
Molecular Epidemiology of Mitochondrial Cardiomyopathy: A Search Among Mitochondrial and Nuclear Genes

 ,
, 
 , ,
, ,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Mutations in Nuclear and Mitochondrial Genes
3. Mitochondrial Cardiomyopathy (MCM)
3.1. Hypertrophic Cardiomyopathy
3.2. More Rare Cardiomyopathies
3.3. Mitochondrial DNA in Conduction System and Coronary Heart Disease
3.4. Other Mitochondrial Diseases with Cardiomyopathy

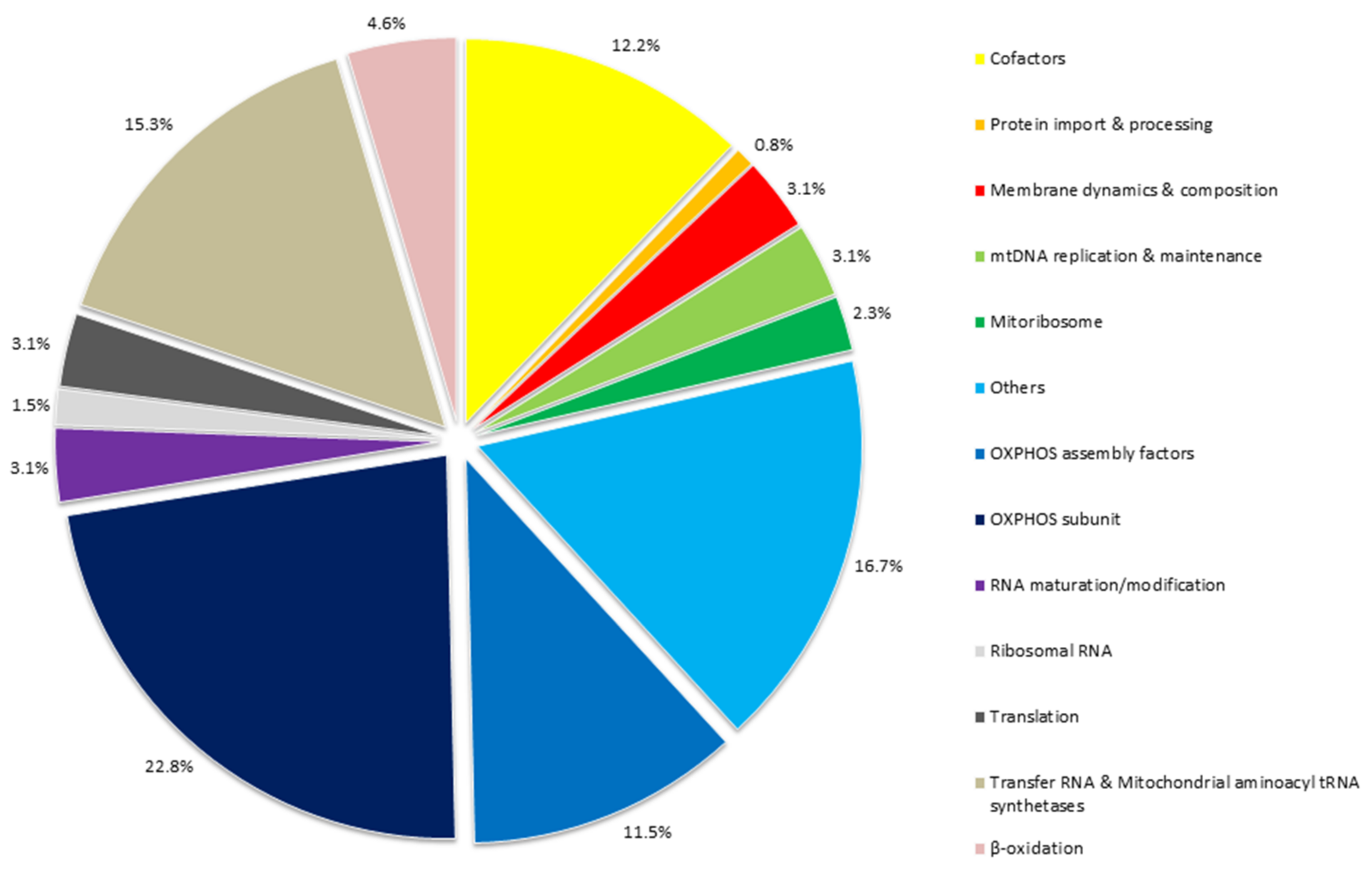{kind=link}
| Cardiological Phenotype | Disorder | Mutations | Amino Acid Change | Ref |
|---|---|---|---|---|
| HCM | MELAS, MERRF, CPEO, LHON, NARP, MIDD, Sengers syndrome, Friedreich ataxia | ACAD9: c.797G>A | p.Arg266Gln | [101] |
| AGK: c.306T>G | p.Tyr102Ter | [102] | ||
| COX6B1: c.58C>T | p.Arg20Cys | [56] | ||
| FXN: GAA repeat expansion | - | [98] | ||
| MRPL3: c.950C>G | p.Pro317Arg | [63] | ||
| MRPL44: c.467T>G | p.Leu156Arg | [103] | ||
| MTCOX2: m.7896G>A | p.Trp104Ter | [58] | ||
| MTCYB: m.14849T>C | p.Ser35Pro | [104] | ||
| MTND1: m.3481G>A | p.Glu59Lys | [105] | ||
| MTND5: m.12338T>C | p.Met1Thr | [106] | ||
| MTRNR2: m.2336T>C | - | [107] | ||
| MTTK: m.8344A>G | - | [60] | ||
| MTTI: m.4300A>G | - | [65] | ||
| MTTL: m.3243A>G | - | [4] | ||
| NDUFS2: c.686C>A | p.Pro229Gln | [108] | ||
| NDUFV2: c.669_670insG | p.Ser224fs | [109] | ||
| NDUFA2: c.208+5G>A | - | [110] | ||
| NDUFAF1: c.631C>T | p.Arg211Cys | [111] | ||
| SCOX2: c.418G>A | p.Asp140Asn | [112] | ||
| SURF1: 845_846delCT | p.Ser282Cysfs | [113] | ||
| SDHD: c.275A>G | p.Asp92Gly | [114] | ||
| TMEM70: c.317-2A>G | - | [115] | ||
| TMEM70: c.366A>T | p.Tyr112Ter | [116] | ||
| TSFM: c.997C>T | p.Arg312Trp | [117] | ||
| DCM | MELAS, MIDD, LHON, Barth syndrome | MTTL: m.3243A>G | - | MITOMAP |
| MTTI: m.4300A>G | - | MITOMAP | ||
| MTTK: m.8344A>G | - | MITOMAP | ||
| MTND4: m.11778G>A | p.Arg340His | MITOMAP | ||
| TAZ: c.527A>G TSFM: c.355G>C | p.His176Arg | [118] | ||
| p.Val119Leu | [119] | |||
| RCM | Hearing loss and multi organ mitochondrial disorder, MELAS, MIDD | MTRNR1: m.1555A>G | - | MITOMAP |
| MTTL: m.3243A>G | - | MITOMAP | ||
| LVNC | MIDD | MTND1: m.3398T>C | p.Met31Thr | MITOMAP |
| HCM/LVNC | Leigh syndrome | MTND1: m.3697G>A | p.Gly131Ser | MITOMAP |
| HICMP | MERF | MTTK: m.8344A>G | - | MITOMAP |
| MTCYB: m.15498G>A | p.Gly251Asp | MITOMAP | ||
| Conduction system disease | KKS, CPEO | MTTL: m.3243A>G | - | MITOMAP |
| MTTK: m.8344A>G | - | MITOMAP | ||
| CHD | carotid atherosclerosis risk, HCM, Leigh syndrome, MELAS | MTCYB: m.15059G>A | p.Gly105Ter | MITOMAP |
| MTCYB: m.15243G>A MTND5: m.13513G>A | p.Gly166Glu | MITOMAP MITOMAP | ||
| p.Asp393Asn | ||||
| MTTL1: m.3256C>T | - | MITOMAP |
3.5. Multiorgan Clinical Expression of Mitochondrial Cardiomyopathy
4. Diagnosis
4.1. Medical Examination
4.2. Laboratory Investigation
4.3. Cardiological and Neuroimaging Investigations
5. Molecular Analysis of Mitochondrial Cardiomyopathies
Variant Interpretation
6. Literature Review
Summary of Evidence
7. Discussion
8. Conclusions
9. Limitation
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AV | Atrioventricular |
| CM | Cardiomyopathies |
| COX | Cytochrome C Oxidase |
| CPEO | Chronic Progressive External Ophthalmoplegia |
| DCM | Dilated Cardiomyopathy |
| DOAJ | Directory Of Open Access Journals |
| HCM | Hypertrophic Cardiomyopathy |
| HICMP | Histiocytoid Cardiomyopathy |
| HMG | High Mobility Group |
| KSS | Kearns–Sayre Syndrome |
| LD | Linear Dichroism |
| LGE | Late Gadolinium Enhancement |
| LHON | Leber Hereditary Optic Neuropathy |
| LV | LeftVentricular |
| LVH | Left Ventricular Hypertrophy |
| LVNC | Left Ventricular Non Compaction |
| MCM | Mitochondrial Cardiomyopathy |
| MDPI | Multidisciplinary Digital Publishing Institute |
| MDs | Mitochondrial Diseases |
| MELAS | Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes |
| MERRF | Myoclonic Epilepsy With Ragged-Red Fibers |
| MIDD | Maternally Inherited Diabetes And Deafness |
| MNGIE | Mitochondrial Neurogastrointestinal Encephalopathy |
| mtDNA | Mitochondrial DNA |
| NARP | Neurogenic Weakness With Ataxia And Retinitis Pigmentosa |
| nDNA | Nuclear DNA |
| NGS | Next-Generation Sequencing |
| NO | Nitric Oxide |
| OXPHOS | Oxidative Phosphorylation |
| PAH | Pulmonary Arterial Hypertension |
| PMD | Primary Mitochondrial Diseases |
| RC | Respiratory Chain |
| RCD | Respiratory chain disease |
| RCM | Restrictive Cardiomyopathy |
| ROS SCD | Reactive Oxygen Species Sudden cardiac death |
| SDH | Succinate Dehydrogenase |
| SMD | Secondary Mitochondrial Dysfunction |
| TFAM | Mitochondrial Transcription Factor A |
| TLA | Three Letter Acronym |
| TTS | Takotsubo Syndrome |
| VUS | Uncertain Significance Variant |
| WES | Whole Exome Sequencing |
References
- Lee, S.R.; Kim, N.; Noh, Y.H.; Xu, Z.; Ko, K.S.; Rhee, B.D.; Han, J. Mitochondrial DNA, mitochondrial dysfunction, and cardiac manifestations. Front. Biosci. 2017, 22, 1177–1194. [Google Scholar] [CrossRef] [Green Version]
- Towbin, J.A.; Jefferies, J.L. Cardiomyopathies Due to Left Ventricular Noncompaction, Mitochondrial and Storage Diseases, and Inborn Errors of Metabolism. Circ. Res. 2017, 121, 838–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limongelli, G.; Masarone, D.; D’Alessandro, R.; Elliott, P.M. Mitochondrial diseases and the heart: An overview of molecular basis, diagnosis, treatment and clinical course. Future Cardiol. 2012, 8, 71–88. [Google Scholar] [CrossRef]
- Meyers, D.E.; Basha, H.I.; Koenig, M.K. Mitochondrial cardiomyopathy: Pathophysiology, diagnosis, and management. Tex. Heart Inst. J. 2013, 40, 385–394. [Google Scholar]
- Mazzaccara, C.; Limongelli, G.; Petretta, M.; Vastarella, R.; Pacileo, G.; Bonaduce, D.; Salvatore, F.; Frisso, G. A common polymorphism in the SCN5A gene is associated with dilated cardiomyopathy. J. Cardiovasc. Med. 2018, 19, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Monda, E.; Tramonte, S.; Gragnano, F.; Masarone, D.; Frisso, G.; Esposito, A.; Gravino, R.; Ammendola, E.; Salerno, G.; et al. Prevalence and clinical significance of red flags in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 299, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Bates, M.G.; Bourke, J.P.; Giordano, C.; d’Amati, G.; Turnbull, D.M.; Taylor, R.W. Cardiac involvement in mitochondrial DNA disease: Clinical spectrum, diagnosis, and management. Eur. Heart J. 2012, 33, 3023–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- Liguori, R.; Mazzaccara, C.; Pasanisi, F.; Buono, P.; Oriani, G.; Finelli, C.; Contaldo, F.; Sacchetti, L. The mtDNA 15497 G/A polymorphism in cytochrome b in severe obese subjects from Southern Italy. Nutr. Metab. Cardiovasc. 2006, 16, 466–470. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, Y.S.; Turnbull, D.M. Mitochondrial disease: Genetics and management. J. Neurol. 2016, 263, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Naing, A.; Kenchaiah, M.; Krishnan, B.; Mir, F.; Charnley, A.; Egan, C.; Bano, G. Maternally inherited diabetes and deafness (MIDD): Diagnosis and management. J. Diabetes Its Complicat. 2014, 28, 542–546. [Google Scholar] [CrossRef]
- Jameson, E.; Morris, A.A.M. Mitochondrial disease—A review. Paediatr. Child. Health 2011, 21, 80–83. [Google Scholar] [CrossRef]
- Schaefer, A.M.; Walker, M.; Turnbull, D.M.; Taylor, R.W. Endocrine disorders in mitochondrial disease. Mol. Cell. Endocrinol. 2013, 379, 2–11. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Mazzaccara, C.; Iafusco, D.; Liguori, R.; Ferrigno, M.; Galderisi, A.; Vitale, D.; Simonelli, F.; Landolfo, P.; Prisco, F.; Masullo, M.; et al. Mitochondrial diabetes in children: Seek and you will find it. PLoS ONE 2012, 7, e34956. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; Turnbull, D.M.; Walker, M.; Hattersley, A.T. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet. Med. A J. Br. Diabet. Assoc. 2008, 25, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Brunel-Guitton, C.; Levtova, A.; Sasarman, F. Mitochondrial Diseases and Cardiomyopathies. Can. J. Cardiol. 2015, 31, 1360–1376. [Google Scholar] [CrossRef]
- Popov, L.D. Mitochondrial networking in diabetic left ventricle cardiomyocytes. Mitochondrion 2017, 34, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Mootha, V.K. The Mitochondrial Proteome and Human Disease. Annu. Rev. Genom. Hum. G 2010, 11, 25–44. [Google Scholar] [CrossRef] [Green Version]
