1. Introduction
Lung cancer is one of the most common malignant tumors and a frequent cause of death worldwide [
1,
2]. Surgical resection and radical radiotherapy can offer patients a high probability of a cure at the presentation stage of lung cancer. Unfortunately, the majority of patients diagnosed with lung cancer are at an advanced stage of their disease, leading to a relatively poor outcome [
3,
4]. The combined treatment of etoposide or irinotecan with platinum for lung cancer therapy is the standard first-line method, although some cytotoxic agents such as paclitaxel, docetaxel, gemcitabine and vinorelbine were used in phase II clinical trials with modest efficacy. To date, very few drugs are approved as effective candidates for lung-cancer therapy [
5]. Therefore, it is very urgent to develop new therapeutic agents for the treatment of lung cancer patients.
Cyclovirobuxine D (CVB-D), a natural bioactive alkaloid component derived from
Buxus microphylla, is primarily used to treat cardiovascular and cerebrovascular disease [
6,
7]. Recently, mounting evidence indicates that CVB-D may possess antitumor effects in various kinds of cancers [
8,
9,
10,
11]. However, the underlying mechanism remains to be investigated.
Autophagy is defined as an evolutionally conserved process of recycling excess intracellular macromolecules, which are engulfed in autophagosomes, subsequently degraded in autolysosomes and ultimately, are broken down into their constituent parts [
12,
13]. Autophagy is known to be a pro-survival stress response through which cells can maintain their own energy homeostasis under certain energy-deficiency conditions by removing aggregated proteins and damaged organelles [
14]. Mitophagy is a kind of well-documented mitochondrial quality-control system that mediates the clearance of dysfunction or damaged mitochondria through the selective autophagy of mitochondria. Two pathways have been well identified to mediate mitophagy initiation in mammalian cells including parkin-Pink1. Parkin is an E3 ubiquitin ligase recruited by Pink1, which translocates to mitochondria to mediate mitophagy initiation [
