1. Introduction
Connexins (Cxs) are transmembrane proteins expressed on almost all mammalian cells. In humans there are 21 connexins named according to their molecular weight that range between 26 and 60 kDa. They are involved in cell-cell and cell-extracellular medium communication, allowing the exchange of small bioactive molecules such as ions, nucleotides, amino acids, sugars, intracellular mediators, and miRNAs. They play such a function by forming canals which are the hemichannels and the gap junction’s channels. Moreover, connexins can participate in various signal transduction pathways that interact individually with intracellular proteins [
1,
2,
3].
The most expressed connexins in the normal adult heart are Cx26, Cx40, Cx43 and Cx45 [
4]. The last three Cxs are mainly involved in forming gap junction structures and are generally located at the intercalated discs, allowing the propagation of electrical activity throughout the heart. This is one of the major reasons why their dysfunction has been linked to a wide variety of cardiac diseases [
5]. In particular, the isotype Cx43 is the most studied and widely expressed as well as the most represented in cardiac myocyte gap junctions [
6]. Cx26 expression has recently been found in mammalian cardiomyocytes by our group and, unlike the other cardiac connexins, it is distributed not in the intercalated discs but in the cytoplasm at the level of mitochondria, myofibrils, and cytoplasmic vesicles [
7]. We demonstrated that the cardiac Cx26 protein is modulated early during aging [
8] but until now, nothing is known about its function or its behaviour in cardiac diseases.
Tyrosine kinases inhibitors (TKIs) are drugs capable of specifically targeting tyrosine kinases receptors that frequently become abnormally hyper-activated in several human cancers. Ponatinib (Iclusig
TM, Wilmignton, DE, USA) is a new orally active multi-tyrosine kinase inhibitor currently approved for patients with chronic myeloid leukaemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukaemia. It specifically targets native and mutated BCR-ABL tyrosine kinases. Moreover, a broader potential of ponatinib in the treatment of tumor diseases can be considered, since its inhibiting action has been demonstrated on other tyrosine kinases in other human malignancies also [
9].
However, despite the impressive power of ponatinib in inhibiting various cancers, the drug is associated with vascular and cardiac toxicity with evidence of congestive heart failure and electrocardiographic abnormalities [
10]. Several mechanisms have been hypothesized to explain the ponatinib-induced toxicity profile, i.e., the simultaneous inhibition of cardiovascular-related kinases [
11], but further studies are needed to fully characterize the ponatinib signaling pathway. It is of interest to understand why the extent of the ponatinib-induced cardiotoxicity observed among the patients is uneven [
12], and whether it is sex-related.
The Notch1 signaling influences multiple cell processes including differentiation, proliferation, apoptosis, migration, and angiogenesis. Notch1 has been shown to have both tumor suppressive and tumorigenic functions in different contexts [
12]. Interestingly, it has been recently shown that a selective blockade of Notch1 can prevent vascular toxicity induced by ponatinib in human aortic endothelial cells [
13]. Therefore, ponatinib treatment could specifically activate Notch signaling on tumour cells and this might represent the “on-target effect” on the tumor.
Against this background, the aim of the present study was to further explore the signaling pathway of ponatinib in a murine model of ponatinib-induced cardiotoxicity, focusing our interest on the connexins. We assessed the involvement of Cx43, Cx26, and some related molecules in ponatinib-induced cardiac toxicity especially considering the Notch1 signaling pathway and sex-related differences.
3. Discussion
Among cardiac Cxs, Cx26 and Cx43 are the ones that are widely expressed throughout the heart. Cx26 is present in vessels, as well as in working and conducting cardiomyocytes, and its localization is scattered all over the cell [
7]. Having been found in cardiomyocytes only recently, its role in cardiac function and disease remains poorly understood. Cx43 is expressed in vessels, stroma cells and working cardiomyocytes. In cardiomyocytes, it is mainly localized at the level of the intercalated discs where it forms gap junctions and, carrying on its canonical function, it allows the propagation of electrical activity throughout the heart. Moreover, it can form hemichannels at intercalated discs, at membrane lateral borders and at mitochondrial membranes. Hemichannels regulate extracellular communications, especially allowing the dissemination of disease. Cx43 is also involved in various signalling transduction pathways that interact individually with intracellular proteins [
1,
2,
3,
18,
19]. Cx43 can undergo some posttranslational modifications, including phosphorylation. Its phosphorylation, which can occur at various specific serine residues, regulates channel assembly/disassembly, localization, and gap junction/hemichannel permeability. Consequently, this regulation will affect cardiac disease development. Several protein kinases are implicated in Cx43 phosphorylation, such as PKC, ERK, and Akt [
14]. Cx43 phosphorylation by ERK and PKC is typically increased during gap junction remodelling and disassembly while Akt activity increases with the gap junction size. Together, they can protect the myocardium from ischemia reperfusion injury. ERK and Akt both phosphorylate Cx43, and, at the same time, they can be controlled themselves by the phosphorylation status of Cx43, in response to ischemic injury. It has been demonstrated that a decreased Cx43 phosphorylation at serine 368 increases arrhythmia occurrence in cultured cardiac myocytes and in vivo in animal models. However, the outcome of Cx43 phosphorylation depends on the type of the kinase, the cell target, and the cellular context [
19].
In the present study, we evaluated the modulation of Cx43, Cx26 and some of their related partners in female and male hearts of a murine model of cardiotoxicity. In particular, we explored a possible Notch1/Cxs signaling pathway triggered by ponatinib, a new multi-tyrosine kinase inhibitor that has been successfully used against human malignancies which, however, causes cardiac toxicity. The results that have been obtained can be summarized as follows. Firstly, analyses of the cardiac function showed a different sex-related susceptibility to ponatinib treatment, higher in males than in females, as demonstrated by the greater cardiac dysfunctions in male rather than in female mice. Moreover, the observed revertant action of siRNA-Notch1 cotreatment evidenced that ponatinib exerts its cardiotoxic effects through the Notch1 pathway, both in females and males. In addition, regarding Cxs, we found that ponatinib influenced the protein expression of both Cx43 and Cx26 as well as related molecules, often through the Notch1 signaling pathway and in a sex-related mode. The results that have been obtained are interesting for their novelty, for their support of previous observations, and for future research inspirations.