- Scaglia, F.; Towbin, J.A.; Craigen, W.J.; Belmont, J.W.; Smith, E.O.; Neish, S.R.; Ware, S.M.; Hunter, J.V.; Fernbach, S.D.; Vladutiu, G.D.; et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 2004, 114, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Bougouin, W.; Behin, A.; Stojkovic, T.; Becane, H.M.; Jardel, C.; Berber, N.; Mochel, F.; Lombes, A.; Eymard, B.; et al. Long-term cardiac prognosis and risk stratification in 260 adults presenting with mitochondrial diseases. Eur. Heart J. 2015, 36, 2886–2893. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, M.; Giordano, C.; Nediani, C.; Travaglini, C.; Borchi, E.; Zani, M.; Feccia, M.; Mancini, M.; Petrozza, V.; Cossarizza, A.; et al. Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. J. Am. Coll. Cardiol. 2007, 50, 1362–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limongelli, G.; Masarone, D.; Pacileo, G. Mitochondrial disease and the heart. Heart 2017, 103, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Legati, A.; Reyes, A.; Nasca, A.; Invernizzi, F.; Lamantea, E.; Tiranti, V.; Garavaglia, B.; Lamperti, C.; Ardissone, A.; Moroni, I.; et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim. Biophys. Acta 2016, 1857, 1326–1335. [Google Scholar] [CrossRef]
- Falkenberg, M.; Larsson, N.G.; Gustafsson, C.M. DNA replication and transcription in mammalian mitochondria. Annu. Rev. Biochem. 2007, 76, 679–699. [Google Scholar] [CrossRef]
- Wong, L.J.C. Next generation molecular diagnosis of mitochondrial disorders. Mitochondrion 2013, 13, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.J.; Brilhante, V.; Suomalainen, A. Next-generation sequencing for mitochondrial disorders. Br. J. Pharmacol. 2014, 171, 1837–1853. [Google Scholar] [CrossRef] [Green Version]
- Dames, S.; Chou, L.S.; Xiao, Y.; Wayman, T.; Stocks, J.; Singleton, M.; Eilbeck, K.; Mao, R. The development of next-generation sequencing assays for the mitochondrial genome and 108 nuclear genes associated with mitochondrial disorders. J. Mol. Diagn. JMD 2013, 15, 526–534. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Petruzzella, V.; Papa, S. Mutations in human nuclear genes encoding for subunits of mitochondrial respiratory complex I: The NDUFS4 gene. Gene 2002, 286, 149–154. [Google Scholar] [CrossRef]
- Sallevelt, S.C.; de Die-Smulders, C.E.; Hendrickx, A.T.; Hellebrekers, D.M.; de Coo, I.F.; Alston, C.L.; Knowles, C.; Taylor, R.W.; McFarland, R.; Smeets, H.J. De novo mtDNA point mutations are common and have a low recurrence risk. J. Med. Genet. 2017, 54, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnery, P.F.; DiMauro, S.; Shanske, S.; Schon, E.A.; Zeviani, M.; Mariotti, C.; Carrara, F.; Lombes, A.; Laforet, P.; Ogier, H.; et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet 2004, 364, 592–596. [Google Scholar] [CrossRef]
- Damas, J.; Samuels, D.C.; Carneiro, J.; Amorim, A.; Pereira, F. Mitochondrial DNA Rearrangements in Health and Disease-A Comprehensive Study. Hum. Mutat. 2014, 35, 1–14. [Google Scholar] [CrossRef]
- Viscomi, C.; Zeviani, M. MtDNA-maintenance defects: Syndromes and genes. J. Inherit. Metab. Dis. 2017, 40, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndromol. 2016, 7, 122–137. [Google Scholar] [CrossRef] [Green Version]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial cytopathies. Cell Calcium 2016, 60, 199–206. [Google Scholar] [CrossRef]
- Schwarz, K.; Siddiqi, N.; Singh, S.; Neil, C.J.; Dawson, D.K.; Frenneaux, M.P. The breathing heart—Mitochondrial respiratory chain dysfunction in cardiac disease. Int. J. Cardiol. 2014, 171, 134–143. [Google Scholar] [CrossRef]
- Dorn, G., II. Mitochondrial fission/fusion and cardiomyopathy. Curr. Opin. Genet. Dev. 2016, 38, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 mutation and late-onset cardiomyopathy: Mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.R.; Han, J. Mitochondrial Mutations in Cardiac Disorders. Adv. Exp. Med. Biol. 2017, 982, 81–111. [Google Scholar] [CrossRef] [PubMed]
- Rotig, A. Human diseases with impaired mitochondrial protein synthesis. BBA Bioenergy 2011, 1807, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Debray, F.G.; Lambert, M.; Chevalier, I.; Robitaille, Y.; Decarie, J.C.; Shoubridge, E.A.; Robinson, B.H.; Mitchell, G.A. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics 2007, 119, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Imai-Okazaki, A.; Kishita, Y.; Kohda, M.; Mizuno, Y.; Fushimi, T.; Matsunaga, A.; Yatsuka, Y.; Hirata, T.; Harashima, H.; Takeda, A.; et al. Cardiomyopathy in children with mitochondrial disease: Prognosis and genetic background. Int. J. Cardiol. 2019, 279, 115–121. [Google Scholar] [CrossRef]
- Brignole, M.; Auricchio, A.; Baron-Esquivias, G.; Bordachar, P.; Boriani, G.; Breithardt, O.A.; Cleland, J.; Deharo, J.C.; Delgado, V.; Elliott, P.M.; et al. 2013 ESC Guidelines on cardiac pacing and cardiac resynchronization therapy: The Task Force on cardiac pacing and resynchronization therapy of the European Society of Cardiology (ESC). Developed in collaboration with the European Heart Rhythm Association (EHRA). Eur. Heart J. 2013, 34, 2281–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.C.; Lin, Y.; Xu, X.F.; Lin, S.Y.; Chen, X.M.; Wang, S.S. The alterations of mitochondrial DNA in coronary heart disease. Exp. Mol. Pathol 2020, 114. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, J.H.; Cui, M.N.; Lee, Y.S.; Jung, M.H.; Yi, J.E.; Jung, H.O.; Youn, H.J. Hypertrophic Cardiomyopathy Attributable to Mitochondrial DNA Mutation Diagnosed by Pathology and Gene Sequencing. Circulation 2016, 133, 1297–1299. [Google Scholar] [CrossRef] [Green Version]
- Govindaraj, P.; Khan, N.A.; Rani, B.; Rani, D.S.; Selvaraj, P.; Jyothi, V.; Bahl, A.; Narasimhan, C.; Rakshak, D.; Premkumar, K.; et al. Mitochondrial DNA variations associated with hypertrophic cardiomyopathy. Mitochondrion 2014, 16, 65–72. [Google Scholar] [CrossRef]
- Limongelli, G.; Tome-Esteban, M.; Dejthevaporn, C.; Rahman, S.; Hanna, M.G.; Elliott, P.M. Prevalence and natural history of heart disease in adults with primary mitochondrial respiratory chain disease. Eur. J. Heart Fail. 2010, 12, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Brecht, M.; Richardson, M.; Taranath, A.; Grist, S.; Thorburn, D.; Bratkovic, D. Leigh Syndrome Caused by the MT-ND5 m.13513G>A Mutation: A Case Presenting with WPW-Like Conduction Defect, Cardiomyopathy, Hypertension and Hyponatraemia. JIMD Rep. 2015, 19, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassone, E.; Rahman, S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulhag, U.N.; Soiferman, D.; Schueler-Furman, O.; Miller, C.; Shaag, A.; Elpeleg, O.; Edvardson, S.; Saada, A. Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet. 2015, 23, 159–164. [Google Scholar] [CrossRef]
- Marin-Garcia, J.; Goldenthal, M.J.; Ananthakrishnan, R.; Pierpont, M.E. The complete sequence of mtDNA genes in idiopathic dilated cardiomyopathy shows novel missense and tRNA mutations. J. Card. Fail. 2000, 6, 321–329. [Google Scholar] [CrossRef]
- Alston, C.; Ceccatelli Berti, C.; Blakely, E.; Olahova, M.; He, L.P.; McMahon, C.; Olpin, S.; Hargreaves, I.; Nolli, C.; McFarland, R.; et al. A recessive homozygous p.Asp92Gly SDHD mutation causes prenatal cardiomyopathy and a severe mitochondrial complex II deficiency. Hum. Genet. 2015, 134, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Ahola, S.; Isohanni, P.; Euro, L.; Brilhante, V.; Palotie, A.; Pihko, H.; Lonnqvist, T.; Lehtonen, T.; Laine, J.; Tyynismaa, H.; et al. Mitochondrial EFTs defects in juvenile-onset Leigh disease, ataxia, neuropathy, and optic atrophy. Neurology 2014, 83, 743–751. [Google Scholar] [CrossRef]
- Wahbi, K.; Larue, S.; Jardel, C.; Meune, C.; Stojkovic, T.; Ziegler, F.; Lombes, A.; Eymard, B.; Duboc, D.; Laforet, P. Cardiac involvement is frequent in patients with the m.8344A > G mutation of mitochondrial DNA. Neurology 2010, 74, 674–677. [Google Scholar] [CrossRef]
- Ng, Y.S.; Grady, J.P.; Lax, N.Z.; Bourke, J.P.; Alston, C.L.; Hardy, S.A.; Falkous, G.; Schaefer, A.G.; Radunovic, A.; Mohiddin, S.A.; et al. Sudden adult death syndrome in m.3243A > G-related mitochondrial disease: An unrecognized clinical entity in young, asymptomatic adults. Eur. Heart J. 2016, 37, 2552–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distelmaier, F.; Haack, T.B.; Catarino, C.B.; Gallenmuller, C.; Rodenburg, R.J.; Strom, T.M.; Baertling, F.; Meitinger, T.; Mayatepek, E.; Prokisch, H.; et al. MRPL44 mutations cause a slowly progressive multisystem disease with childhood-onset hypertrophic cardiomyopathy. Neurogenetics 2015, 16, 319–323. [Google Scholar] [CrossRef]