15]. Both BNIP3 (BCL2 and adenovirus E1B 19-kDa-interacting protein 3) and BNIP3L (also known as NIX) are localized to mitochondria and are atypical BH3-only members of the BCL2 family [
16]. BNIP3 was first identified in a yeast two-hybrid screen as BCL2 and adenovirus E1B-19 K-interacting protein. The transcription of BNIP3 can be activated by transcription factor HIF1α [
17] and FOXO3a [
18] and transcriptionally silenced by p65 [
19,
20,
21]. BNIP3 and BNIP3L have been reported to participate in hypoxia-induced mitophagy [
22]. Mechanistically, BNIP3 interacts with microtubule-associated protein LC3 to form a mitochondria-BNIP3-LC3-autophagosome complex and mediates mitophagy initiation [
23,
24].
Under certain conditions, the occurrence of mitophagy is of great importance for cells to preserve a population of normal-function mitochondria and protect cells from cellular damage [
25,
26]. There have been reports that mitophagy can not only protect cells from death but also demonstrates pro-death functions in response to various chemicals. It is perceived that mitophagy may play dual roles in response to anticancer treatment, depending on the property of chemical substances. Therefore, the dissection of the underlying role of mitophagy involved in cancer therapy is crucial for improving anticancer efficiency through the pro-survival or pro-death role of mitophagy.
In this study, we demonstrated that CVB-D-induced lung-cancer cell death is closely associated with mitophagy activation. CVB-D treatment causes significant depletion of mitochondria, which is accompanied by the upregulation of the mitophagy receptors BNIP3 and BNIP3L. Our results further demonstrated that the upregulation of BNIP3 is due to the downregulation of p65, which relieves its inhibition on BNIP3 expression. Notably, mitophagy induced by CVB-D leads to a further increase of apoptotic death in lung cancer cells. These findings provide a cross link between mitophagy and apoptosis upon CVB-D treatment, which indicates that CVB-D is a potential candidate and is of particular clinical relevance for lung-cancer treatment.
3. Discussion