More specifically, the results have demonstrated:
(1) A higher expression of cardiac Cx43, pS368-Cx43 and Cx26 in female mice rather than in male mice under basal conditions. Interestingly, the fact that there was a higher amount of Cx43 in female control groups than in their male counterparts is in agreement with previous studies and demonstrates that this sex-dependent difference in Cx43 expression under basal conditions was preserved during aging. Indeed, the mice in our study were very old and were older than the animals used in previous works [
20,
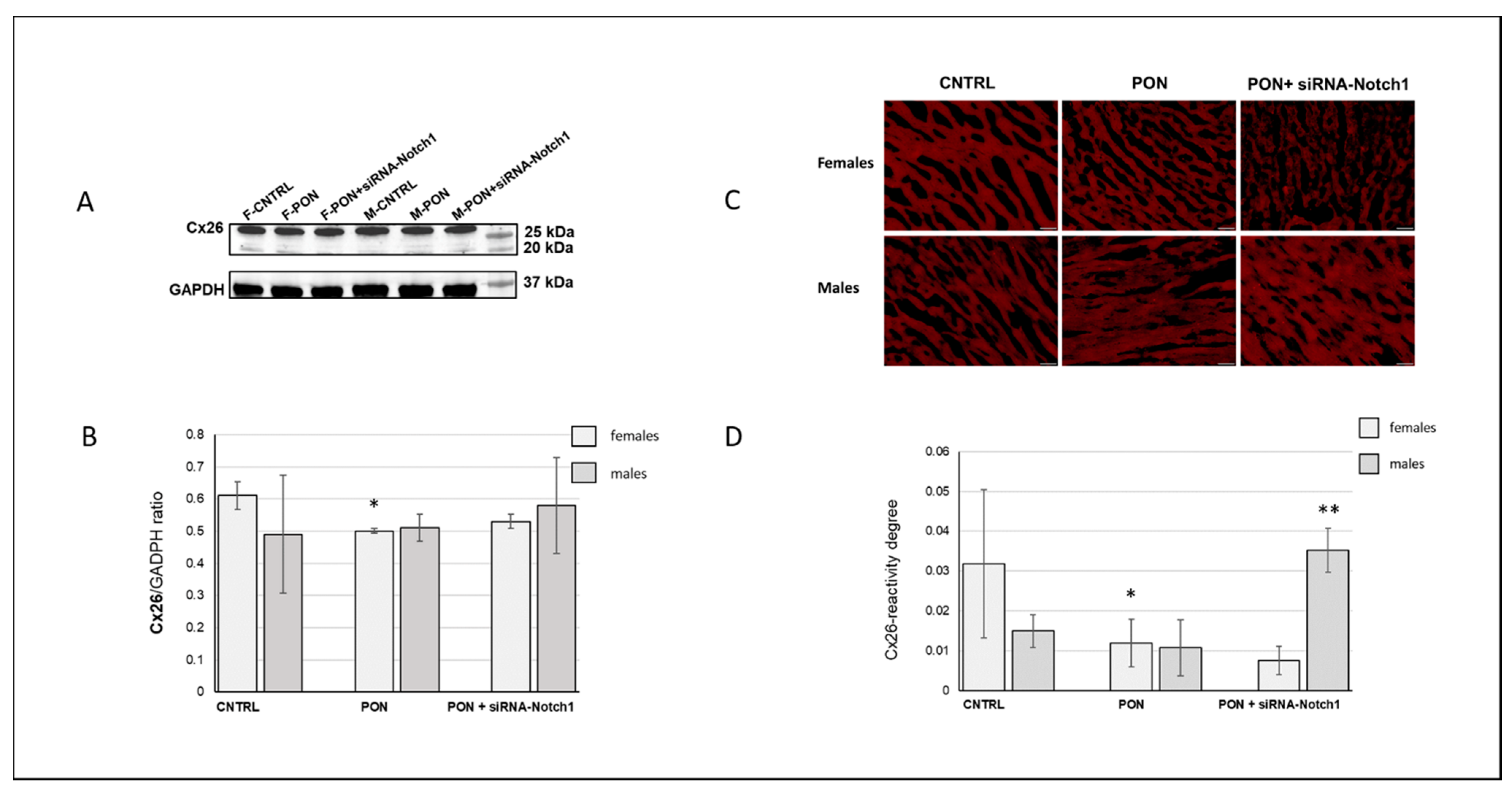
21]. On the other hand, the higher basal cardiac Cx26 expression in female mice compared to male mice has been demonstrated in the present study for the first time. In this regard, it would be interesting to investigate if the sex-dependent Cx expression can be extended to the other cardiac Cxs isotypes.