- Galmiche, L.; Serre, V.; Beinat, M.; Assouline, Z.; Lebre, A.S.; Chretien, D.; Nietschke, P.; Benes, V.; Boddaert, N.; Sidi, D.; et al. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum. Mutat. 2011, 32, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Song, Y.; Li, D.; He, X.; Li, S.; Wu, B.; Wang, W.; Gu, S.; Zhu, X.; Wang, X.; et al. The novel mitochondrial 16S rRNA 2336T>C mutation is associated with hypertrophic cardiomyopathy. J. Med. Genet. 2014, 51, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.W.; Giordano, C.; Davidson, M.M.; d’Amati, G.; Bain, H.; Hayes, C.M.; Leonard, H.; Barron, M.J.; Casali, C.; Santorelli, F.M.; et al. A homoplasmic mitochondrial transfer ribonucleic acid mutation as a cause of maternally inherited hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2003, 41, 1786–1796. [Google Scholar] [CrossRef] [Green Version]
- Govindaraj, P.; Rani, B.; Sundaravadivel, P.; Vanniarajan, A.; Indumathi, K.P.; Khan, N.A.; Dhandapany, P.S.; Rani, D.S.; Tamang, R.; Bahl, A.; et al. Mitochondrial genome variations in idiopathic dilated cardiomyopathy. Mitochondrion 2019, 48, 51–59. [Google Scholar] [CrossRef]
- Bates, M.G.; Nesbitt, V.; Kirk, R.; He, L.; Blakely, E.L.; Alston, C.L.; Brodlie, M.; Hasan, A.; Taylor, R.W.; McFarland, R. Mitochondrial respiratory chain disease in children undergoing cardiac transplantation: A prospective study. Int. J. Cardiol. 2012, 155, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, F.; Tanaka, M.; Kawamura, K.; Kanzaki, Y.; Okabe, M.; Hayashi, T.; Shimomura, H.; Ito, T.; Suwa, M.; Gong, J.S.; et al. A case of cardiomyopathy showing progression from the hypertrophic to the dilated form: Association of Mt8348A-->G mutation in the mitochondrial tRNA(Lys) gene with severe ultrastructural alterations of mitochondria in cardiomyocytes. Jpn. Circ. J. 2001, 65, 691–694. [Google Scholar] [CrossRef] [Green Version]
- Khogali, S.S.; Mayosi, B.M.; Beattie, J.M.; McKenna, W.J.; Watkins, H.; Poulton, J. A common mitochondrial DNA variant associated with susceptibility to dilated cardiomyopathy in two different populations. Lancet 2001, 357, 1265–1267. [Google Scholar] [CrossRef]
- Thebault, C.; Ollivier, R.; Leurent, G.; Marcorelles, P.; Langella, B.; Donal, E. Mitochondriopathy: A rare aetiology of restrictive cardiomyopathy. Eur. J. Echocardiogr. J. Work. Group Echocardiogr. Eur. Soc. Cardiol. 2008, 9, 840–845. [Google Scholar] [CrossRef] [Green Version]
- Santorelli, F.M.; Tanji, K.; Manta, P.; Casali, C.; Krishna, S.; Hays, A.P.; Mancini, D.M.; DiMauro, S.; Hirano, M. Maternally inherited cardiomyopathy: An atypical presentation of the mtDNA 12S rRNA gene A1555G mutation. Am. J. Hum. Genet. 1999, 64, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Batra, A.; Zhang, Y.; Ebenroth, E.S.; Huang, T.S. Left ventricular noncompaction is associated with mutations in the mitochondrial genome. Mitochondrion 2010, 10, 350–357. [Google Scholar] [CrossRef]
- Ye, Z.; Gu, X. A mitochondrial mutation A8701G is associated with maternally inherited hypertension and dilated cardiomyopathy in a Chinese pedigree of a consanguineous marriage. J. Am. Coll. Cardiol. 2016, 68, C132. [Google Scholar] [CrossRef]
- Alila, O.F.; Rebai, E.M.; Tabebi, M.; Tej, A.; Chamkha, I.; Tlili, A.; Bouguila, J.; Tilouche, S.; Soyah, N.; Boughamoura, L.; et al. Whole mitochondrial genome analysis in two families with dilated mitochondrial cardiomyopathy: Detection of mutations in MT-ND2 and MT-TL1 genes. Mitochondrial DNA A 2016, 27, 2873–2880. [Google Scholar] [CrossRef] [PubMed]
- Gilbert-Barness, E. Review: Metabolic cardiomyopathy and conduction system defects in children. Ann. Clin. Lab. Sci. 2004, 34, 15–34. [Google Scholar] [PubMed]
- Vallance, H.D.; Jeven, G.; Wallace, D.C.; Brown, M.D. A case of sporadic infantile histiocytoid cardiomyopathy caused by the A8344G (MERRF) mitochondrial DNA mutation. Pediatr. Cardiol. 2004, 25, 538–540. [Google Scholar] [CrossRef]
- Andreu, A.L.; Checcarelli, N.; Iwata, S.; Shanske, S.; DiMauro, S. A missense mutation in the mitochondrial cytochrome b gene in a revisited case with histiocytoid cardiomyopathy. Pediatr. Res. 2000, 48, 311–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterer, J. Histiocytoid cardiomyopathy: A mitochondrial disorder. Clin. Cardiol. 2008, 31, 225–227. [Google Scholar] [CrossRef]
- Zhou, L.F.; Solhjoo, S.; Millare, B.; Plank, G.; Abraham, M.R.; Cortassa, S.; Trayanova, N.; O’Rourke, B. Effects of Regional Mitochondrial Depolarization on Electrical Propagation Implications for Arrhythmogenesis. Circ. Arrhythmia Elec. 2014, 7, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Majamaa-Voltti, K.; Peuhkurinen, K.; Kortelainen, M.L.; Hassinen, I.E.; Majamaa, K. Cardiac abnormalities in patients with mitochondrial DNA mutation 3243A>G. BMC Cardiovasc. Disord. 2002, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Lev, D.; Nissenkorn, A.; Leshinsky-Silver, E.; Sadeh, M.; Zeharia, A.; Garty, B.Z.; Blieden, L.; Barash, V.; Lerman-Sagie, T. Clinical presentations of mitochondrial cardiomyopathies. Pediatr. Cardiol. 2004, 25, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Pentiah, A.D. Mitochondrial cardiomyopathy and related arrhythmias. Card. Electrophysiol. Clin. 2015, 7, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.F.; Hu, H.C.; Lin, Y.; Huang, F.Z.; Ji, H.H.; Yin, L.; Lin, S.Y.; Chen, X.M.; Duan, S.W. Differences in Leukocyte Telomere Length between Coronary Heart Disease and Normal Population: A Multipopulation Meta-Analysis. BioMed Res. Int. 2019, 2019. [Google Scholar] [CrossRef]
- Andreassi, M.G. Coronary atherosclerosis and somatic mutations: An overview of the contributive factors for oxidative DNA damage. Mutat Res. Rev. Mutat 2003, 543, 67–86. [Google Scholar] [CrossRef]
- Mueller, E.E.; Brunner, S.M.; Mayr, J.A.; Stanger, O.; Sperl, W.; Kofler, B. Functional Differences between Mitochondrial Haplogroup T and Haplogroup H in HEK293 Cybrid Cells. PLoS ONE 2012, 7, e52367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kofler, B.; Mueller, E.E.; Eder, W.; Stanger, O.; Maier, R.; Weger, M.; Haas, A.; Winker, R.; Schmut, O.; Paulweber, B.; et al. Mitochondrial DNA haplogroup T is associated with coronary artery disease and diabetic retinopathy: A case control study. BMC Med. Genet. 2009, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.Y.; Sun, L.; Chen, X.P.; Zhang, D.S. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural. Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress and Mitochondrial Damage in Neurodegenerative Diseases: From Molecular Mechanisms to Targeted Therapies. Oxid. Med. Cell Longev. 2020, 2020. [Google Scholar] [CrossRef]
- Ait-Aissa, K.; Blaszak, S.C.; Beutner, G.; Tsaih, S.W.; Morgan, G.; Santos, J.H.; Flister, M.J.; Joyce, D.L.; Camara, A.K.S.; Gutterman, D.D.; et al. Mitochondrial Oxidative Phosphorylation defect in the Heart of Subjects with Coronary Artery Disease. Sci. Rep. 2019, 9, 7623. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Barinova, V.A.; Ryzhkova, A.I.; Zhelankin, A.V.; Postnov, A.Y.; Sobenin, I.A.; Bobryshev, Y.V.; Orekhov, A.N. Mosaicism of mitochondrial genetic variation in atherosclerotic lesions of the human aorta. Biomed. Res. Int. 2015, 2015, 825468. [Google Scholar] [CrossRef] [Green Version]
- Mitrofanov, K.Y.; Zhelankin, A.V.; Shiganova, G.M.; Sazonova, M.A.; Bobryshev, Y.V.; Postnov, A.Y.; Sobenin, I.A.; Orekhov, A.N. Analysis of mitochondrial DNA heteroplasmic mutations A1555G, C3256T, T3336C, C5178A, G12315A, G13513A, G14459A, G14846A and G15059A in CHD patients with the history of myocardial infarction. Exp. Mol. Pathol. 2016, 100, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L. Barth syndrome. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2013, 163C, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Mazurova, S.; Tesarova, M.; Magner, M.; Houstkova, H.; Hansikova, H.; Augustinova, J.; Tomek, V.; Vondrackova, A.; Zeman, J.; Honzik, T. Novel mutations in the TAZ gene in patients with Barth syndrome. Prague Med. Rep. 2013, 114, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Guleray, N.; Kosukcu, C.; Taskiran, Z.E.; Simsek Kiper, P.O.; Utine, G.E.; Gucer, S.; Tokatli, A.; Boduroglu, K.; Alikasifoglu, M. Atypical Presentation of Sengers Syndrome: A Novel Mutation Revealed with Postmortem Genetic Testing. Fetal Pediatric Pathol. 2020, 39, 163–171. [Google Scholar] [CrossRef]
