition si-MEG3 (small interfering RNA) in lentiviruses | C57BL/6 male mice, MI by LAD ligation |
| [167] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowara, M.; Borodzicz-Jazdzyk, S.; Rybak, K.; Kubik, M.; Cudnoch-Jedrzejewska, A. Therapies Targeted at Non-Coding RNAs in Prevention and Limitation of Myocardial Infarction and Subsequent Cardiac Remodeling—Current Experience and Perspectives. Int. J. Mol. Sci. 2021, 22, 5718. https://doi.org/10.3390/ijms22115718
Kowara M, Borodzicz-Jazdzyk S, Rybak K, Kubik M, Cudnoch-Jedrzejewska A. Therapies Targeted at Non-Coding RNAs in Prevention and Limitation of Myocardial Infarction and Subsequent Cardiac Remodeling—Current Experience and Perspectives. International Journal of Molecular Sciences. 2021; 22(11):5718. https://doi.org/10.3390/ijms22115718
Chicago/Turabian StyleKowara, Michal, Sonia Borodzicz-Jazdzyk, Karolina Rybak, Maciej Kubik, and Agnieszka Cudnoch-Jedrzejewska. 2021. "Therapies Targeted at Non-Coding RNAs in Prevention and Limitation of Myocardial Infarction and Subsequent Cardiac Remodeling—Current Experience and Perspectives" International Journal of Molecular Sciences 22, no. 11: 5718. https://doi.org/10.3390/ijms22115718
APA StyleKowara, M., Borodzicz-Jazdzyk, S., Rybak, K., Kubik, M., & Cudnoch-Jedrzejewska, A. (2021). Therapies Targeted at Non-Coding RNAs in Prevention and Limitation of Myocardial Infarction and Subsequent Cardiac Remodeling—Current Experience and Perspectives. International Journal of Molecular Sciences, 22(11), 5718. https://doi.org/10.3390/ijms22115718