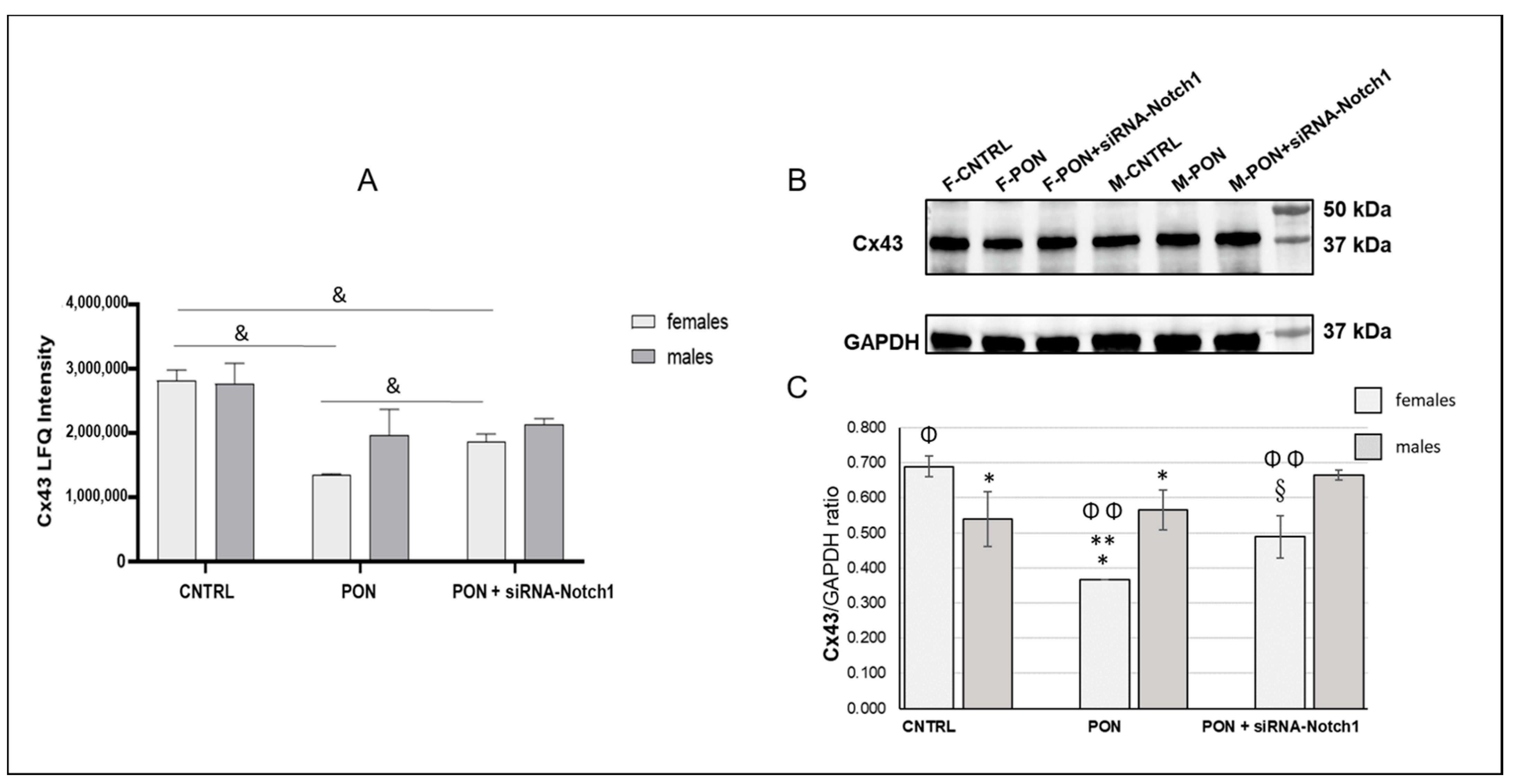
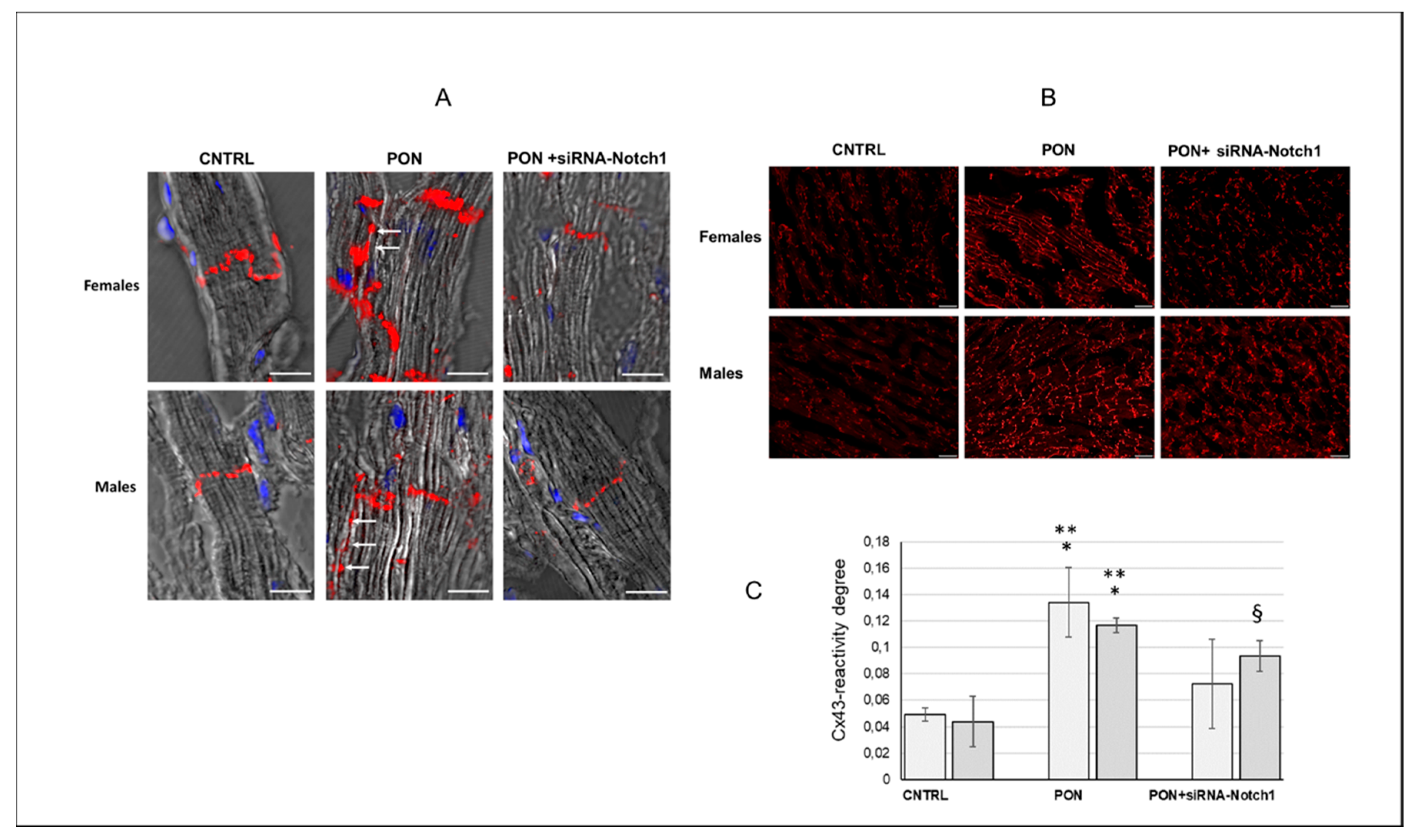
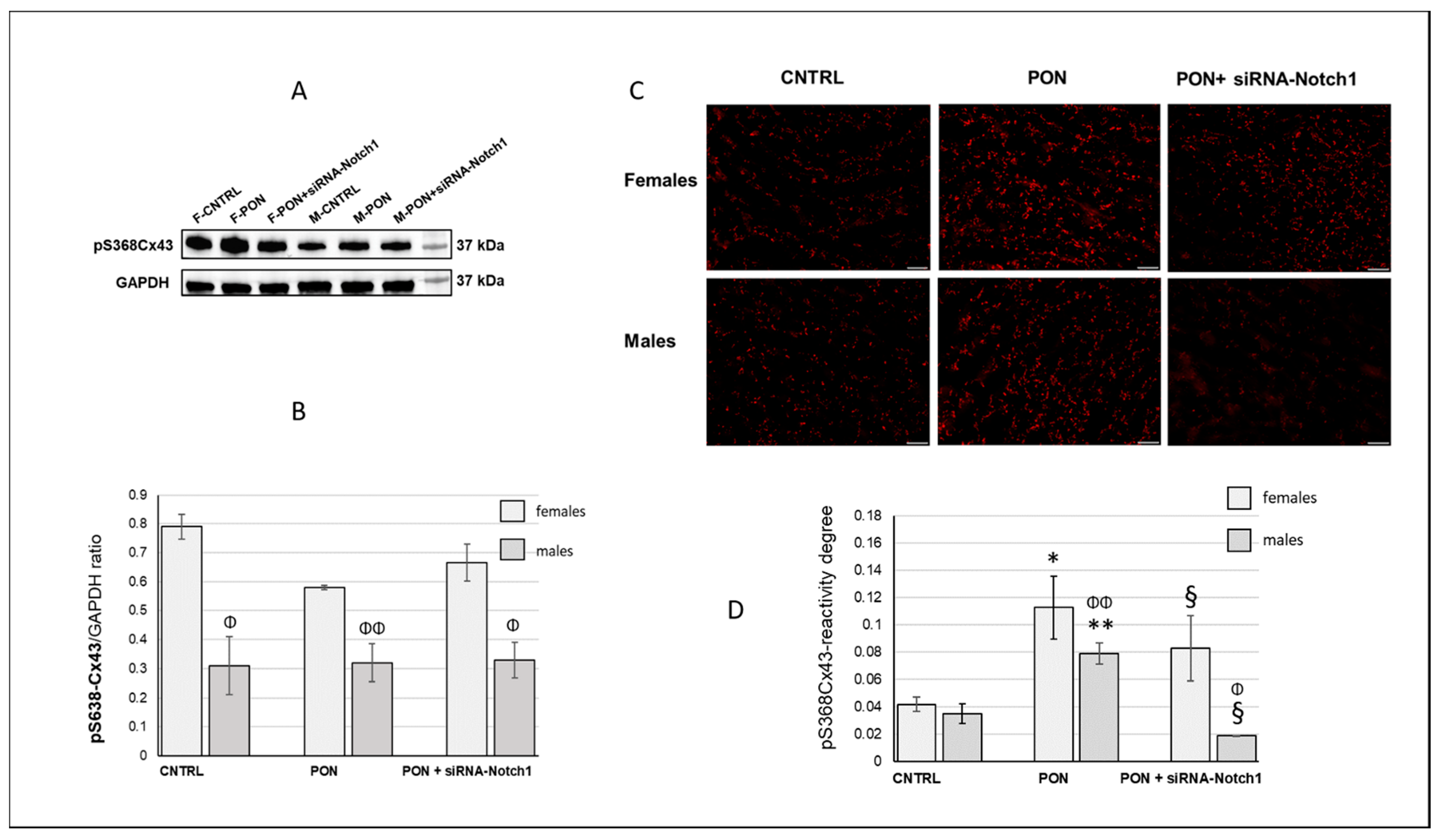
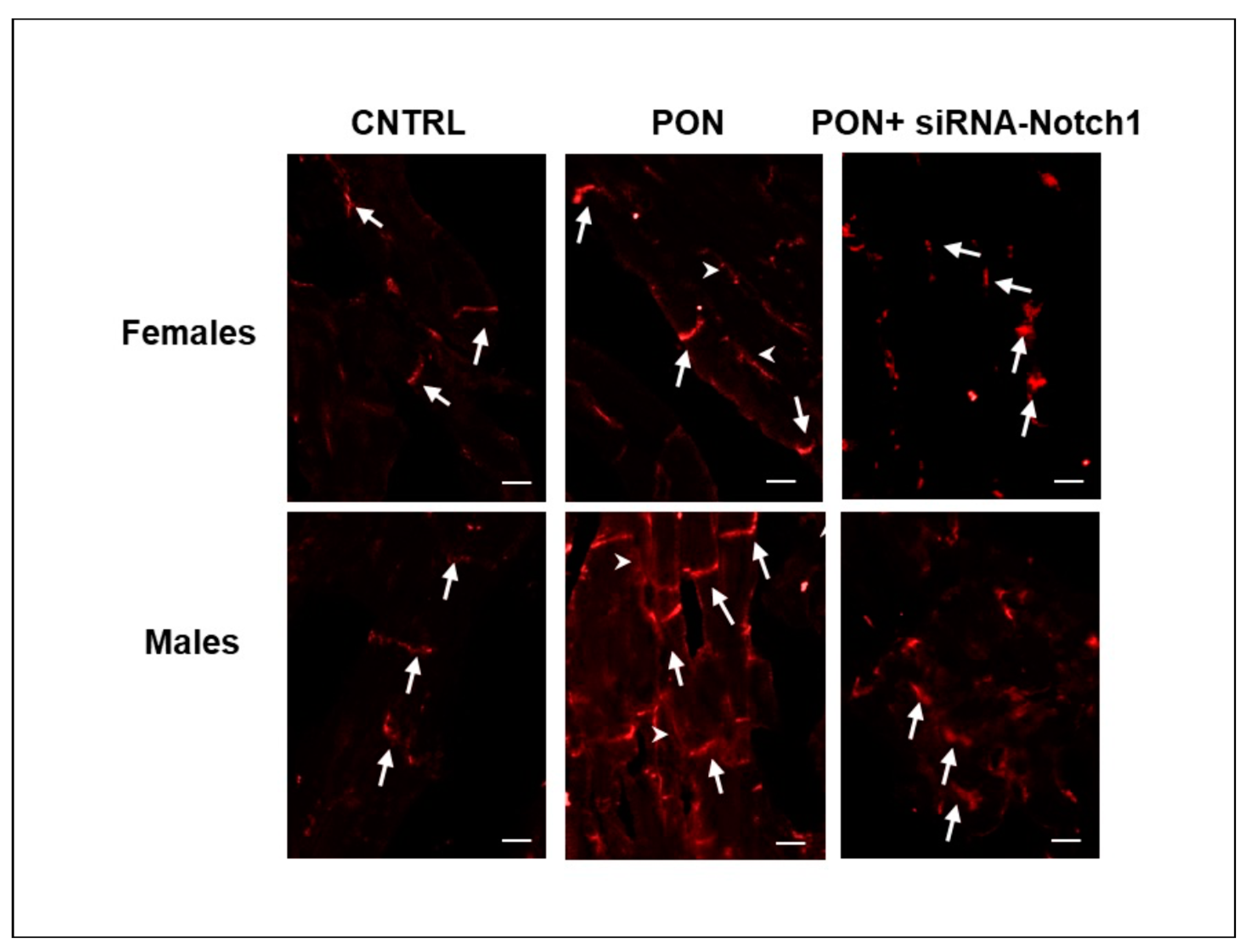
(2) The cardiotoxic effect of ponatinib influenced Cx43 and Cx26 expression, often in a sex-related mode. Indeed, in ponatinib treated groups compared to controls, Cx43 and its S368 phosphorylated form increased at the level of the membrane cardiomyocytes and exhibited a disordered distribution. Again, the increase in Cx43 and pS368Cx43 was higher in females than in males. An improved expression of Cx43 and its pS368-phosphorylated form as well as their disordered distribution has been found previously in myocarditis and it has been suggested that this could favour a cardioprotective phenotype, especially in females [
20,
21,
22,
23]. Ponatinib affected cardiomyocyte Cx26 expression differently as it induced its decrease, but only in females. However, the amounts of total cardiac and cardiomyocyte Cx26 were always higher in females than in males both for controls and for ponatinib treated groups. It has to be noted that the Cx26 expression was often highly variable in the samples from the same group, as shown by the large error bars (