- Sanchez-Caballero, L.; Elurbe, D.M.; Baertling, F.; Guerrero-Castillo, S.; van den Brand, M.; van Strien, J.; van Dam, T.J.P.; Rodenburg, R.; Brandt, U.; Huynen, M.A.; et al. TMEM70 functions in the assembly of complexes I and V. Biochim. Biophys. Acta. Bioenerg. 2020, 1861, 148202. [Google Scholar] [CrossRef]
- Cizkova, A.; Stranecky, V.; Mayr, J.A.; Tesarova, M.; Havlickova, V.; Paul, J.; Ivanek, R.; Kuss, A.W.; Hansikova, H.; Kaplanova, V.; et al. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat. Genet. 2008, 40, 1288–1290. [Google Scholar] [CrossRef]
- Cameron, J.M.; Levandovskiy, V.; Mackay, N.; Ackerley, C.; Chitayat, D.; Raiman, J.; Halliday, W.H.; Schulze, A.; Robinson, B.H. Complex V TMEM70 deficiency results in mitochondrial nucleoid disorganization. Mitochondrion 2011, 11, 191–199. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Ramirez, R.L.; Becker, A.B.; Bjork, S.T.; Levi, S.; Santambrogio, P.; Parsons, P.J.; Kruger, P.C.; Yang, K.X.; Feustel, P.J.; et al. The Pathogenesis of Cardiomyopathy in Friedreich Ataxia. PLoS ONE 2015, 10, 0116396. [Google Scholar] [CrossRef]
- Weidemann, F.; Stork, S.; Liu, D.; Hu, K.; Herrmann, S.; Ertl, G.; Niemann, M. Cardiomyopathy of Friedreich Ataxia. J. Neurochem. 2013, 126, 88–93. [Google Scholar] [CrossRef]
- Cook, A.; Giunti, P. Friedreich’s ataxia: Clinical features, pathogenesis and management. Brit. Med. Bull. 2017, 124, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Danhauser, K.; Haberberger, B.; Hoser, J.; Strecker, V.; Boehm, D.; Uziel, G.; Lamantea, E.; Invernizzi, F.; Poulton, J.; et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat. Genet. 2010, 42, 1131–1134. [Google Scholar] [CrossRef]
- Mayr, J.A.; Haack, T.B.; Graf, E.; Zimmermann, F.A.; Wieland, T.; Haberberger, B.; Superti-Furga, A.; Kirschner, J.; Steinmann, B.; Baumgartner, M.R.; et al. Lack of the Mitochondrial Protein Acylglycerol Kinase Causes Sengers Syndrome. Am. J. Hum. Genet. 2012, 90, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Carroll, C.J.; Isohanni, P.; Poyhonen, R.; Euro, L.; Richter, U.; Brilhante, V.; Gotz, A.; Lahtinen, T.; Paetau, A.; Pihko, H.; et al. Whole-exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J. Med. Genet. 2013, 50, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, M.; Krude, H.; Finckh, B.; Mayatepek, E.; Janssen, A.; Schmelz, M.; Trefz, F.; Trijbels, F.; Smeitink, J. Septo-optic dysplasia associated with a new mitochondrial cytochrome b mutation. Ann. Neurol. 2002, 51, 388–392. [Google Scholar] [CrossRef]
- Moslemi, A.R.; Darin, N.; Tulinius, M.; Wiklund, L.M.; Holme, E.; Oldfors, A. Progressive encephalopathy and complex I deficiency associated with mutations in MTND1. Neuropediatrics 2008, 39, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Song, Y.R.; Gu, S.L.; He, X.Y.; Zhu, X.Y.; Shen, Y.Y.; Wu, B.F.; Wang, W.; Li, S.S.; Jiang, P.P.; et al. Mitochondrial ND5 12338T > C variant is associated with maternally inherited hypertrophic cardiomyopathy in a Chinese pedigree. Gene 2012, 506, 339–343. [Google Scholar] [CrossRef]
- Li, D.; Sun, Y.P.; Zhuang, Q.Q.; Song, Y.R.; Wu, B.F.; Jia, Z.X.; Pan, H.Y.; Zhou, H.; Hu, S.Y.; Zhang, B.T.; et al. Mitochondrial dysfunction caused by m.2336T > C mutation with hypertrophic cardiomyopathy in cybrid cell lines. Mitochondrion 2019, 46, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Loeffen, J.; Elpeleg, O.; Smeitink, J.; Smeets, R.; Stockler-Ipsiroglu, S.; Mandel, H.; Sengers, R.; Trijbels, F.; van den Heuvel, L. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann. Neurol. 2001, 49, 195–201. [Google Scholar] [CrossRef]
- Cameron, J.M.; MacKay, N.; Feigenbaum, A.; Tarnopolsky, M.; Blaser, S.; Robinson, B.H.; Schulze, A. Exome sequencing identifies complex I NDUFV2 mutations as a novel cause of Leigh syndrome. Eur. J. Paediatr. Neuro. 2015, 19, 525–532. [Google Scholar] [CrossRef]
- Hoefs, S.J.G.; Dieteren, C.E.J.; Distelmaier, F.; Janssen, R.J.R.J.; Epplen, A.; Swarts, H.G.P.; Forkink, M.; Rodenburg, R.J.; Nijtmans, L.G.; Willems, P.H.; et al. NDUFA2 complex I mutation leads to Leigh disease. Am. J. Hum. Genet. 2008, 82, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Fassone, E.; Taanman, J.W.; Hargreaves, I.P.; Sebire, N.J.; Cleary, M.A.; Burch, M.; Rahman, S. Mutations in the mitochondrial complex I assembly factor NDUFAF1 cause fatal infantile hypertrophic cardiomyopathy. J. Med. Genet. 2011, 48, 691–697. [Google Scholar] [CrossRef]
- Hallas, T.; Eisen, B.; Shemer, Y.; Ben Jehuda, R.; Mekies, L.N.; Naor, S.; Schick, R.; Eliyahu, S.; Reiter, I.; Vlodavsky, E.; et al. Investigating the cardiac pathology of SCO2-mediated hypertrophic cardiomyopathy using patients induced pluripotent stem cell-derived cardiomyocytes. J. Cell. Mol. Med. 2018, 22, 913–925. [Google Scholar] [CrossRef] [Green Version]
- Piekutowska-Abramczuk, D.; Magner, M.; Popowska, E.; Pronicki, M.; Karczmarewicz, E.; Sykut-Cegielska, J.; Kmiec, T.; Jurkiewicz, E.; Szymanska-Debinska, T.; Bielecka, L.; et al. SURF1 missense mutations promote a mild Leigh phenotype. Clin. Genet. 2009, 76, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Courage, C.; Jackson, C.B.; Hahn, D.; Euro, L.; Nuoffer, J.M.; Gallati, S.; Schaller, A. SDHA Mutation with Dominant Transmission Results in Complex II Deficiency with Ocular, Cardiac, and Neurologic Involvement. Am. J. Med. Genet. A 2017, 173, 225–230. [Google Scholar] [CrossRef]
- Diodato, D.; Invernizzi, F.; Lamantea, E.; Fagiolari, G.; Parini, R.; Menni, F.; Parenti, G.; Bollani, L.; Pasquini, E.; Donati, M.A.; et al. Common and Novel TMEM70 Mutations in a Cohort of Italian Patients with Mitochondrial Encephalocardiomyopathy. JIMD Rep. 2015, 15, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, R.; Khayat, M.; Shalev, S.A.; Horovitz, Y.; Mandel, H.; Hershkovitz, E.; Barghuti, F.; Shaag, A.; Saada, A.; Korman, S.H.; et al. TMEM70 mutations are a common cause of nuclear encoded ATP synthase assembly defect: Further delineation of a new syndrome. J. Med. Genet. 2011, 48, 177–182. [Google Scholar] [CrossRef]
- Smeitink, J.A.M.; Elpeleg, O.; Antonicka, H.; Diepstra, H.; Saada, A.; Smits, P.; Sasarman, F.; Vriend, G.; Jacob-Hirsch, J.; Shaag, A.; et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am. J. Hum. Genet. 2006, 79, 869–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Guo, Y.; Huang, M.R.; Zhang, Z.; Zhu, J.X.; Liu, T.L.; Shi, L.; Li, F.; Huang, H.M.; Fu, L.J. Identification of TAZ mutations in pediatric patients with cardiomyopathy by targeted next-generation sequencing in a Chinese cohort. Orphanet J. Rare Dis. 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Perli, E.; Pisano, A.; Glasgow, R.I.C.; Carbo, M.; Hardy, S.A.; Falkous, G.; He, L.; Cerbelli, B.; Pignataro, M.G.; Zacara, E.; et al. Novel compound mutations in the mitochondrial translation elongation factor (TSFM) gene cause severe cardiomyopathy with myocardial fibro-adipose replacement. Sci. Rep. 2019, 9, 5108. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Finsterer, J.; Kothari, S. Cardiac manifestations of primary mitochondrial disorders. Int. J. Cardiol. 2014, 177, 754–763. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Koolen, D.; Smeitink, J.A.; van den Heuvel, L.; Rodenburg, R.J. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J. Inherit. Metab. Dis. 2015, 38, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Morava, E.; van den Heuvel, L.; Hol, F.; de Vries, M.C.; Hogeveen, M.; Rodenburg, R.J.; Smeitink, J.A.M. Mitochondrial disease criteria—Diagnostic applications in children. Neurology 2006, 67, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Bernier, F.P.; Boneh, A.; Dennett, X.; Chow, C.W.; Cleary, M.A.; Thorburn, D.R. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002, 59, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, G.; Steinberg, C.; Dubois, M.; Senechal, M. What the Cardiologist Should Know About Mitochondrial Cardiomyopathy? Can. J. Cardiol. 2019, 35, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.H.G.; Margeta, M. Educational Case: Mitochondrial Myopathy. Acad Pathol. 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, M.C.; Grady, J.P.; Grunewald, A.; Vincent, A.; Dobson, P.F.; Taylor, R.W.; Turnbull, D.M.; Rygiel, K.A. A novel immunofluorescent assay to investigate oxidative phosphorylation deficiency in mitochondrial myopathy: Understanding mechanisms and improving diagnosis. Sci Rep. Uk 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, R.; Onoue, N.; Rikimaru, H.; Tsukita, K.; Fujita, H.; Yamaguchi, N.; Ishizuka, T.; Suzuki, Y.; Suzuki, H.; Shinozaki, T. Mitochondrial Cardiomyopathy with a Unique (99m)Tc-MIBI/(123)I-BMIPP Mismatch Pattern. Intern. Med. 2017, 56, 321–325. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Florian, A.; Ludwig, A.; Stubbe-Drager, B.; Boentert, M.; Young, P.; Waltenberger, J.; Rosch, S.; Sechtem, U.; Yilmaz, A. Characteristic cardiac phenotypes are detected by cardiovascular magnetic resonance in patients with different clinical phenotypes and genotypes of mitochondrial myopathy. J. Cardiovasc. Magn. Reson. Off. J. Soc. Cardiovasc. Magn. Reson. 2015, 17, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delvecchio, M.; Salzano, G.; Bonura, C.; Cauvin, V.; Cherubini, V.; d’Annunzio, G.; Franzese, A.; Giglio, S.; Grasso, V.; Graziani, V.; et al. Can HbA1c combined with fasting plasma glucose help to assess priority for GCK-MODY vs HNF1A-MODY genetic testing? Acta Diabetol. 2018, 55, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef] [Green Version]