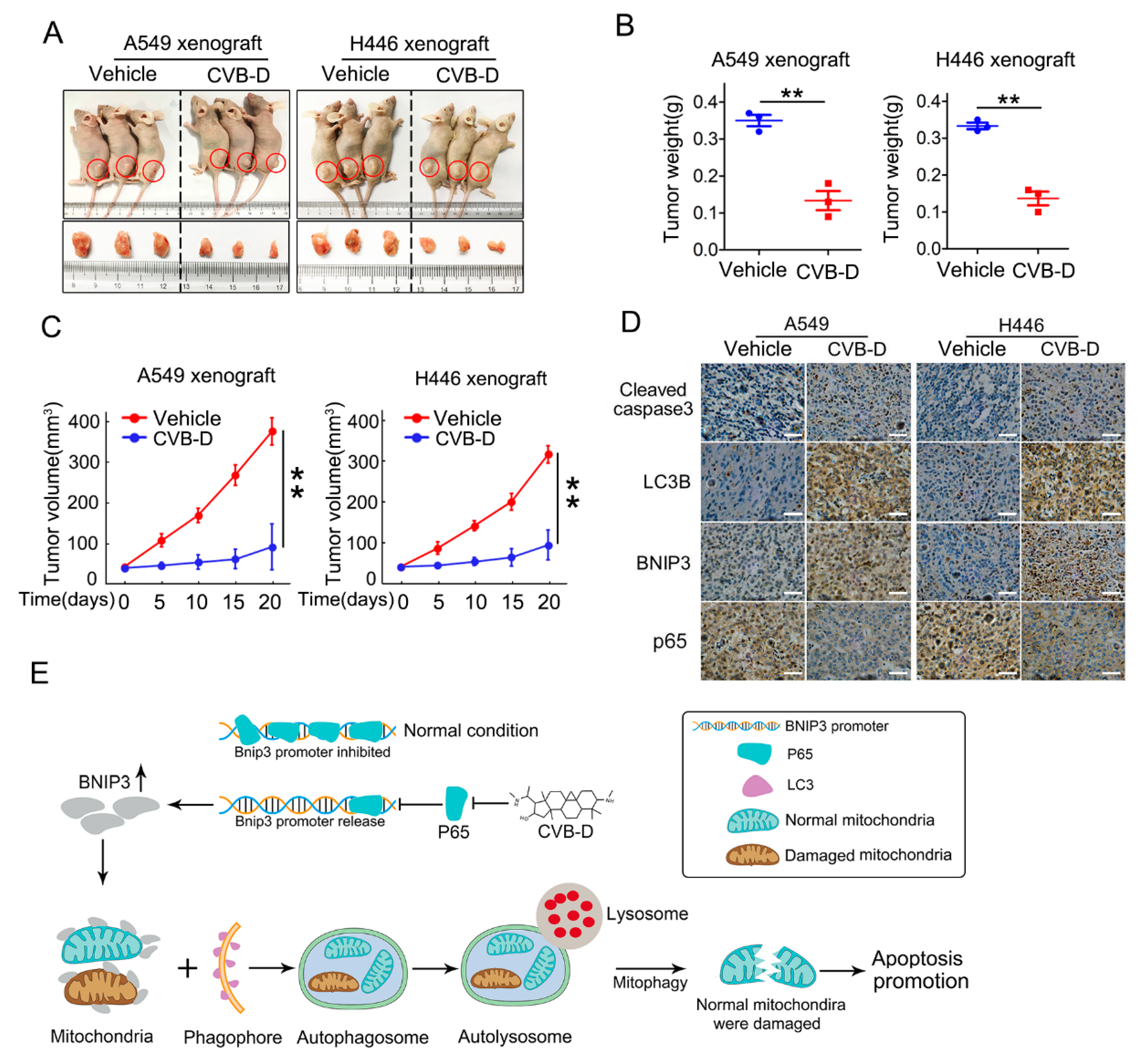
In this study, we revealed that CVB-D, a natural compound derived from Buxus microphylla, can provoke apoptotic death in lung cancer cells. In addition to apoptotic death, CVB-D treatment can also significantly provoke mitophagy activation. Under certain conditions, mitophagy serves a protective role by removing dysfunction or damaged mitochondria to alleviate cellular stress. The extent and duration of mitophagy activation is a crucial impact factor for cell survival or death decisions, so we conducted a series of experiments to address the role of CVB-D-induced mitophagy in the apoptosis of lung cancer cells. Interestingly, we found that mitophagy induced by CVB-D serves a pro-death role in CVB-D-treated lung cancer cells by promoting mitophagy-mediated mitochondrial dysfunction. It is apparent that mitophagy induced by CVB-D is more likely to be overactivated and that this kind of mitophagy serves as a destroyer of mitochondria in lung cancer cells and ultimately potentiates CVB-D-induced apoptotic death. Our results further demonstrated that the p65/BNIP3/LC3 axis is a potential mediator for mitophagy activation upon CVB-D treatment. Treatment of lung cancer cells with CVB-D can significantly suppress the expression of p65, a transcriptional suppressor of BNIP3, and the downregulation of p65 can significantly relieve its inhibition on BNIP3 transcription upon CVB-D treatment and can cause enhanced expression of BNIP3, thus enhancing its interaction with LC3 and mediating mitophagy activation. We demonstrated that CVB-D exhibits a kind of favorable anti-lung-cancer activity in vitro and in vivo and revealed a novel antitumor mechanism of CVB-D in lung cancer cells, namely that mitophagy-mediated mitochondrial dysfunction is a contributor to CVB-D-induced apoptotic death through the targeting of the p65/BNIP3/LC3 axis.
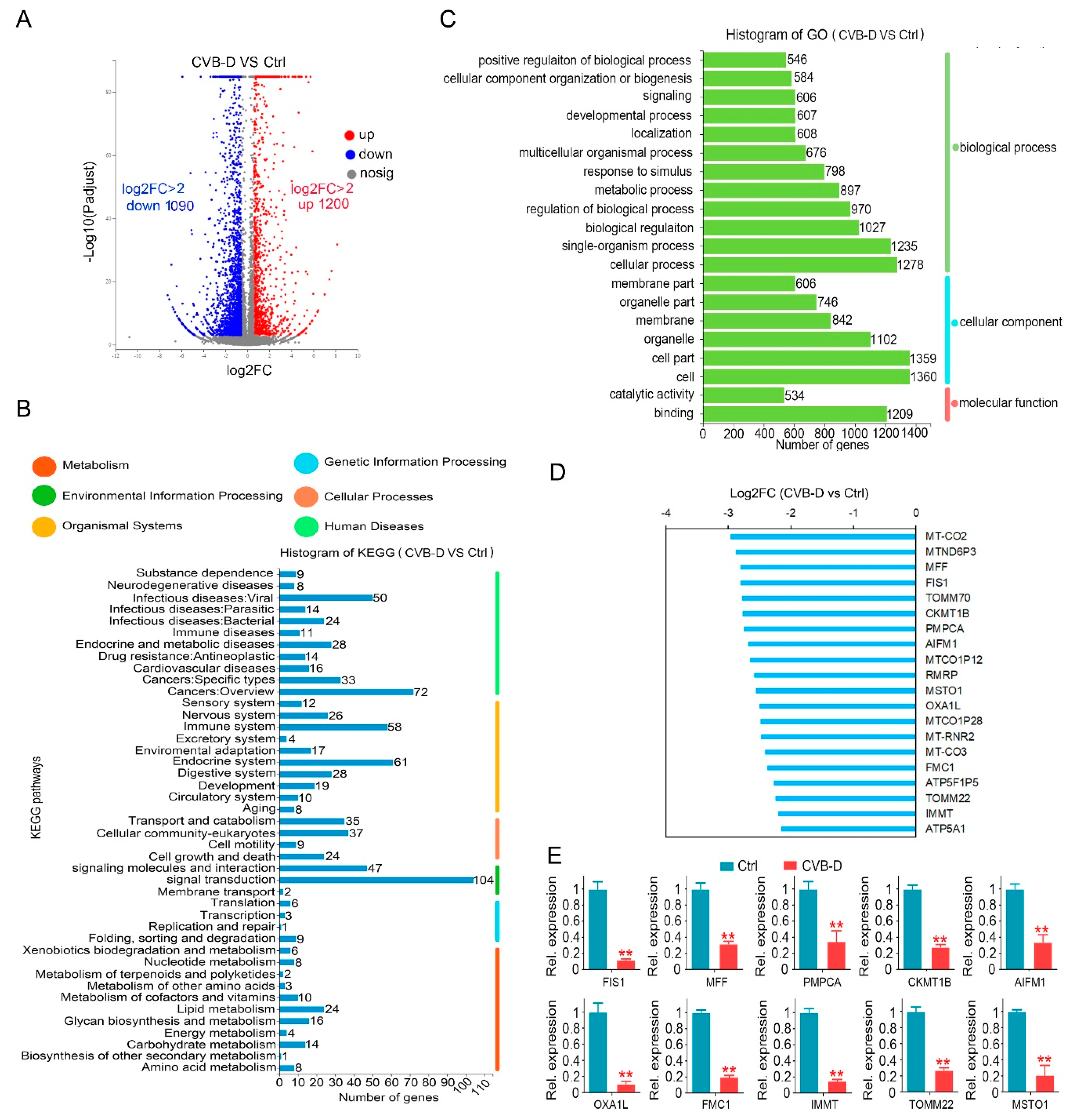
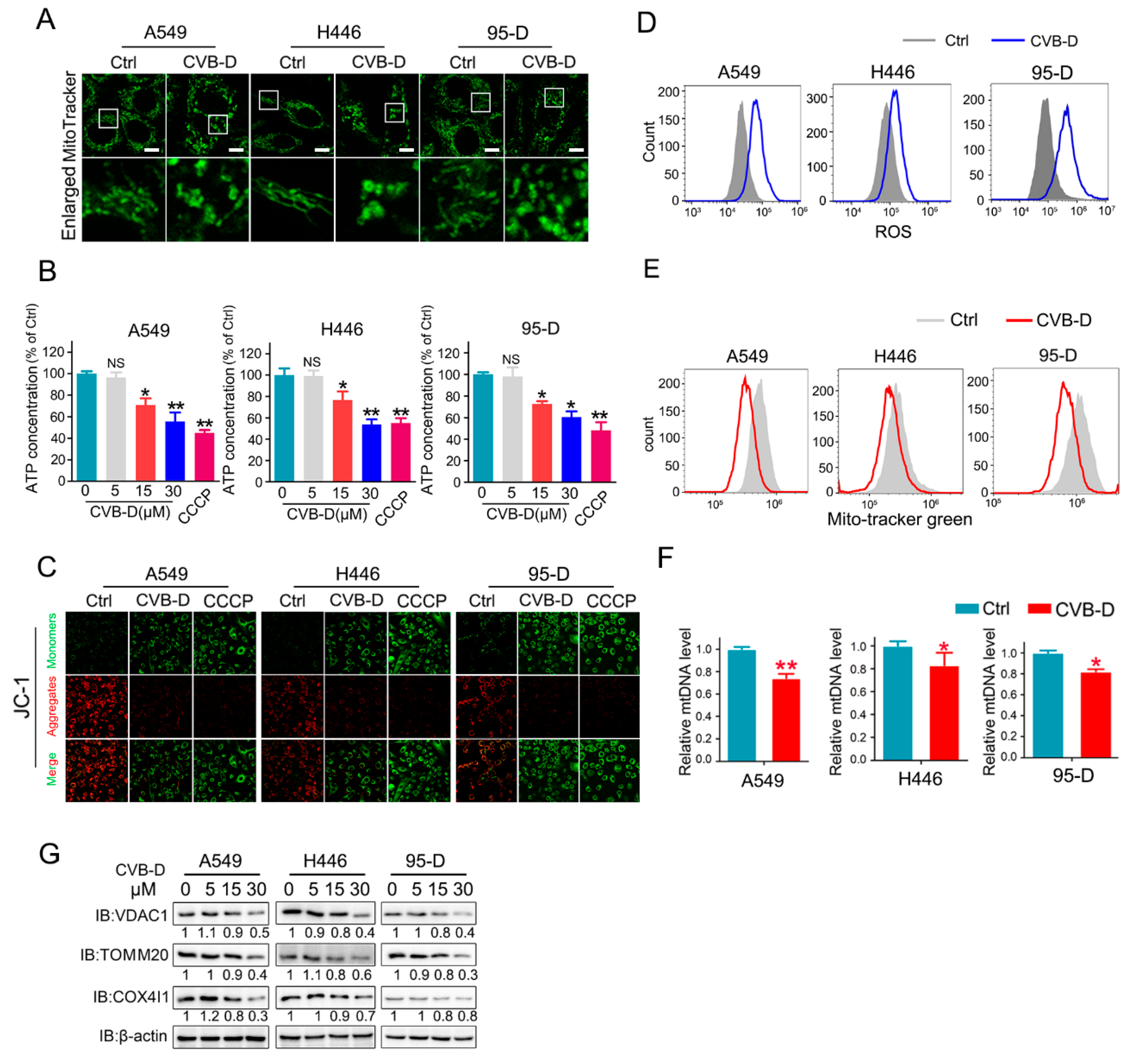
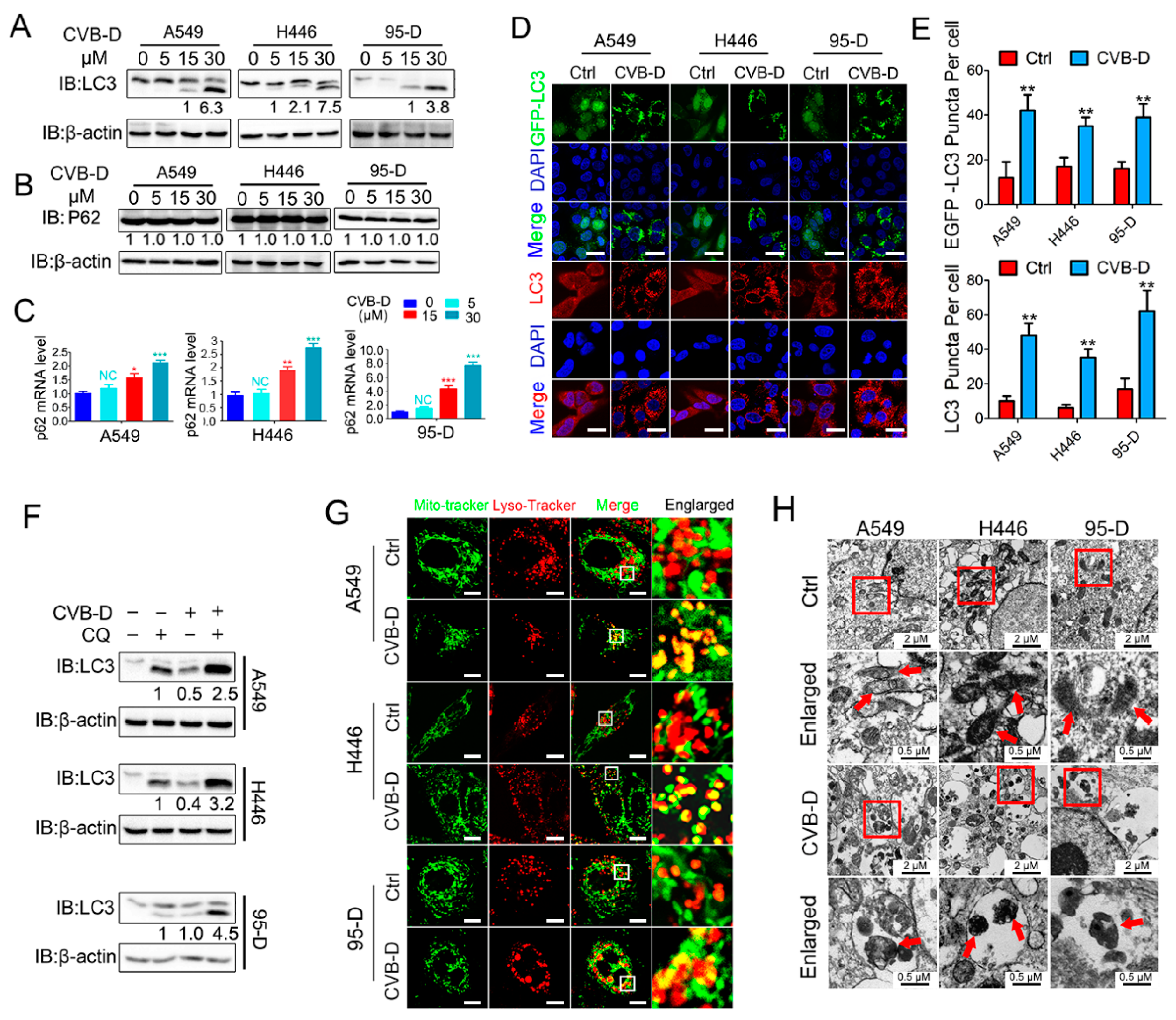
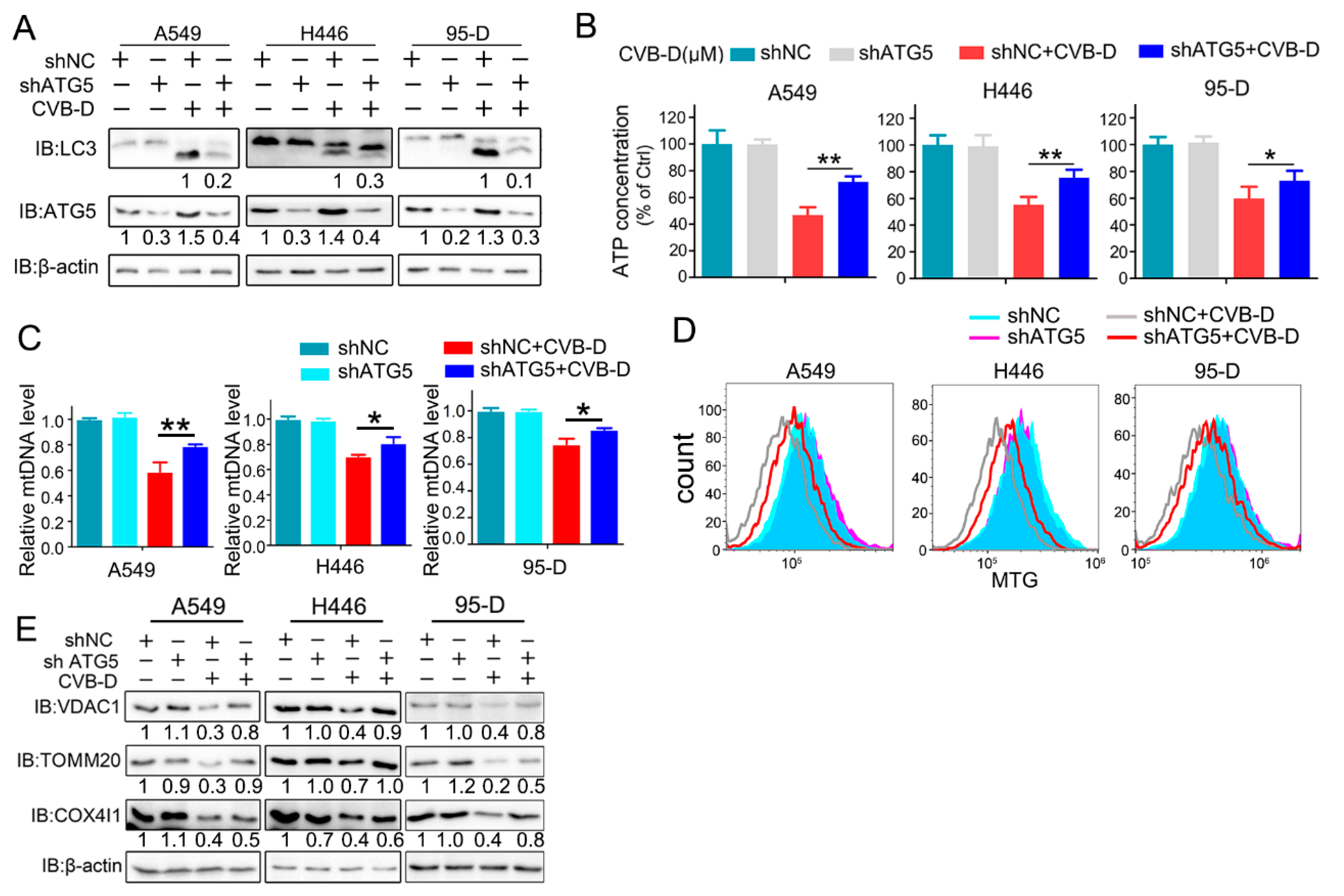