Figure 7B,D). Consequently, Cx26 expression could be influenced also by factors on a case-by-case basis. Therefore, further studies will be necessary to ascertain the real involvement of Cx26 in heart injury. Due to the poor literature data on cardiac Cx26, this result is very important as it demonstrates a modulation of Cx26 expression in the response to heart injury.
In conclusion, the lower susceptibility to ponatinib shown by females could be due, at least in part, to Cx regulation which appeared always present or more marked in females than males.
(3) The results on the amounts of Cx43 and pS368-Cx43, which were obtained from whole heart samples, were different from those obtained from the cardiomyocytes analysis performed by immunofluorescence microscopy. Indeed, total cardiac Cx43, in a different way from cardiomyocyte Cx43, decreased after ponatinib treatment in females while it did not change in males. The same was shown for total cardiac pS368-Cx43 that appeared to not be influenced by ponatinib treatment either in females or in males. Instead, total cardiac Cx26 decreased after ponatinib treatment, in a similar way to cardiomyocytes Cx26 in females. The differences between total cardiac and cardiomyocyte Cx43 expression as well as those between female and male Cx43 modulation could be due to the presence of this connexin in vessels and stromal cells too. Thus, ponatinib could influence Cx43 both by reducing its expression in cardiac stroma cells and enhancing its expression in cardiomyocyte membranes, however in a more marked manner in females. The same can happen to pS368-Cx43, as ponatinib could stimulate Cx43 phosphorylation mainly at the level of intercalated discs and to a minor extent at the level of vessels and stroma cells, compared to controls, and always in more marked way for females. The observed increase in PKC activity at the intercalated discs in the cardiomyocytes of mice treated with ponatinib compared to controls could corroborate this assumption. However, to better characterize this interpretation, further studies are needed to examine the specific Cx modulation also in cardiac vessels and stromal cells.
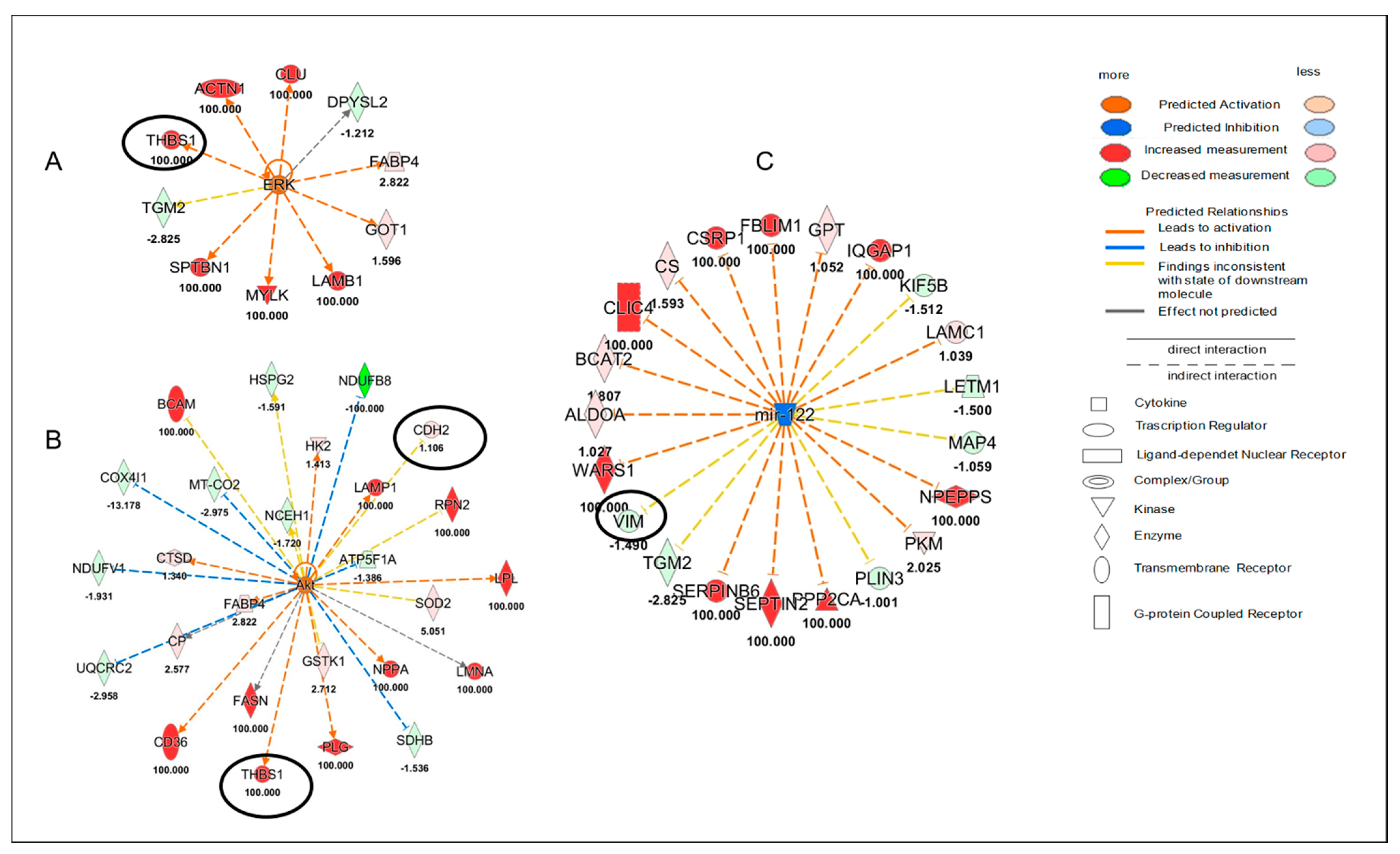
(4) Ponatinib action was sex-dependent concerning Akt, ERK and miR-122 molecules that were modulated after ponatinib treatment but only in female hearts. Akt and ERK were upregulated, while miR-122 was downregulated. This miRNA appears to be directly implicated in the development of cardiovascular diseases and its inhibition plays antifibrotic, anti-apoptotic, anti-inflammatory, and antioxidant functions [