- Chinault, A.C.; Shaw, C.A.; Brundage, E.K.; Tang, L.Y.; Wong, L.J. Application of dual-genome oligonucleotide array-based comparative genomic hybridization to the molecular diagnosis of mitochondrial DNA deletion and depletion syndromes. Genet. Med. Off. J. Am. Coll. Med. Genet. 2009, 11, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhan, H.L.; Li, F.Y.; Pursley, A.N.; Schmitt, E.S.; Wong, L.J. Targeted array CGH as a valuable molecular diagnostic approach: Experience in the diagnosis of mitochondrial and metabolic disorders. Mol. Genet. Metab. 2012, 106, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.J.; Dimmock, D.; Geraghty, M.T.; Quan, R.; Lichter-Konecki, U.; Wang, J.; Brundage, E.K.; Scaglia, F.; Chinault, A.C. Utility of oligonucleotide array-based comparative genomic hybridization for detection of target gene deletions. Clin. Chem. 2008, 54, 1141–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinwiddie, D.L.; Smith, L.D.; Miller, N.A.; Atherton, A.M.; Farrow, E.G.; Strenk, M.E.; Soden, S.E.; Saunders, C.J.; Kingsmore, S.F. Diagnosis of mitochondrial disorders by concomitant next-generation sequencing of the exome and mitochondrial genome. Genomics 2013, 102, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, S.H. Targeted next-generation sequencing expands the spectrum of mitochondrial disorders. Genome Med. 2012, 4, 22. [Google Scholar] [CrossRef]
- Liu, Z.; Fang, F.; Ding, C.; Zhang, W.; Li, J.; Yang, X.; Wang, X.; Wu, Y.; Wang, H.; Liu, L.; et al. [Diagnosis of mitochondrial disorders in children with next generation sequencing]. Zhonghua Er Ke Za Zhi Chin. J. Pediatr. 2015, 53, 747–753. [Google Scholar]
- Zhang, W.; Cui, H.; Wong, L. Development and Validation of Next Generation Sequencing for Clinical Molecular Diagnosis of Mitochondrial Disorders. J. Mol. Diagn. 2012, 14, 637–638. [Google Scholar]
- Zhang, V.W.; Cui, H.; Wong, L.J. Implementation of next generation sequencing for clinical molecular diagnosis of mitochondrial disorders. Mitochondrion 2012, 12, 555–556. [Google Scholar] [CrossRef]
- Vasta, V.; Ng, S.B.; Turner, E.H.; Shendure, J.; Hahn, S.H. Next generation sequence analysis for mitochondrial disorders. Genome Med. 2009, 1. [Google Scholar] [CrossRef] [Green Version]
- Matthijs, G.; Souche, E.; Alders, M.; Corveleyn, A.; Eck, S.; Feenstra, I.; Race, V.; Sistermans, E.; Sturm, M.; Weiss, M.; et al. Guidelines for diagnostic next-generation sequencing. Eur. J. Hum. Genet. 2016, 24, 1515. [Google Scholar] [CrossRef]
- Tang, S.; Wang, J.; Zhang, V.W.; Li, F.Y.; Landsverk, M.; Cui, H.; Truong, C.K.; Wang, G.; Chen, L.C.; Graham, B.; et al. Transition to next generation analysis of the whole mitochondrial genome: A summary of molecular defects. Hum. Mutat 2013, 34, 882–893. [Google Scholar] [CrossRef]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucinska-Wieckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosinska, J.; et al. New perspective in diagnostics of mitochondrial disorders: Two years’ experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med. 2016, 14, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, B.; D’Argenio, V.; Monda, E.; Vitale, A.; Caiazza, M.; Sacchetti, L.; Pastore, L.; Limongelli, G.; Frisso, G.; Mazzaccara, C. Genetic analysis resolves differential diagnosis of a familial syndromic dilated cardiomyopathy: A new case of Alstrom syndrome. Mol. Genet. Genom. Med. 2020, 8, e1260. [Google Scholar] [CrossRef]
- Frisso, G.; Detta, N.; Coppola, P.; Mazzaccara, C.; Pricolo, M.R.; D’Onofrio, A.; Limongelli, G.; Calabro, R.; Salvatore, F. Functional Studies and In Silico Analyses to Evaluate Non-Coding Variants in Inherited Cardiomyopathies. Int. J. Mol. Sci. 2016, 17, 1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suay-Corredera, C.; Pricolo, M.R.; Herrero-Galán, E.; Velázquez-Carreras, D.; Sánchez-Ortiz, D.; García-Giustiniani, D.; Delgado, J.; Galano-Frutos, J.J.; García-Cebollada, H.; Vilches, S.; et al. Protein haploinsufficiency drivers identify MYBPC3 mutations that cause hypertrophic cardiomyopathy. MedRxiv 2020. [Google Scholar] [CrossRef]
- Girolami, F.; Frisso, G.; Benelli, M.; Crotti, L.; Iascone, M.; Mango, R.; Mazzaccara, C.; Pilichou, K.; Arbustini, E.; Tomberli, B.; et al. Contemporary genetic testing in inherited cardiac disease: Tools, ethical issues, and clinical applications. J. Cardiovasc. Med. 2018, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Correia, S.P.; Moedas, M.F.; Naess, K.; Bruhn, H.; Maffezzini, C.; Calvo-Garrido, J.; Lesko, N.; Wibom, R.; Schober, F.A.; Jemt, A.; et al. Severe congenital lactic acidosis and hypertrophic cardiomyopathy caused by an intronic variant in NDUFB7. Hum. Mutat. 2021. [Google Scholar] [CrossRef]
- Chung, H.; Kim, Y.; Cho, S.M.; Lee, H.J.; Park, C.H.; Kim, J.Y.; Lee, S.H.; Min, P.K.; Yoon, Y.W.; Lee, B.K.; et al. Differential contributions of sarcomere and mitochondria-related multigene variants to the endophenotype of hypertrophic cardiomyopathy. Mitochondrion 2020, 53, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Chau, E.M.C.; Ma, E.S.K.; Chan, A.O.O.; Tsio, T.H.; Law, W.L. Mitochondrial cardiomyopathy due to m.3243A > G mitochondrial DNA mutation presenting in late adulthood: A case report. Hong Kong Med. J. 2020, 26, 240–242. [Google Scholar] [CrossRef]
- Kamps, R.; Szklarczyk, R.; Theunissen, T.E.; Hellebrekers, D.; Sallevelt, S.; Boesten, I.B.; de Koning, B.; van den Bosch, B.J.; Salomons, G.S.; Simas-Mendes, M.; et al. Genetic defects in mtDNA-encoded protein translation cause pediatric, mitochondrial cardiomyopathy with early-onset brain disease. Eur. J. Hum. Genet. 2018, 26, 537–551. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Olahova, M.; Kishita, Y.; Garone, C.; Kremer, L.S.; Yagi, M.; Uchiumi, T.; Jourdain, A.A.; Thompson, K.; D’Souza, A.R.; et al. Biallelic C1QBP Mutations Cause Severe Neonatal-, Childhood-, or Later-Onset Cardiomyopathy Associated with Combined Respiratory-Chain Deficiencies. Am. J. Hum. Genet. 2017, 101, 525–538. [Google Scholar] [CrossRef]
- Haack, T.B.; Kopajtich, R.; Freisinger, P.; Wieland, T.; Rorbach, J.; Nicholls, T.J.; Baruffini, E.; Walther, A.; Danhauser, K.; Zimmermann, F.A.; et al. ELAC2 Mutations Cause a Mitochondrial RNA Processing Defect Associated with Hypertrophic Cardiomyopathy. Am. J. Hum. Genet. 2013, 93, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Malfatti, E.; Laforet, P.; Jardel, C.; Stojkovic, T.; Behin, A.; Eymard, B.; Lombes, A.; Benmalek, A.; Becane, H.M.; Berber, N.; et al. High risk of severe cardiac adverse events in patients with mitochondrial m.3243A > G mutation. Neurology 2013, 80, 100–105. [Google Scholar] [CrossRef]
- Hollingsworth, K.G.; Gorman, G.S.; Trenell, M.I.; McFarland, R.; Taylor, R.W.; Turnbull, D.M.; MacGowan, G.A.; Blamire, A.M.; Chinnery, P.F. Cardiomyopathy is common in patients with the mitochondrial DNA m.3243A > G mutation and correlates with mutation load. Neuromuscul. Disord. 2012, 22, 592–596. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, P.; Engelstad, K.; Wei, Y.; Kulikova, R.; Oskoui, M.; Sproule, D.M.; Battista, V.; Koenigsberger, D.Y.; Pascual, J.M.; Shanske, S.; et al. Natural history of MELAS associated with mitochondrial DNA m.3243A > G genotype. Neurology 2011, 77, 1965–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozieres, C.; Quillasi, V.; Mouly-Bertin, C.; Thomson, V.; Gachon-Lanier, E.; Lantelme, P. [Diabetes and hypokinetic cardiopathy: When to consider mitochondrial disease?]. Ann. De Cardiol. Et D’angeiologie 2011, 60, 176–178. [Google Scholar] [CrossRef]