In our preliminary experiments, we found that CVB-D exhibited favorable anti-lung-cancer potential both in vitro and in vivo. Next, we investigated the underlying mechanism by which CVB-D is involved in lung-cancer cell death. A RNA-seq analysis was conducted to detect the change in gene expression upon CVB-D treatment, and we found that numerous mitochondria-related genes were significantly downregulated, so it revealed that CVB-D treatment causes direct or indirect damage to mitochondria in lung cancer cells. In line with RNA-seq results, we also provided novel evidence that CVB-D treatment induces mitochondria dysfunction and loss by testing cellular ATP concentration, mitochondrial mass and mitochondrial DNA copy-numbers, as well as the degradation of mitochondrial proteins. Along with these findings, we also revealed that mitophagy was significantly activated upon CVB-D treatment. Subsequently a series of ATG5 knockdown experiments were conducted to confirm our results. These observations strongly supported the idea that CVB-D treatment promotes mitophagy activation and that mitophagy is a contributing factor involved in mitochondria loss in lung cancer cells.
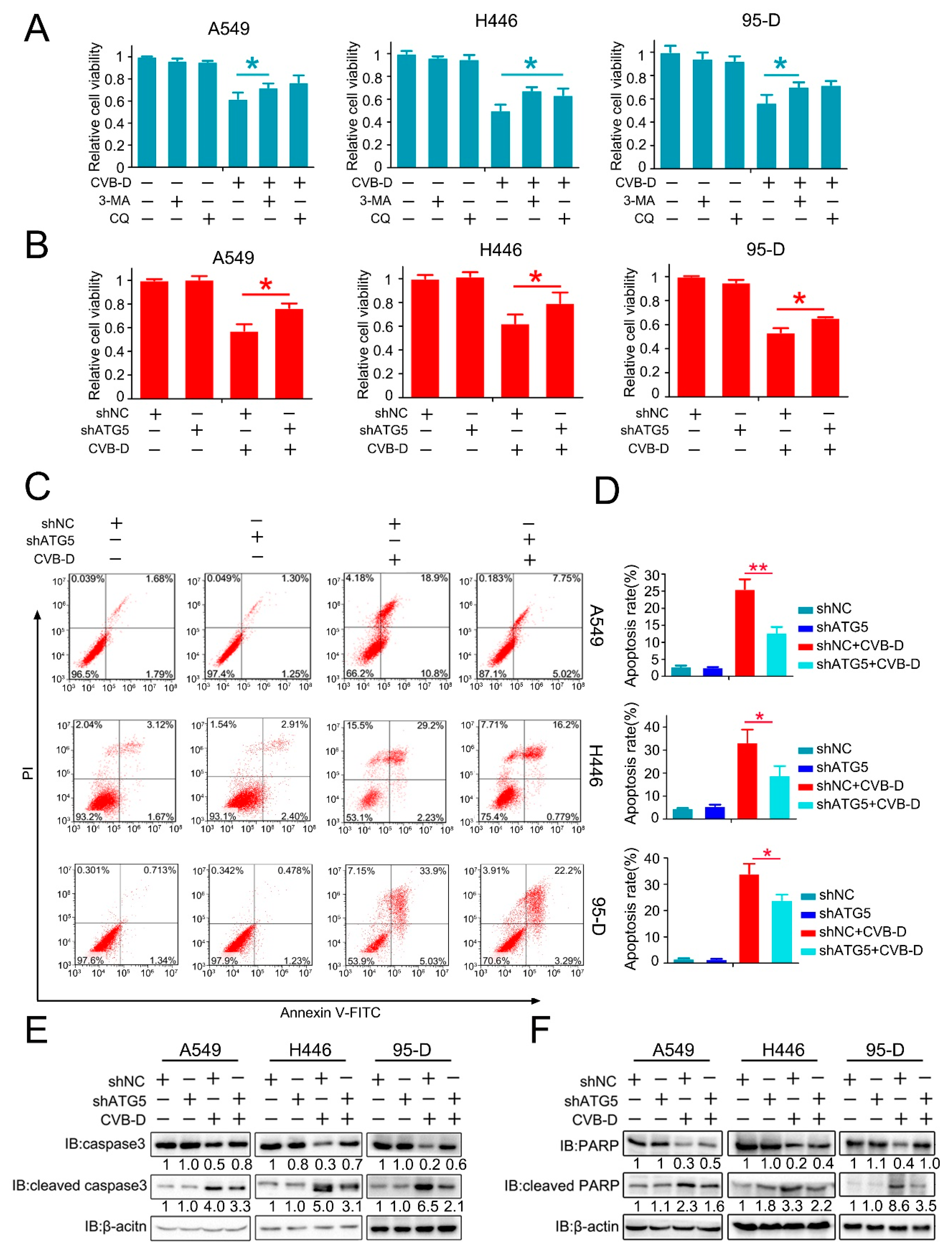
Although mitophagy is a specific autophagy that selectively removes dysfunctional or damaged mitochondria to protect cells from death [
32], abnormal or overactivated mitophagy is toxic to cells. Our observation revealed that CVB-D-induced mitophagy was seemingly overactivated, as evidenced by the knockdown of ATG5 significantly relieving the CVB-D-induced mitochondrial ATP reduction, which is similar to previous reports that mitochondrial dysfunction mediated by mitophagy is a switch for cell death fate decisions [
33,
34]. Indeed, these conclusions can be verified by the observations that the genetic abolishing of the mitophagy process can significantly alleviate cell apoptosis upon CVB-D treatment; therefore, CVB-D-induced mitophagy contributes to apoptosis activation. Based on the above, we concluded that CVB-D treatment causes the overactivation of mitophagy and that this kind of mitophagy contributes to cell apoptosis induction by causing dysfunction and damage to healthy mitochondria.