16]. MiR-122 could be linked to Cxs for their own involvement in miRNA intercellular transfer [
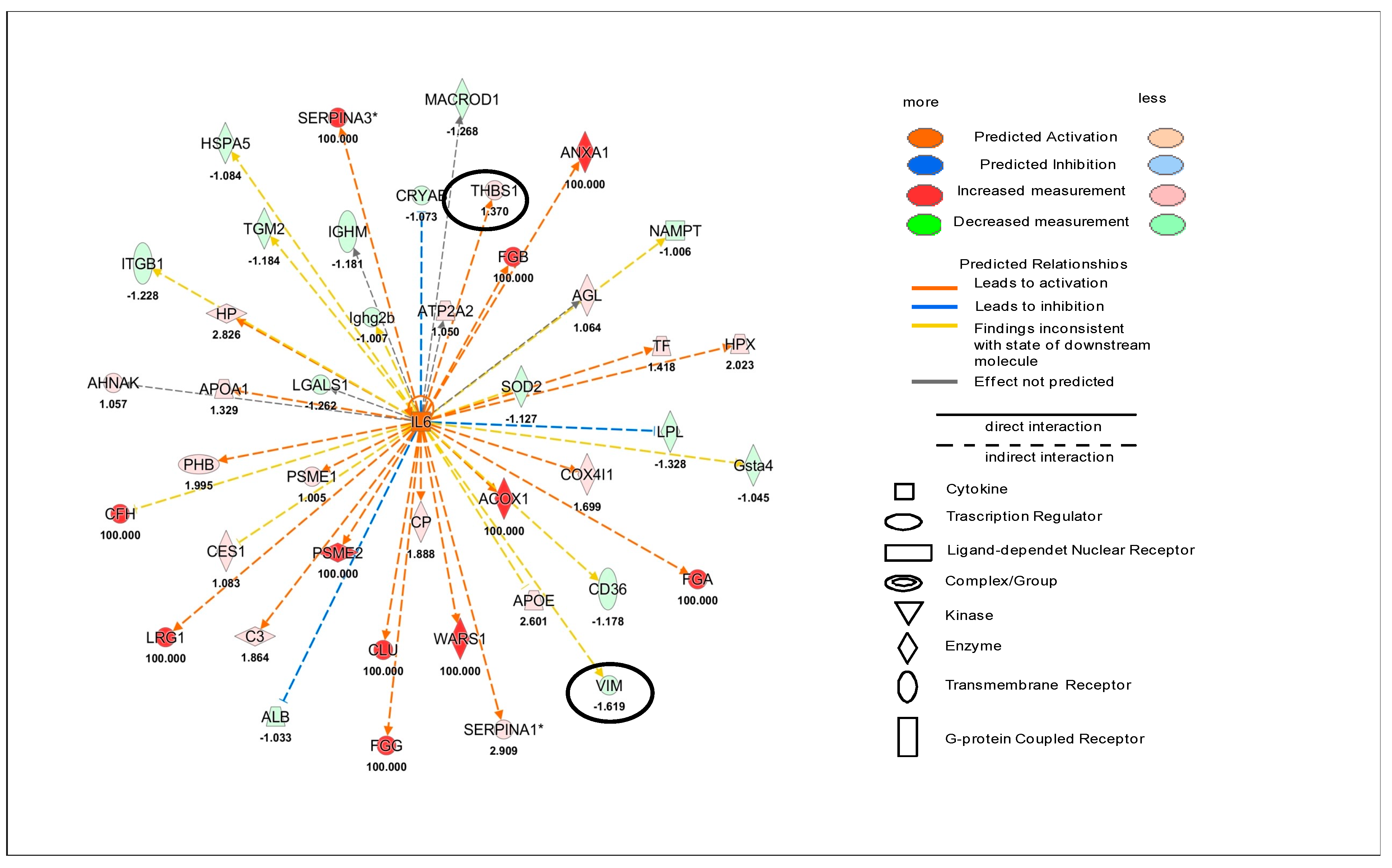
15]. Thus, we can speculate that the observed reduction in cardiac Cx43 and Cx26 in female hearts could be, in part, responsible for miR-122 down regulation. Akt and ERK could regulate Cx43 phosphorylation and consequently modulate its expression in an independent way at the level of various heart components. The proteomic analysis revealed that the regulation of Akt, ERK and miR-122 indirectly involved Cx43. Indeed, among the several proteins regulating them, there were VIM, CDH2 and THBS1, which were identified as Cx43 target molecules. However, THBS1 was only significantly upregulated and this happened in females only. Interestingly, it has been suggested that an increase in THBS signaling in the heart during stress and haemodynamic overload might be beneficial [
24].
In addition to the above-described potential cardio protective mechanisms, female mice also showed an activation of Cx43-related cell functions at the cardiac level, such as cell viability and survival, as well as the transport of molecules, which could be consistent with an adaptative/compensatory reaction to ponatinib cardiotoxic action.