- Yajima, N.; Yazaki, Y.; Yoshida, K.; Sano, K.; Takahashi, W.; Sasaki, Y.; Ikeda, U. A case of mitochondrial cardiomyopathy with pericardial effusion evaluated by Tc-99m-MIBI myocardial scintigraphy. J. Nucl. Cardiol. 2009, 16, 989–994. [Google Scholar] [CrossRef]
- Majamaa-Voltti, K.A.M.; Winqvist, S.; Remes, A.M.; Tolonen, U.; Pyhtinen, J.; Uimonen, S.; Karppa, M.; Sorri, M.; Peuhkurinen, K.; Majamaa, K. A 3-year clinical follow-up of adult patients with 3243A > G in mitochondrial DNA. Neurology 2006, 66, 1470–1475. [Google Scholar] [CrossRef]
- Chae, J.H.; Hwang, H.; Lim, B.C.; Cheong, H.I.; Hwang, Y.S.; Kim, K.J. Clinical features of A3243G mitochondrial tRNA mutation. Brain Dev. 2004, 26, 459–462. [Google Scholar] [CrossRef]
- Holmgren, D.; Wahlander, H.; Eriksson, B.O.; Oldfors, A.; Holme, E.; Tulinius, M. Cardiomyopathy in children with mitochondrial disease—Clinical course and cardiological findings. Eur. Heart J. 2003, 24, 280–288. [Google Scholar] [CrossRef]
- Momiyama, Y.; Furutani, M.; Suzuki, Y.; Ohmori, R.; Imamura, S.; Mokubo, A.; Asahina, T.; Murata, C.; Kato, K.; Anazawa, S.; et al. A mitochondrial DNA variant associated with left ventricular hypertrophy in diabetes. Biochem. Bioph Res. Co 2003, 312, 858–864. [Google Scholar] [CrossRef]
- Shin, W.S.; Tanaka, M.; Suzuki, J.; Hemmi, C.; Toyo-oka, T. A novel homoplasmic mutation in mtDNA with a single evolutionary origin as a risk factor for cardiomyopathy. Am. J. Hum. Genet. 2000, 67, 1617–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okajima, Y.; Tanabe, Y.; Takayanagi, M.; Aotsuka, H. A follow up study of myocardial involvement in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS). Heart 1998, 80, 292–295. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Howell, N.; Lightowlers, R.N.; Turnbull, D.M. Molecular pathology of MELAS and MERRF - The relationship between mutation load and clinical phenotypes. Brain 1997, 120, 1713–1721. [Google Scholar] [CrossRef] [Green Version]
- Anan, R.; Nakagawa, M.; Miyata, M.; Higuchi, I.; Nakao, S.; Suehara, M.; Osame, M.; Tanaka, H. Cardiac Involvement in Mitochondrial Diseases—A Study on 17 Patients with Documented Mitochondrial-DNA Defects. Circulation 1995, 91, 955–961. [Google Scholar] [CrossRef]
- Guenthard, J.; Wyler, F.; Fowler, B.; Baumgartner, R. Cardiomyopathy in Respiratory-Chain Disorders. Arch. Dis. Child. 1995, 72, 223–226. [Google Scholar] [CrossRef]
- Ciafaloni, E.; Ricci, E.; Shanske, S.; Moraes, C.T.; Silvestri, G.; Hirano, M.; Simonetti, S.; Angelini, C.; Donati, M.A.; Garcia, C.; et al. Melas—Clinical-Features, Biochemistry, and Molecular-Genetics. Ann. Neurol. 1992, 31, 391–398. [Google Scholar] [CrossRef]
- Zeviani, M.; Gellera, C.; Antozzi, C.; Rimoldi, M.; Morandi, L.; Villani, F.; Tiranti, V.; Didonato, S. Maternally Inherited Myopathy and Cardiomyopathy—Association with Mutation in Mitochondrial-DNA Transfer Rnaleu(Uur). Lancet 1991, 338, 143–147. [Google Scholar] [CrossRef]
- Ozawa, T.; Tanaka, M.; Sugiyama, S.; Hattori, K.; Ito, T.; Ohno, K.; Takahashi, A.; Sato, W.; Takada, G.; Mayumi, B.; et al. Multiple Mitochondrial-DNA Deletions Exist in Cardiomyocytes of Patients with Hypertrophic or Dilated Cardiomyopathy. Biochem. Bioph. Res. Commun. 1990, 170, 830–836. [Google Scholar] [CrossRef]
- Thompson, K.; Collier, J.J.; Glasgow, R.I.C.; Robertson, F.M.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Olahova, M.; McFarland, R.; Taylor, R.W. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J. Inherit. Metab. Dis. 2020, 43, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Bayona-Bafaluy, M.P.; Iglesias, E.; Lopez-Gallardo, E.; Emperador, S.; Pacheu-Grau, D.; Labarta, L.; Montoya, J.; Ruiz-Pesini, E. Genetic aspects of the oxidative phosphorylation dysfunction in dilated cardiomyopathy. Mutat. Res. 2020, 786, 108334. [Google Scholar] [CrossRef]
- Rafiq, M.A.; Chaudhry, A.; Care, M.; Spears, D.A.; Morel, C.F.; Hamilton, R.M. Whole exome sequencing identified 1 base pair novel deletion in BCL2-associated athanogene 3 (BAG3) gene associated with severe dilated cardiomyopathy (DCM) requiring heart transplant in multiple family members. Am. J. Med. Genet. Part. A 2017, 173, 699–705. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danese, E.; Raimondi, S.; Montagnana, M.; Tagetti, A.; Langaee, T.; Borgiani, P.; Ciccacci, C.; Carcas, A.J.; Borobia, A.M.; Tong, H.Y.; et al. Effect of CYP4F2, VKORC1, and CYP2C9 in Influencing Coumarin Dose: A Single-Patient Data Meta-Analysis in More Than 15,000 Individuals. Clin. Pharmacol. Ther. 2019, 105, 1477–1491. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, B.; Izzo, V.; Terracciano, D.; Ranieri, A.; Mazzaccara, C.; Fimiani, F.; Cesaro, A.; Gentile, L.; Leggiero, E.; Pero, R.; et al. Laboratory medicine: Health evaluation in elite athletes. Clin. Chem. Lab. Med. 2019, 57, 1450–1473. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Redi, A.; Lemme, E.; Pelliccia, A.; Salvatore, F.; Frisso, G. Impact of molecular diagnostics in an asymptomatic amateur athlete found to be affected by hypertrophic cardiomyopathy. Med. Sport 2018, 71, 405–412. [Google Scholar] [CrossRef]
- Barretta, F.; Mirra, B.; Monda, E.; Caiazza, M.; Lombardo, B.; Tinto, N.; Scudiero, O.; Frisso, G.; Mazzaccara, C. The Hidden Fragility in the Heart of the Athletes: A Review of Genetic Biomarkers. Int. J. Mol. Sci. 2020, 21, 6682. [Google Scholar] [CrossRef]
- Mazzaccara, C.; D’Argenio, V.; Nunziato, M.; Esposito, M.V.; Salvatore, F.; Frisso, G. Clinical molecular biology in the assessment and prevention of cardiological risk in case of participation in sports activity and intense physical activity. Biochim. Clin. 2019, 43, 24–43. [Google Scholar] [CrossRef]
- Limongelli, G.; Nunziato, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Mazzaccara, C.; Caiazza, M.; D’Aponte, A.; D’Andrea, A.; Bossone, E.; et al. Yield and clinical significance of genetic screening in elite and amateur athletes. Eur. J. Prev. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Nunziato, M.; Mazzaccara, C.; Intrieri, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Di Maggio, F.; Frisso, G.; Salvatore, F. Genotype-Phenotype Correlation: A Triple DNA Mutational Event in a Boy Entering Sport Conveys an Additional Pathogenicity Risk. Genes 2020, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Pricolo, M.R.; Herrero-Galan, E.; Mazzaccara, C.; Losi, M.A.; Alegre-Cebollada, J.; Frisso, G. Protein Thermodynamic Destabilization in the Assessment of Pathogenicity of a Variant of Uncertain Significance in Cardiac Myosin Binding Protein C. J. Cardiovasc. Transl. Res. 2020. [Google Scholar] [CrossRef] [PubMed]

| Authors | Type of Study | Aim of the Study | Inclusion Criteria | RESULTS | Ref. | ||
|---|---|---|---|---|---|---|---|
| N. of Patients | N. of Patients with Cardiac Phenotype | N. of Patients with Positive Genotype § and MCM | |||||
| SP. Correia et al. 2021 | CR | To investigate a patient with congenital lactic acidosis and HCM | Patients with HCM, lactic acidosis | 1 | 1 | 1 | [151] |
| H. Chung et al. 2020 | RS | To investigate phenotype-based clinical and genetic characteristics of HCM patients using comprehensive genetic tests and rare variant association analysis | Patients with HCM | 212 | 212 | 41 | [152] |
| EMC. Chau et al. 2020 | CR | To investigate MCM | MD phenotype | 1 | 1 | 1 | [153] |
| A. Imai-Okazaki et al. 2019 | RS | To investigate CM in children with MD | MD genotype | 137 | 29 | 29 | [48] |
| R. Kamps et al. 2018 | CR | To identify gene defects in pediatric CM and early-onset brain disease with OXPHOS deficiencies | Patients with CM | 3 | 3 | 3 | [154] |
| RG. Feichtinger et al. 2017 | CR | To investigate by NGS four unrelated patients | Patients with combined RCD | 4 | 4 | 4 | [155] |
| K. Wahbi et al. 2015 | RS | To assess the long-term cardiac prognosis of patients with MD | MD genotype | 260 | 108 | 108 | [23] |
| TB. Haack et al. 2013 | CR | To investigate by NGS three unrelated family | Patients with HCM, lactic acidosis, isolated complex I deficiency on muscle biopsy | 5 | 5 | 5 | [156] |
| E. Malfatti et al. 2013 | RS | To determine the long-term incidence of cardiac life-threatening complications and death | MT-TL1 m.3243A>G | 41 | 10 | 10 | [157] |
| KG. Hollingsworth et al. 2012 | CCS | To determine whether cardiac complications in patients with MD are sufficiently common to warrant prospective screening in all mutation carriers | MT-TL1 m.3243A>G | 10 | 10 | 10 | [158] |