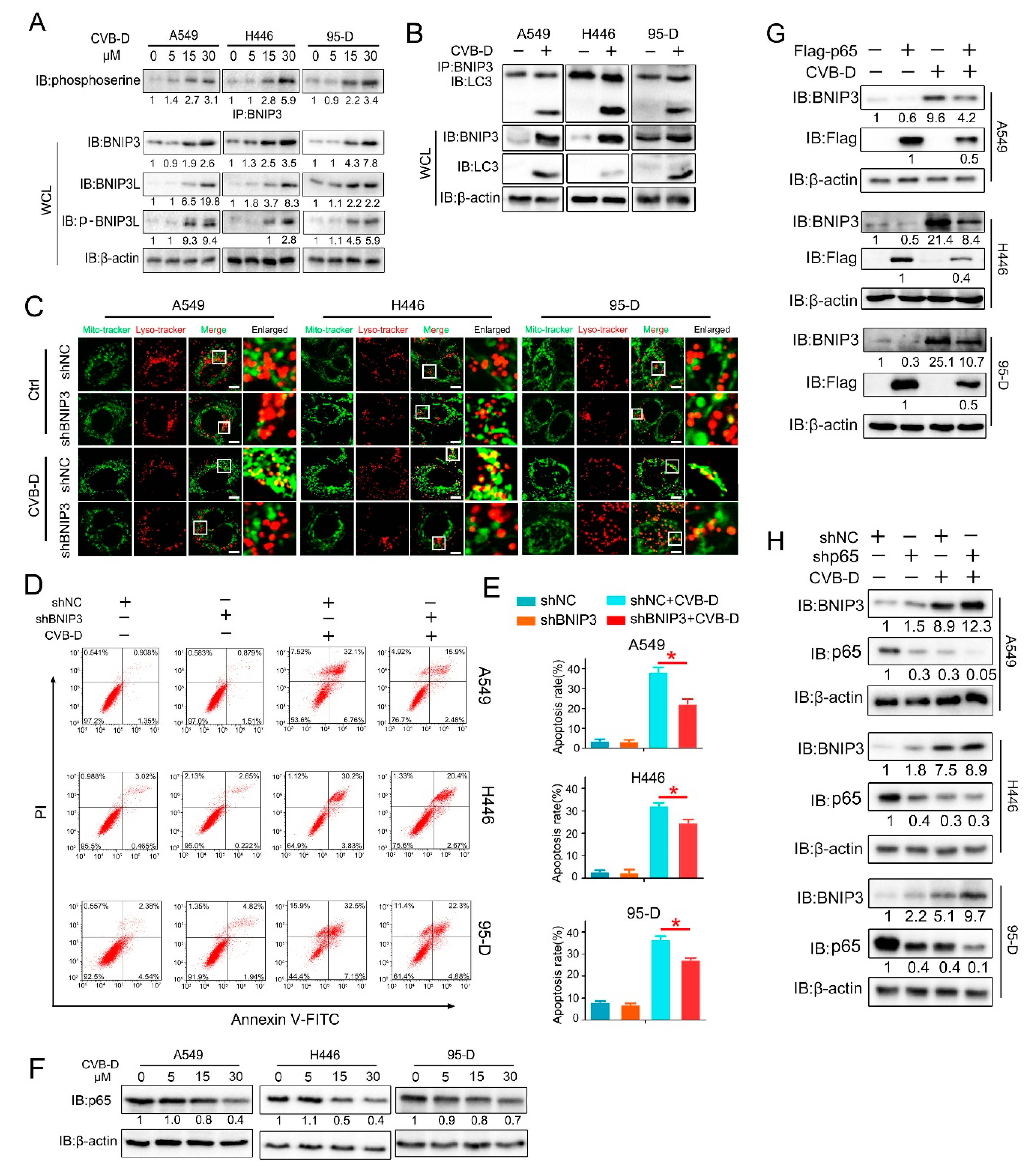
Notably, we found that mitophagy induced by CVB-D is not associated with the parkin-Pink1 pathway, a well-studied pathway involved in mitophagy activation. CVB-D treatment caused a significant decrease in parkin protein in all lung cancer cell lines upon CVB-D treatment in a dose-dependent manner for 24 h, suggesting that CVB-D-induced mitophagy in lung cancer cells is independent of parkin, at least at our time point. A previous study showed that chemical-induced mitophagy is not accompanied by the enhanced expression of parkin but with the enhanced expression of Pink1 recruited to outer membrane mitochondria [
35]. Next we examined whether Pink1 is involved in CVB-D-induced mitophagy. In our time point, CVB-D treatment caused a significant decrease in Pink1 expression in H446 and 95-D cells but no visible change in A549. These differential expressions upon CVB-D treatment in lung cancer cells may depend on the cellular content. Given this, we concluded that Pink1 is also not involved in CVB-D-induced mitophagy. But for a short time of CVB-D stimulation (within 24 h), the manner of change in the expressions of parkin and Pink1 needs further clarification. P62 is a kind of adaptor protein. In this study, we detected no significant p62 protein expression change, but a significantly enhanced expression of p62 mRNA was seen, suggesting that the degradation of the SQSTM1/p62 protein in autophagy was counterbalanced by the increase in SQSTM1/p62 translation upon CVB-D treatment, but whether p62 was involved in CVB-D induced mitophagy is also largely unknown and needs to be further clarified. In sharp contrast to the expression of parkin and Pink1, we detected a strong increase in the expression of the mitophagy receptor BNIP3. Based on this, we hypothesized that the BNIP3 receptor may be a major candidate to mediate mitophagy activation upon CVB-D treatment, and further evidences confirmed our hypothesis that the reduced expression of BNIP3 by knocking down significantly abolished the CVB-D-induced colocalization of mitochondria and lysosomes. BNIP3-mediated mitophagy is closely related to the interaction of BNIP3 and LC3 (23). In analogy to previous findings, our results revealed that CVB-D treatment induced the upregulation of BNIP3 expression and resulted in tighter interaction of BNIP3 and LC3. Altogether, based on the above results, we concluded that CVB-D-induced mitophagy is associated with the enhanced expression of the BNIP3 receptor, which mediated the activation of mitophagy. Several studies showed that BNIP3 and BNIP3L could cause the opening of the mitochondrial permeability transition pore (mPTP). Therefore, the overexpression of BNIP3 and BNIP3L contributes to the collapse of MMP (mitochondrial membrane potential) (16,22). In this study, CVB-D may also function as a destroyer by directly causing mitochondrial damage by enhancing the expression of BNIP3. These intricate relationships between BNIP3-mediated apoptosis-promoting mitophagy and the BNIP3-mediated collapse of MMP deserves further exploration. BNIP3 can be transcriptionally activated by several transcription factors, including HIF1α and FOXO3a. Unexpectedly, these regulators are not involved in the CVB-D-induced upregulation of the BNIP3 protein and our data further showed that the downregulation of p65 (BNIP3 transcriptional suppressor) was closely associated with CVB-D-induced BNIP3 upregulation.