On the other hand, male hearts did not present the activation or inactivation of specific Cx43-related cell functions after ponatinib treatment. Il-6 up regulation was found at cardiac level only in male mice. This condition could be involved in the higher susceptibility to ponatinib treatment of males compared to females. Indeed, it has been demonstrated that cardiac long-term IL-6 signalling or cardiac over production of IL-6R have a causal role in cardiovascular disease [
25].
(5) Silencing the Notch1 signaling obtained by using siRNA-Notch1 treatment allowed us to demonstrate that the Notch1/Cxs pathway is involved in the cardiotoxic effect of ponatinib. Indeed, a ponatinib-induced modulation of Cx43 expression and phosphorylation as well as some specific cell functions were prevented by siRNA-Notch1 cotreatment. However, silencing Notch1 did not reverse Cx26 modulation, total cardiac Cx43 in males, pS368-Cx43, Akt, ERK, miR122, and IL-6 modulation, suggesting that Notch1 is not the only mediator of the ponatinib effect on Cxs regulation. On the other hand, cardiac dysfunctions observed in mice treated with ponatinib were prevented by siRNA-Notch1 treatment confirming the involvement of the other molecules (
Table S1), in addition to Cxs, in ponatinib/Notch1 signaling pathway.
4. Materials and Methods
4.1. Materials, Animals and Treatments
Ponatinib (PON) was acquired by Incyte S.r.l (Wilmington, DE, USA). SiRNA-Notch1 and siRNA-scrambled were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA).
Male and female C57BL/6 mice (body weight: 30 ± 4 g, 24 months-old) were purchased from Charles River Italia (Lecco, Italy). Mice were housed under a 12 h light/dark cycle in temperature and humidity-controlled rooms and were provided with ad libitum rodent chow (Teklad 7001, 4.4%; Harlan Teklad Global Diets, Madison, WI, USA) and water. Animals were randomized into 3 groups (
n = 12,
n = 6 male and
n = 6 female for each treatment group): control (CNTRL), PON + siRNA-scrambled (PON), PON + siRNA-Notch1 (PON + siRNot). PON was dissolved in DMSO and administered to mice in the experimental groups by oral gavage daily (30 mg/kg/d for 28 days, corresponding to the oral doses clinically used in humans:
https://go.drugbank.com/drugs/DB08901, accessed on 25 February 2019), with siRNA-Notch1 or siRNA-scrambled administered via tail vein every 3 days. The control group was given the same dosage of DMSO dissolved in the same volume of water for 28 days. Mice were then euthanized by deep anesthesia under 2% isoflurane. A terminal blood sample was immediately drawn from the left ventricle and later it was used for cardiac troponin (cTn) dosage. Hearts were excised, snap-frozen in liquid N
2 and stored at −80 °C for protein extraction or embedded in an optical cutting temperature (OCT) medium and stored at −80 °C for immunohistological analyses. All procedures were approved by the local Institutional Ethics Committee for Animal Research (Protocol number 176/2019-R released in 25 February 2019). All studies complied with the Guidelines from Directive 2010/63 EU of the European Parliament on the protection of animals used for scientific purpose of the NIH guidelines.
Non-viral siRNA delivery in vivo was performed by tail vein injection. For monitoring the transfection efficacy, we used scrambled siRNA conjugated with a Cy3 fluorochrome provided by Mirus (Mirus Bio, Madison, WI, USA). Accordingly, 1.33 μL (25 nM) of Cy3-conjugated scrambled siRNA or Cy3-conjugated siRNA-Notch (NM_008714.3) were combined with 0.5 μL of Transit-TKO (Mirus) in a final volume 10 μL sterile H
2O, and incubated for 30 min at room temperature, according to the protocol previously published [
26]. According to mice randomization, tail vein injections of 10 μL phosphate-buffered saline (PBS) (
n = 6 male mice and
n = 6 female mice), or 10 μL siRNA-scrambled (
n = 6 male mice and
n = 6 female mice), or 10 μL siRNA-Notch1 (
n = 6 male mice and
n = 6 female mice) were performed with a 32 gauge needle attached to a 1 mL syringe (Beckton Dickinson, Franklin Lake, NJ, USA). Injections were repeated after 72 h and every three days from the first injection.
4.2. Echocardiography
Using a portable ultrasound apparatus (Esaote; Genoa, Italy) equipped with a 21-MHz linear probe, we performed transthoracic echocardiography 28 days after treatments to assess the functional effects of each treatment. The mice were anesthetized (ketamine, 100 mg/kg) and placed in the left lateral decubitus position. To evaluate LV structural changes, the following parameters were measured: LV end-diastolic diameter [LVEDD] and LV end-systolic diameter [LVESD; left ventricular fractional shortening (FS%), calculated as: FS (%) = ([LVEDD–LVESD)/LVEDD] × 100; left ventricular ejection fraction (EF%), calculated as an index of systolic function, ([LVEDD3–LVESD3)/LVEDD3] × 100. The same parameters were measured on control, non-manipulated healthy animals. Passive LV filling peak velocity (E, mm/s) and atrial contraction flow peak velocity (A, mm/s) were obtained by Pulsed-wave Doppler through the mitral valve. Each measurement was obtained by averaging the results of 3 consecutive heart beats. Individuals conducting the echocardiography were blinded to the animal treatment groups.