| Z. Liu et al. 2012 | CR | To investigate clinical, genetic and molecular characterization of a family with a likely maternally transmitted HCM | Patients with HCM | 7 | 4 | 4 | [106] |
| P. Kaufmann et al. 2011 | PS | To describe clinical and laboratory features associated with mtDNA mutation in 35 unrelated family | MT-TL1 m.3243A>G | 85 | 4 | 4 | [159] |
| C. Nozières et al. 2011 | CR | To investigate a patient with diabetes and hypokinetic cardiomyopathy | MD-phenotype | 1 | 1 | 1 | [160] |
| G. Limongelli et al. 2010 | RS | To determine the frequency and natural history of cardiac disease in patients with primary respiratory chain disease | RCD phenotype | 32 | 26 | 16 | [53] |
| K. Wahbi et al. 2010 | RS | To determine the prevalence and prognostic value of cardiac abnormalities in unrelated families with mtDNA mutation | MT-TK m.8344A>G | 18 | 9 | 9 | [60] |
| N. Yajima et al. 2009 | CR | To investigate a patient with pericardial effusion and heart failure in whom MCM was definitively diagnosed | Patient with LVH | 1 | 1 | 1 | [161] |
| KA. Majamaa-Voltti et al. 2006 | RS | To follow the clinical course of patients with the MD | MT-TL1 m.3243A>G | 33 | 18 | 18 | [162] |
| JH. Chae et al. 2004 | RS | To evaluate the incidence and clinical heterogeneity of m.3243A>G mutation in Korean population | MD phenotype | 85 | 2 | 0* | [163] |
| D. Holmgren et al. 2003 | RS | To determine the frequency of CM in children with MD and describe their clinical course, prognosis and cardiological manifestations | Genotype or phenotype of MD | 101 | 17 | 4 | [164] |
| Y. Momiyama et al. 2003 | CCS | To investigate the prevalence of several mtDNA mutations in diabetic patients with LVH | Patients with type 2 DM with or without LVH | 168 | 68 | 38 | [165] |
| F. Terasaki et al. 2001 | CR | To investigate CM showing progression from the HCM to DCM | HCM phenotype | 1 | 1 | 1 | [68] |
| WS. Shin et al. 2000 | CCS | To clarify the relationship between variation in mtDNA and the development of CM | 2 brothers with maternally inherited CM, 126 HCM and 55 DCM patients | 183 | 6 | 6 | [166] |
| Y. Okajima et al. 1998 | PS | To investigate cardiac function in children with MELAS and clarify the clinical features of CM | MELAS phenotype | 11 | 9 | 9 | [167] |
| PF. Chinnery et al. 1997 | RS | To evaluate the relationship between the incidence of specific clinical features and the level of mutant mtDNA in blood and/or skeletal muscle | MT-TL1 m.3243A>G and or MT-TK m.8344A>G | 245 | 3 | 3 | [168] |
| R. Anan et al. 1995 | RS | To determine the cardiac involvement in MD | MDs phenotype | 17 | 10 | 10 | [169] |
| J. Guenthard et al. 1995 | CR | To evaluate the CM in a patient with RCD | RCD phenotype | 1 | 1 | 0 | [170] |
| E. Ciafaloni et al. 1992 | CCS | To determine the relationship of m.3243A>G mutation to the MELAS phenotype | MELAS phenotype | 23 | 4 | 4 | [171] |
| M. Zeviani et al. 1991 | CR | To identify a maternally inherited disorder characterized by CM | MD phenotype | 21 | 5 | 5 | [172] |
| T. Ozawa et al. 1990 | CR | To identify mitochondrial abnormalities in CM | Cardiac tissue specimens from patients with CM | 5 | 5 | 3 | [173] |
| Function | Gene OMIM ID | Gene | Transcript ID | Length bps | Protein | Inheritance |
|---|---|---|---|---|---|---|
| 612035 | AARS2 | ENST00000244571.5 | 4798 | Alanyl-tRNA synthetase 2 | AR | |
| 611103 | ACAD9 | ENST00000308982.12 | 2445 | Acyl-CoA dehydrogenase family member 9 | AR | |
| 609575 | ACADVL | ENST00000356839.10 | 2184 | Acyl-CoA dehydrogenase very long chain | AR | |
| 610345 | AGK | ENST00000649286.2 | 3628 | Acylglycerol kinase | AR | |
| 613183 | BOLA3 | ENST00000327428.10 | 555 | BOLA family member 3 | AR | |
| 606158 | BSCL2 | ENST00000360796.10 | 1710 | BSCL2 lipid droplet biogenesis associated, seipin | AR | |
| 614775 | COA3 | ENST00000328434.8 | 780 | Cytochrome C oxidase assembly factor 3 | AR | |
| 613920 | COA5 | ENST00000328709.8 | 1770 | Cytochrome C oxidase assembly factor 5 | AR | |
| 614772 | COA6 | ENST00000366615.10 | 1741 | Cytochrome C oxidase assembly factor 6 | AR | |
| 609825 | COQ2 | ENST00000647002.2 | 1525 | Coenzyme Q2, polyprenyltransferase | AR | |
| 612898 | COQ4 | ENST00000300452.8 | 1245 | Coenzyme Q4 | AD/AR | |
| 614647 | COQ6 | ENST00000334571.7 | 2109 | Coenzyme Q6, monooxygenase | AR | |
| 601683 | COQ7 | ENST00000321998.10 | 2642 | Coenzyme Q7, hydroxylase | AR | |
| 606980 | COQ8A | ENST00000366777.4 | 2866 | Coenzyme Q8A | AR | |
| 612837 | COQ9 | ENST00000262507.11 | 1630 | Coenzyme Q9 | AR | |
| 602125 | COX10 | ENST00000261643.8 | 2898 | Cytochrome C oxidase assembly factor COX10 | AR | |
| 614478 | COX14 | ENST00000550487.6 | 484 | Cytochrome C oxidase assembly factor COX14 | AR | |
| 603646 | COX15 | ENST00000016171.6 | 5030 | Cytochrome C oxidase assembly homolog COX15 | AR | |
| 614698 | COX20 | ENST00000411948.7 | 2295 | Cytochrome C oxidase assembly factor COX20 | AR | |
| 124089 | COX6B1 | ENST0000649813.2 | 488 | Cytochrome C oxidase subunit 6B1 | AR | |
| 601269 | C1QBP | ENST00000225698.8 | 1169 | Complement component C1q-binding protein | AR | |
| 600650 | CPT2 | ENST00000371486.4 | 2699 | Carnitine palmitoyltransferase 2 | AD/AR | |
| 608977 | DNAJC19 | ENST00000382564.8 | 1416 | DNAJ heat shock protein family (Hsp40) member C19 | AR | |
| 602462 | DRP-1 | ENST00000324989.12 | 3174 | Collapsin response mediator protein 1 | - | |
| 602292 | ECHS1 | ENST00000368547.4 | 1277 | Enoyl-CoA hydratase, short chain 1 | AR | |
| 605367 | ELAC2 | ENST00000338034.9 | 3767 | ELAC ribonuclease Z 2 | AR | |
| 608253 | ETFA | ENST00000557943.6 | 2289 | Electron transfer flavoprotein subunit alpha | AR | |
| 130410 | ETFB | ENST00000309244.9 | 872 | Electron transfer flavoprotein subunit beta | AR | |
| 231675 | ETFDH | ENST00000511912.6 | 3111 | Electron transfer flavoprotein dehydrogenase | AR | |
| 609003 | FIS1 | ENST00000223136.5 | 870 | Fission, mitochondrial 1 | - | |
| 613622 | FOXRED1 | ENST00000263578.10 | 1951 | FAD dependent oxidoreductase domain containing protein 1 | AR | |
| 606829 | FXN | ENST00000484259.3 | 6978 | Frataxin | AR | |
| 608536 | GTPBP3 | ENST00000324894.13 | 2566 | GTP binding protein 3 | AR | |
| 600890 | HADHA | ENST00000380649.8 | 2943 | Hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha | AR | |
| 143450 | HADHB | ENST00000317799.10 | 1997 | Hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit beta | AR | |
| 601421 | KARS1 | ENST00000302445.8 | 1991 | Lysyl-tRNA synthetase 1 | AR | |
| 607544 | LRPPRC | ENST00000260665.12 | 6603 | Leucine rich pentatricopeptide repeat containing | AR | |
| 608419 | MCEE | ENST00000244217.6 | 824 | Methylmalonyl-CoA epimerase | AR | |
| 608506 | MFN1 | ENST00000471841.6 | 5212 | Mitofusin 1 | - | |
| 608507 | MFN2 | ENST00000235329.10 | 4407 | Mitofusin 2 | - | |
| 607481 | MMAA | ENST00000649156.2 | 5944 | Metabolism of cobalamin associated A | AR | |
| 607568 | MMAB | ENST00000545712.7 | 4090 | Metabolism of cobalamin associated B | AR | |
| 609831 | MMACHC | ENST00000401061.9 | 5049 | Metabolism of cobalamin associated C | AR | |
| 611935 | MMADHC | ENST00000303319.10 | 1392 | Metabolism of cobalamin associated D | AR | |
| 609058 | MMUT | ENST00000274813.4 | 3811 | Methylmalonyl-CoA mutase | AR | |
| 607118 | MRPL3 | ENST00000264995.8 | 1708 | Mitochondrial ribosomal protein L3 | AR | |
| 611849 | MRPL44 | ENST00000258383.4 | 1689 | Mitochondrial ribosomal protein L44 | AR | |
| 605810 | MRPS22 | ENST00000680020.1 | 1205 | Mitochondrial ribosomal protein S22 | AR | |
| 614667 | MTO1 | ENST00000498286.6 | 10698 | Mitochondrial tRNA translation optimization 1 | AR | |
| 605299 | NCOA6 | ENST00000359003.7 | 7083 | Nuclear receptor coactivator 6 | - | |
| 300078 | NDUFA1 | ENST00000371437.5 | 421 | NADH:Ubiquinone oxidoreductase subunit A1 | XLR | |
| 603835 | NDUFA10 | ENST00000252711.7 | 4855 | NADH:Ubiquinone oxidoreductase subunit A10 | AR | |
| 612638 | NDUFA11 | ENST00000308961.5 | 575 | NADH:Ubiquinone oxidoreductase subunit A11 | AR | |
| 602137 | NDUFA2 | ENST00000252102.9 | 649 | NADH:Ubiquinone oxidoreductase subunit A2 | AR | |
| 606934 | NDUFAF1 | ENST00000260361.9 | 1509 | NADH:Ubiquinone oxidoreductase complex assembly factor 1 | AR | |
| 609653 | NDUFAF2 | ENST00000296597.10 | 632 | NADH:Ubiquinone oxidoreductase complex assembly factor 2 | AR | |