In physiological conditions, mitochondrial quality and mass are accurately controlled by undergoing mitochondrial biogenesis and mitophagy constantly [
26]. Generally, mitophagy is a kind of survival strategy for cells using the degradation of dysfunctional or damaged mitochondria. In certain physiological conditions, for example, in the process of ischemic preconditioning, the activation of mitophagy could significantly mediate the degradation of damaged mitochondria and protect kidneys from damage [
36]. In oxidative stress conditions, the activation of mitophagy is also of great importance to maintain bone-marrow mesenchymal stem-cell survival [
37]. In screening potential antitumor drugs, many chemicals were also found to provoke mitophagy. Usually, the kind of mitophagy induced by chemicals serves a protective role for cancer cells to alleviate cytotoxicity, for example, retigeric acid B-induced mitophagy can significantly antagonize cell death, and the inhibition of mitophagy can improve the chemotherapeutic effects against prostate cancer cells [
38]. In the
125I seeds brachytherapy process,
125I seeds irradiation causes both mitochondrial damage and mitophagy activation, and the activated mitophagy contributes to the clearance of damaged mitochondria to maintain cellular homeostasis and survival [
39]. Contrary to the above results, our data demonstrated that CVB-D-induced mitophagy serves a pro-death role in lung cancer cells by causing mitochondrial dysfunction, thus promoting apoptosis activation. Previous reports demonstrated that ceramide targets autophagosomes to mitochondria, causing lethal mitophagy [
40] and that mitophagy contributes to pulmonary epithelial cells’ necroptosis in response to cigarette smoke exposure [
41]. BAY 87-2243 (inhibitor of complex I (CI) of the mitochondrial respiratory chain)-induced Pink1-dependent mitophagy is responsible for melanoma cell death [
42]. Ketoconazole-induced mitophagy is a crucial contributor to provoking apoptosis [
34]. Similar to previous reports, our results demonstrated that mitophagy induced by chemicals exhibits cytotoxicity to cells (abnormal or excessive mitophagy). Although the pro-survival or pro-death role of mitophagy induced by chemicals depends on the property of the chemicals, there remains controversy about the roles of chemicals-induced autophagy/mitophagy in cell death and survival, and it seems that it is cellular-content-dependent. For example, AT 101 triggers mitophagic cell death in glioma cells [
33] but triggers protective autophagy in MCF7 cells [
43].
Based on our findings that mitophagy induced by CVB-D could significantly potentiate the apoptotic death of lung cancer cells, it is certain that mitophagy induced by CVB-D is excessive and toxic to lung cancer cells. Despite our experimental evidence proving that CVB-D induced mitophagy and promoted apoptotic cell death, the knockdown of ATG5 or BNIP3 led to a partial alleviation of cytotoxicity. Therefore, it is undeniable that other factors may be involved in CVB-D-triggered cell death and need to be further elucidated.
In this study, we showed that CVB-D demonstrated favorable anti-lung-cancer activity both in vitro and in vivo and that it has potential application in the treatment of lung cancer. Based on its autophagy/mitophagy induction activity, its potential application in the treatment of other diseases also deserves to be explored. Under certain conditions, the activation of autophagy may serve as a treatment strategy to defend against intracellular bacteria, parasites and virus infection by targeting intracellular microorganisms (xenophagy) and mediating the clearance of microorganisms or via modulating the antiviral immune response of organisms to defend against infection [
44]. In terms of anti-infection, whether CVB-D possesses potential applications is largely unknown and deserves to be explored.
4. Materials and Methods
4.1. Cell Culture, Reagents and Antibodies
Human lung cancer cell lines (A549, H446 and 95-D) were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), and cultured in DMEM containing 10% fetal bovine serum (FBS) and a 100 U/mL mixture of penicillin and streptomycin. All cells were kept at 37 °C and 5% CO2. Cyclovirobuxine D (CVB-D) was purchased from the Chengdu Alpha Biotechnology Incorporation and dissolved in methanol to generate a 70 mM stock solution. The primary antibodies for Western blotting, immunoprecipitation and immunohistochemistry against target proteins are as follows: caspase3 (Zen Bioscience, 300968, Chengdu, China), cleaved-caspase3 (Zen Bioscience, 380169), PARP (Zen Bioscience, 380451), cleaved-PARP (Zen Bioscience, 380374), VDAC1 (Sangon Biotech, D124100, Shanghai, China), TOMM20 (Sangon Biotech, D153158), COX4I1 (Sangon Biotech, D262690), LC3 (Sangon Biotech, D163557), ATG5 (Sangon Biotech, D121650), SQSTM1/p62 (Beyotime, AF5312), BNIP3 (Zen Bioscience, 381756), BNIP3L (Zen Bioscience, 381891), β-actin (Santa Cruz, sc-47778), p65 (Cell Signaling Technology, 8242S, Danvers, MA, USA), CDC2 (Cell Signaling Technology, 9116S), cyclinB1 (Cell Signaling Technology, 4135S), p-BNIP3L (phosphoS81) (abcam, ab208190, Cambridge, UK) and phosphoserine (Millipore, AB1603, Burlington, MA, USA).