4.3. Label-Free Proteomics Analysis and Bioinformatics
Cardiac tissues from each treatment group were prepared according to the Filter Aided Sample Preparation (FASP) method. Briefly, cardiac tissues were lysed by sonication in a RIPA buffer and centrifuged to remove cell debris. A total of 50 µg of proteins for each treatment were digested by trypsin (Promega, Madison, WI, USA). For protein label free identification and quantification, tryptic peptides from each sample were analyzed in triplicate with LC-MS/MS using the UltiMateTM 3000 UPLC (Thermo Fisher Scientific, Waltham, MA, USA) chromatographic system coupled to the Orbitrap FusionTM TribridTM (Thermo Fisher Scientific) mass spectrometer. Peptides were loaded on the Trap Cartridge C18 (0.3 mm ID, 5 mm L, 5 μm PS, Thermo Fisher Scientific) and then separated on an EASY-spray AcclaimTM PepMapTM C18 (75 μm ID, 25 cm L, 2 μm PS, Thermo Fisher Scientific) nanoscale chromatographic column. Mobile phase A was 0.1% formic acid in H2O and mobile phase B was 0.1% formic acid in acetonitrile. The flow rate was set at 300 nL/min, with a total run time of 120 min using a chromatographic gradient from 2 to 90% of phase B. Proteomics data were acquired in positive-ion polarity with a Data Dependent Acquisition (DDA) mode to trigger precursor isolation and the MS2 sequence using N2 as a collision gas for CID fragmentation. MS1 scans were performed in the Orbitrap analyzer covering a m/z range of 375–1500 with 120,000 of resolution. The signal intensity threshold was set to 5 × 103 and the MS2 spectra were acquired using a Top Speed method of 3 s. In particular, precursor ions with charges of +2 to +7 were used for MS2 sequencing and scanned in the ion trap by setting the following parameters: MS2 isolation window of 1.6 Da, AGC target of 2 × 103, a dynamic exclusion time of 60 s and mass tolerance of ± 10 ppm were used. We performed a CID fragmentation with a fixed collision energy of 35% and an activation time of 10 ms.
Proteomics raw data were processed using a free computational platform, MaxQuant v1.6.6.0 (Max-Planck Institute for Biochemistry, Martinsried, Germany). Peak lists, generated in MaxQuant, were searched using the Andromeda peptide search engine against the UniProt database (released 2019_11, taxonomy Mus Musculus, 21,990 entries) supplemented with frequently observed contaminants and containing forward and reverse sequences. LFQ Intensity was used to quantify protein abundance in each sample. Bioinformatics analysis was performed with Perseus v1.6.10.50 (Max-Planck Institute for Biochemistry, Martinsried, Germany). LFQ intensities were log2 transformed to facilitate the calculation of the protein expression. The minimum number of valid values accepted was set at 2 in at least one group.
Finally, the protein ratio was uploaded for “Core Analysis” through the Ingenuity Pathway Analysis tool, (IPA, Qiagen, Hilden, Germany). IPA is able to statistically map the modulated proteins for their functional annotations, such as Canonical Pathways, Upstream Regulators Analysis and downstream effects networks, through Gene Ontology and pathway analysis. In this way, it is possible to identify the metabolic pathways and the secondary genes/proteins inhibited (z-score ≤ −2.00) and/or stimulated (z-score ≥ 2.00) for a specific phenotype and consequently classify potential effectors molecules and/or a pharmacological target.
4.4. Western Blotting
Cardiac tissue samples were obtained and homogenized in a RadioImmuno Precipitation Assay (RIPA) buffer containing a protease inhibitor cocktail 1× (S8830, Sigma Aldrich, Merck KGaA, Darmstadt, Germany) at 4 °C. After centrifugation at 15,000 rpm for 20 min at 4 °C, supernatants were collected and the protein concentration was evaluated by the bicinchoninic acid assay (BCA) (Pierce™, Thermo Fisher Scientific) microplate method.
Proteins (30 µg/lane) were separated under reducing conditions on a 4–20% polyacrylamide gel (BioRad, Hercules, CA, USA) and electroblotted onto a nitrocellulose membrane using the Trans Turbo Blot system (BioRad). After blocking in 5% dry fat milk (Sigma Aldrich) in Tris Buffered Saline solution containing 0.1% Tween 20 (T-TBS) for 1 h at room temperature (RT), the membranes were incubated overnight at 4 °C with the following antibodies diluted in T-TBS: (1) anti-Cx43; (2) anti pS368-Cx43; (3) anti-Cx26; (4) anti-GAPDH as protein loading control (see
Table 3). Anti-Rabbit-HRP-conjugated secondary antibodies (BioRad), diluted in T-TBS containing 5% of dry fat milk, were used and the chemiluminescent signal (ECL clarity, BioRad) was detected using Chemi-Doc XR (BioRad). The intensity of the bands was measured using the Image Lab Software (BioRad) on
n = 3 different series of samples.
4.5. Immunofluorescence Analysis
Immunofluorescence was performed on 5–8 μm thick sections from OCT-embedded hearts as previously described [
8]. Briefly, the sections were treated with 0.2% triton-X100/PBS for antigen retrieval and successively with blocking solution (BS, 0.1% Tween, 0.25% BSA in PBS) for 1 h at room temperature (RT). Then, the samples were incubated overnight at 4 °C with previously tested primary antibodies [
7,
19,
27] diluted in BS (see
Table 3).
After washing in BS, sections were incubated for 90 min at RT in the dark with fluorescent anti-rabbit secondary antibody (see
Table 3). Samples on slides were finally mounted with a mounting medium (Prolong
TM Diamond Antifade Mountant with DAPI, Thermo Fisher Scientific) and observed under a fluorescence microscope (BX43, Olympus, Hamburg, Germany). Images were captured at 200× total magnification (20× objective magnification and 10× ocular magnification) by a SC50 digital camera (Olympus; pixel dimension photocamera sensor 2.2 × 2.2 μm, LED light). Cx43 immunofluorescence images were also captured by a confocal laser scanning microscopy (TC SSP8 Leica Microsystems, Mannheim, Germany) using 40× oil immersion lenses by with 488 nm and 561 nm excitation wavelength lasers. Negative controls for secondary antibodies were performed by omitting primary antibodies and incubating the specimens with nonimmune serum or BS.
4.6. Image Analysis
Immunofluorescence positivity was quantified by image analysis on cardiomyocytes from female and male mice hearts. Three nonconsecutive sections were examined per sample and the microscopic fields were objectively selected in order to cover the whole section. Moreover, in every microscopic field of the sections, tissue area containing only cardiomyocytes was selected. The cardiomyocyte reacting areas were quantified with CellSens Imaging Software (Olympus).
The medium colour threshold (the level above which the pictures were considered to be reacting) was evaluated for every sample on negative controls. The image analysis was performed by considering the percentage of the reacting area and the respective level of pixel colour intensity per field. The final rate of reactivity degree of each group was calculated as the product between the average of the positive area percentage and the mean value of the pixel color intensity per microscopic field.
4.7. Statistical Analysis
Data are expressed as mean ± standard deviation (SD). Two group comparisons were performed by using the Student’s t-test for unpaired values. Multiple test correction was completed using the ANOVA test with a p-value threshold of 0.05 and Tukey’s HSD test was used as post-hoc test to determine statistical significance within and between groups.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}