| 612911 | NDUFAF3 | ENST00000326925.11 | 902 | NADH:Ubiquinone oxidoreductase complex assembly factor 3 | AR | |
| 611776 | NDUFAF4 | ENST00000316149.8 | 2407 | NADH:Ubiquinone oxidoreductase complex assembly factor 4 | AR | |
| 612360 | NDUFAF5 | ENST00000378106.10 | 5449 | NADH:Ubiquinone oxidoreductase complex assembly factor 5 | AR | |
| 603839 | NDUFB3 | ENST00000237889.9 | 493 | NADH:Ubiquinone oxidoreductase subunit B3 | AR | |
| 603842 | NDUFB7 | ENST00000215565.3 | 535 | NADH:Ubiquinone oxidoreductase subunit B7 | - | |
| 601445 | NDUFB9 | ENST00000276689.8 | 691 | NADH:Ubiquinone oxidoreductase subunit B9 | AR | |
| 157655 | NDUFS1 | ENST00000233190.11 | 11660 | NADH:Ubiquinone oxidoreductase core subunit S1 | AR | |
| 602985 | NDUFS2 | ENST00000676972.1 | 1613 | NADH:Ubiquinone oxidoreductase core subunit S2 | AR | |
| 603846 | NDUFS3 | ENST00000263774.9 | 894 | NADH:Ubiquinone oxidoreductase core subunit S3 | AR | |
| 602694 | NDUFS4 | ENST00000296684.10 | 669 | NADH:Ubiquinone oxidoreductase core subunit S4 | AR | |
| 603848 | NDUFS6 | ENST00000274137.10 | 518 | NADH:Ubiquinone oxidoreductase core subunit S6 | AR | |
| 601825 | NDUFS7 | ENST00000233627.14 | 758 | NADH:Ubiquinone oxidoreductase core subunit S7 | AR | |
| 602141 | NDUFS8 | ENST00000313468.10 | 737 | NADH:Ubiquinone oxidoreductase core subunit S8 | AR | |
| 161015 | NDUFV1 | ENST00000322776.11 | 1560 | NADH:Ubiquinone oxidoreductase core subunit V1 | AR | |
| 600532 | NDUFV2 | ENST00000318388.11 | 857 | NADH:Ubiquinone oxidoreductase core subunit V2 | AR | |
| 608100 | NFU1 | ENST00000410022.7 | 898 | NFU1 iron-sulfur cluster scaffold | AR | |
| 139139 | NR4A1 | ENST00000394825.6 | 2485 | Nuclear receptor subfamily 4 group A member 1 | - | |
| 613621 | NUBPL | ENST00000281081.12 | 3040 | Nucleotide binding protein like | AR | |
| 605290 | OPA1 | ENST00000361510.8 | 6429 | OPA1, mitochondrial dynamin like GTPase | AD/AR | |
| 612036 | PARS2 | ENST00000371279.4 | 2356 | Prolyl-tRNA synthetase 2 | AR | |
| 232000 | PCCA | ENST00000376285.6 | 2484 | Propionyl-CoA carboxylase subunit alpha | AR | |
| 232050 | PCCB | ENST00000251654.9 | 1799 | Propionyl-CoA carboxylase subunit beta | AR | |
| 607429 | PDSS1 | ENST00000376215.10 | 1584 | Prenyl diphosphate synthase subunit 1 | AR | |
| 610564 | PDSS2 | ENST00000369037.9 | 3536 | Prenyl diphosphate synthase subunit 2 | AR | |
| 174763 | POLG | ENST00000268124.11 | 4462 | Polymerase, DNA, gamma | AD/AR | |
| 617209 | QRSL1 | ENST00000369046.8 | 4106 | Glutaminyl-tRNA amidotransferase subunit QRSL1 | AR | |
| 179710 | RCC1 | ENST00000373833.10 | 2844 | Regulator of chromosome condensation 1 | - | |
| 614917 | RMND1 | ENST00000444024.3 | 1948 | Required for meiotic nuclear division 1 HOMOLOG | AR | |
| 603644 | SCO1 | ENST00000255390.10 | 9577 | Synthesis of Cytochrome C oxidase 1 | AR | |
| 604272 | SCO2 | ENST00000395693.8 | 984 | SCO2, Cytochrome C oxidase assembly protein | AR | |
| 600857 | SDHA | ENST00000264932.11 | 2693 | Succinate dehydrogenase complex flavoprotein subunit A | AR | |
| 613019 | SDHAF2 | ENST00000301761.7 | 1186 | Succinate dehydrogenase complex assembly factor 2 | - | |
| 602690 | SDHD | ENST00000375549.7 | 1439 | Succinate dehydrogenase complex subunit D | AR | |
| 603941 | SLC19A2 | ENST00000236137.10 | 3612 | Solute carrier family 19 member 2 | AR | |
| 603377 | SLC22A5 | ENST00000245407.8 | 3277 | Solute carrier family 22 member 5 | AR | |
| 600370 | SLC25A3 | ENST00000228318.8 | 5987 | Solute carrier family 25 member 3 | AR | |
| 103220 | SLC25A4 | ENST00000281456.11 | 4415 | Solute carrier family 25 member 4 | AD/AR | |
| 613698 | SLC25A20 | ENST00000319017.5 | 1778 | Solute carrier family 25 member 20 | AR | |
| 185620 | SURF1 | ENST00000371974.8 | 1092 | SURF1, Cytochrome C oxidase assembly factor | AR | |
| 300394 | TAZ | ENST00000601016.6 | 1906 | Tafazzin | XLR | |
| 612418 | TMEM70 | ENST00000312184.6 | 2032 | Transmembrane protein 70 | AR | |
| 601243 | TOP3A | ENST00000321105.10 | 6596 | Topoisomerase, DNA, III, alpha | AR | |
| 611023 | TRMT5 | ENST00000261249.7 | 5304 | tRNA methyltransferase 5 | AR | |
| 604723 | TSFM | ENST00000652027.2 | 1997 | Ts translation elongation factor, mitochondrial | AR | |
| 602389 | TUFM | ENST00000313511.8 | 2011 | Tu translation elongation factor, mitochondrial | AR | |
| 131222 | TYMP | ENST00000252029.8 | 1586 | Thymidine phosphorylase | AR | |
| 610957 | YARS2 | ENST00000324868.13 | 2117 | Tyrosyl-tRNA synthetase 2 | AR | |
| 516060 | MT-ATP6 | ENST00000361899.2 | 681 | Mitochondrially encoded ATP synthase membrane subunit 6 | M | |
| 516070 | MT-ATP8 | ENST00000361851.1 | 207 | Mitochondrially encoded ATP synthase membrane subunit 8 | M | |
| 516030 | MT-CO1 | ENST00000361624.2 | 1542 | Mitochondrially encoded Cytochrome C oxidase I | M | |
| 516040 | MT-CO2 | ENST00000361739.1 | 684 | Mitochondrially encoded Cytochrome C oxidase II | M | |
| 516050 | MT-CO3 | ENST00000362079.2 | 784 | Mitochondrially encoded Cytochrome C oxidase III | M | |
| 516020 | MT-CYB | ENST00000361789.2 | 1141 | Mitochondrially encoded cytochrome b | M | |
| 516000 | MT-ND1 | ENST00000361390.2 | 956 | Mitochondrially encoded NADH-ubiquinone oxidoreductase core subunit 1 | M | |
| 516001 | MT-ND2 | ENST00000361453.3 | 1042 | Mitochondrially encoded NADH-Ubiquinone oxidoreductase core subunit 2 | M | |
| 516003 | MT-ND4 | ENST00000361381.2 | 1378 | Mitochondrially encoded NADH-Ubiquinone oxidoreductase core subunit 4 | M | |
| 516005 | MT-ND5 | ENST00000361567.2 | 1812 | Mitochondrially encoded NADH-Ubiquinone oxidoreductase core subunit 5 | M | |
| 516006 | MT-ND6 | ENST00000361681.2 | 525 | Mitochondrially encoded NADH-Ubiquinone oxidoreductase core subunit 6 | M | |
| 561000 | MT-RNR1 | ENST00000389680.2 | 954 | Mitochondrially encoded 12S rRNA | M | |
| 561010 | MT-RNR2 | ENST00000387347.2 | 1559 | Mitochondrially encoded 16S rRNA | M | |
| 590025 | MT-TE | ENST00000387459.1 | 69 | Mitochondrially encoded tRNA glutamic acid | M | |
| 590070 | MT-TF | ENST00000387314.1 | 71 | Mitochondrially encoded tRNA phenylalanine | M | |
| 590035 | MT-TG | ENST00000387429.1 | 68 | Mitochondrially encoded tRNA glycine | M | |
| 590040 | MT-TH | ENST00000387441.1 | 69 | Mitochondrially encoded tRNA histidine | M | |
| 590045 | MT-TI | ENST00000387365.1 | 69 | Mitochondrially encoded tRNA isoleucine | M | |
| 590060 | MT-TK | ENST00000387421.1 | 70 | Mitochondrially encoded tRNA lysine | M | |
| 590050 | MT-TL1 | ENST00000386347.1 | 75 | Mitochondrially encoded tRNA leucine 1 | M | |
| 590055 | MT-TL2 | ENST00000387456.1 | 71 | Mitochondrially encoded tRNA leucine 2 | M | |
| 590010 | MT-TN | ENST00000387400.1 | 73 | Mitochondrially encoded tRNA asparagine | M | |
| 590075 | MT-TP | ENST00000387461.2 | 68 | Mitochondrially encoded tRNA proline | M | |
| 590005 | MT-TR | ENST00000387439.1 | 65 | Mitochondrially encoded tRNA arginine | M | |
| 590090 | MT-TT | ENST00000387460.2 | 66 | Mitochondrially encoded tRNA threonine | M | |
| 590105 | MT-TV | ENST00000387342.1 | 69 | Mitochondrially encoded tRNA valine | M | |
| 590095 | MT-TW | ENST00000387382.1 | 68 | Mitochondrially encoded tRNA tryptophan | M |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzaccara, C.; Mirra, B.; Barretta, F.; Caiazza, M.; Lombardo, B.; Scudiero, O.; Tinto, N.; Limongelli, G.; Frisso, G. Molecular Epidemiology of Mitochondrial Cardiomyopathy: A Search Among Mitochondrial and Nuclear Genes. Int. J. Mol. Sci. 2021, 22, 5742. https://doi.org/10.3390/ijms22115742
Mazzaccara C, Mirra B, Barretta F, Caiazza M, Lombardo B, Scudiero O, Tinto N, Limongelli G, Frisso G. Molecular Epidemiology of Mitochondrial Cardiomyopathy: A Search Among Mitochondrial and Nuclear Genes. International Journal of Molecular Sciences. 2021; 22(11):5742. https://doi.org/10.3390/ijms22115742
Chicago/Turabian StyleMazzaccara, Cristina, Bruno Mirra, Ferdinando Barretta, Martina Caiazza, Barbara Lombardo, Olga Scudiero, Nadia Tinto, Giuseppe Limongelli, and Giulia Frisso. 2021. "Molecular Epidemiology of Mitochondrial Cardiomyopathy: A Search Among Mitochondrial and Nuclear Genes" International Journal of Molecular Sciences 22, no. 11: 5742. https://doi.org/10.3390/ijms22115742
APA StyleMazzaccara, C., Mirra, B., Barretta, F., Caiazza, M., Lombardo, B., Scudiero, O., Tinto, N., Limongelli, G., & Frisso, G. (2021). Molecular Epidemiology of Mitochondrial Cardiomyopathy: A Search Among Mitochondrial and Nuclear Genes. International Journal of Molecular Sciences, 22(11), 5742. https://doi.org/10.3390/ijms22115742