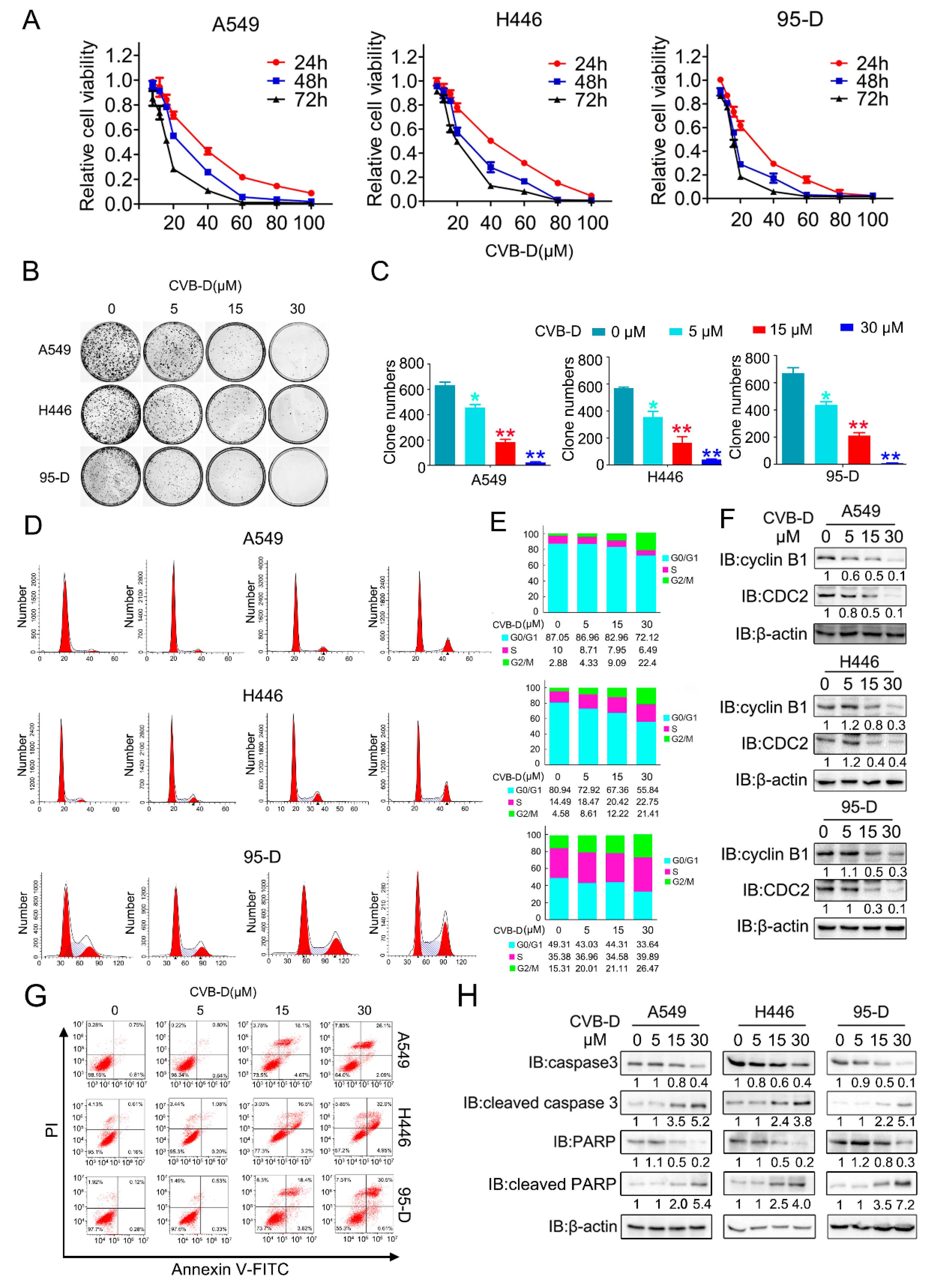
4.2. Cell Viability and Colony Formation Assay
Cell viability was assessed through a cell counting kit-8 (CCK8) assay (MedChemExpress, HY-K0301, Monmouth Junction, NJ, USA). Briefly, cells were seeded in 96-well plates at a concentration of 5 × 103 per well, incubated overnight, then subjected to different concentrations of CVB-D treatments and cultured for 24, 48 or 72 h. The CVB-D-contained medium was replaced with 100 μL fresh DMEM; CCK8 (10 μL/well) was added, and then incubated at 37 °C for 2 h, and the absorbance value was measured at a 450 nm wavelength (BioTek, Synergy/HTX, Winooski, VT, USA). For clonogenic assay, 1 × 103 cells were seeded in 35 mm cell culture dishes, cultured overnight, and then cells were pretreated with various concentrations of CVB-D for 6 h, followed by replacing the CVB-D-containing medium with a fresh medium to form colonies in 2–3 weeks. Colonies were fixed with glutaraldehyde (6.0% v/v) and stained with crystal violet (0.5% w/v) (Beyotime, C0121, Shanghai, China).
4.3. Flow Cytometric Analysis
A549, H446 and 95-D cells were seeded into 6-well plates, cultured overnight, and then the cells were treated with CVB-D for 24 h. Next, cells were harvested and stained with an Annexin V-FITC/PI apoptosis detection kit (CWBIO, CW2574, Beijing, China) to perform a apoptosis assay or stained with 100 nM mitotracker green (Beyotime, C1048) to perform a mitochondria mass assay. Reactive oxygen species (ROS) levels were assessed by staining with 10 μM fluorescent dye 2′7′-dichlorfluorescein-diacetate (DCFH-DA) (Beyotime, S0033S) for 20 min. For a cell-cycle analysis, cells were incubated with various concentrations of CVB-D for 24 h, rinsed in precooled PBS, followed by fixing with 75% alcohol overnight at 4 °C. Then cells were digested with 1% RNase A (Beyotime, ST579) and stained with 1mg/mL propidium iodide (PI) (Beyotime, ST511) for 30 min. All experiments were performed according to the corresponding manufacturer’s instructions. At least 10,000 live cells were collected and analyzed on the flow cytometer (Beckman, CytoFLEX, Brea, CA, USA). Experimental data were analyzed using Flow Jo 7.6 and Modifit software.
4.4. Immunoprecipitation and Western Blotting Analysis
For the interaction analysis of BNIP3 and LC3, lung cancer cells were treated with or without 30 μM CVB-D for 24 h, rinsed in precooled PBS (2.7 mM KCl, 137 mM NaCl, 2 mM KH2PO4 and 10 mM Na2HPO4), and total proteins were extracted using a RIPA lysis buffer (150 mM NaCl, 50 mM Tris-HCl, pH 8, 0.5% sodium deoxycholate, 1% NP-40, 0.2% SDS) containing a protease inhibitor cocktail (EDTA-free) (MedChemExpress, HY-K0011, Monmouth Junction, NJ, USA). After centrifugation (13,000× g, 10 min), total proteins were quantified with a BCA Protein Assay Kit (CWBIO, CW0014, Shanghai, China), and 30 μL of protein A-agarose beads (Santa Cruz, SC-2001) and 2 μg BNIP3 primary antibody were added into equal amounts of lysates and then subjected to immunoprecipitation assay overnight at 4 °C. Subsequently, the precipitates were centrifuged (1000 r/min, 3 min), resuspended and washed with washing buffer, and this process was repeated three times. Then immune complexes were eluted with SDS-PAGE loading buffer (5×, 250 mM Tris-HCL (pH 6.8), 50% (w/v) glycerol, 5% (v/v), β-mercaptoethanol, 10% (w/v) SDS, 0.5% (w/v) bromophenol blue) at 100 °C for 10 min and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis. Subsequently it was transferred onto 0.45 μm PVDF membranes (GE Healthcare, A29280264) and blocked with 5% (w/v) skimmed-milk dissolved in a TBST buffer (150 mM NaCl, 0.05% (v/v) Tween 20, 20 mM Tris-HCL) for 1 h and then incubated with the corresponding primary antibodies overnight at 4 °C. The PVDF membranes were washed three times (10 min/time) with the TBST buffer. Secondary HRP-labeled goat anti-Mouse lgG (H+L) (Beyotime, A0216) or HRP-labeled goat anti-Rabbit lgG (H+L) antibodies were applied (Beyotime, A0208) at 1:5000 for 1 h at room temperature, then washed three times (10 min/time) with the TBST buffer. The immunoreactions were visualized with the Western BrightTM ECL reagent (Advansta, 191026-11, San Jose, CA, USA) using ChemiDocTM XRS+ (Bio Rad, Hercules, CA, USA). For the analysis of protein expression upon CVB-D treatment, cells were rinsed in precooled PBS, and total proteins were extracted using a RIPA lysis buffer containing protease inhibitors. After centrifugation (13,000× g, 10min), total proteins were quantified with a BCA Protein Assay Kit (CWBIO, CW0014, Jiangsu, China). Equal amounts of protein samples (30–80 μg) were subjected to SDS-PAGE analysis. For analysis of the phosphorylation level of BNIP3, briefly, lung cancer cells were treated with the indicated CVB-D concentration for 24 h, and total proteins were extracted using a RIPA lysis buffer containing a protease inhibitor cocktail and phosphorylase inhibitor (Sangon Biotech, C500019). After centrifugation, total proteins were quantified, and 30 μL of protein A-agarose beads and 2 μg BNIP3 primary antibody were added into equal amounts of lysates and then subjected to immunoprecipitation assay overnight at 4 °C, and the subsequent steps were conducted as in the above description. The phosphorylation modification of BNIP3 was detected using a phosphoserine antibody.
4.5. Cellular ATP Detection
Cellular ATP concentration was detected using an ATP Assay Kit (Beyotime, S0026B) according to the corresponding manufacturer’s standard instructions. Briefly, cells were seeded into 60 mm dishes, cultured overnight, and then cells were treated with CVB-D for 24 h. Total proteins were extracted using an ATP detection lysis buffer and quantified with a BCA Protein Assay Kit (CWBIO, CW0014). A total of 100 μg protein for each sample was subjected to the detection of ATP concentration, and chemiluminescence intensity was detected using a multi-mode ELISA reader (BioTek, Synergy/HTX, Winooski, VT, USA). ATP concentration was calculated according to the standard curve, and the relative ATP level for each sample was normalized to the control group.
4.6. Plasmids and Transfection
The full length of LC3 cDNA was cloned into pEGFP-C1 vector (Addgene Company, USA) to generate a recombinant pEGFP-LC3 plasmid. To generate knockdown cell lines, shRNA against ATG5, BNIP3 and P65 was constructed into the PLKO.1 vector. Lentivirus particles were produced using a three-plasmid packaging system. pMD2G, psPAX2 and pLKO.1 (Addgene Company, Watertown, MA, USA); the 293T cells and the supernatant containing lentivirus particles were collected after transfection for 48 h. A549, H446 and 95-D cells were infected with lentivirus in the presence of 8 μg/mL polybrene (MedChemExpress, HY-112735) for 24 h and were selected with 2 μg/mL puromycin (Solarbio, IP1280, Beijing, China) for two weeks. Stable expression clones were validated by Western blotting and used in the following experiments. The target sequences were synthesized by the Sangon company (Shanghai, China) and are as follows: ATG5, forward: CCGGCCTGAACAGAATCATCCTTAACTCGAGTTAAGGATGATTCTGTTCAGGTTTTT, reverse: AATTAAAAACCTGAACAGAATCATCCTTAA CTCGAGTTAAGGATGATTCTGTTCAGG. BNIP3: forward: CCGGGCCTCGGTTTCTATTTATAATCTCGAGATTATAAATAGAAACCGAGGCTTTTT, reverse: AATTAAAAAGCCTCGGTTTCTATTTATAATCTCGAGATTATAAATAGAAACCGAGGC. P65 forward: CCGGCGGATTGAGGAGAAACGTAAACTCGAGTTTACGTTTCTCCTCAATCCGTTTTT, reverse: AATTAAAAACGGATTGAGGAGAAACGTAAACTCGAGTTTACGTTTCTCCTCAATCCG. To generate P65 overexpression cell lines, the full length of p65 cDNA was inserted into pCMV-Flag vector (Addgene Company) and transfected into A549, H446 and 95-D cells, then selected with 500 μg/mL G418 (Solarbio, IG0010) to generate p65 stable expression cell lines.
4.7. Determination of Gene Expression by qPCR
Total RNA was extracted using the trizol reagent (Takara, 9108, Kyoto, Japan); a total of 1 µg RNA for each sample was reverse-transcribed using SuperRT cDNA Synthesis Kit (CWBIO, CW0741), and quantitative real-time PCR was performed using the SYBR-Green qPCR master mix (CWBIO, CW0659). The qRT-PCR primers in this study were synthesized by the Sangon company (Shanghai, China) and used as follows: p62, forward: CCAGAGAGTTCCAGCACAGA, reverse: TGAGGAACAGATGGAGTCGG, β-actin, forward: TGACGTGGACATCCGCAAAG, reverse: CTGGAAGGTGGACAGCGAGG, FIS1 forward: AGCGGGATTACGTCTTCTACC, reverse: CATGCCCACGAGTCCATCTTT, MFF forward: ACTGAAGGCATTAGTCAGCGA, reverse: TCCTGCTACAACAATCCTCTCC, CKMT1B forward: CTGGTTGGACGCTAGATCAGT, reverse: GGTCAGCAAATACCTCATAGGTC, PMPCA forward: CTCCTGCGTACAGACGGTTTA, reverse: CGAAGCCCATTATCCAATGTGG, AIFM1 forward: CCCAATGCTATTGTGCAATCCG, reverse: GAAGCCACCAAAATCTGAGTCTA, OXA1L forward: GCCTCAGTACCTCTGCTATCT, reverse: TCCCACTGGGGTGTATGACC, FMC1 forward: TCCCCGTCGCACACTTTTC, reverse: TGAAGCTCATGTTGGGCTCTG, IMMT forward: CGATTCAGTCGGGTCCACTAA, reverse: AGCTGGAGTATCTCCCTTTTGT, TOMM22 forward: GGCCGGAGCCACTTTTGAT, reverse: AGGTCCTAGAAGTATCTGCCG, MSTO1 forward: ATCCCAAGAACCCTTATCTCCA, reverse: GGGAGTGGTGAGGAACCTTT. The relative gene level was normalized to β-actin using the 2−ΔΔCT method. For mitochondrial DNA quantitation, total DNA was isolated with a DNA Isolation kit (CWBIO, CW0565), followed by a qPCR analysis using the mtDNA primers (synthesized by Sangon company, Shanghai, China): MT-ND1 (mitochondrially encoded NADH: ubiquinone oxidoreductase core subunit 1), forward: CCCTAAAACCCGCCACATCT, reverse: GAGCGATGGTGAGAGCTAAGGT. Nuclear target gene LPL (lipoprotein lipase), forward: ATGAGAGTTGGGTGCCAAAAC, reverse: AGTCCACCACAATGACATTGG. All experiments were performed with the standard amplification protocol according to the corresponding manufacturer’s instructions. Relative mitochondria DNA copy numbers were normalized to LPL using the 2−ΔΔCT method.
4.8. Confocal Microscopy Imaging
Cells were cultured in confocal dishes (Biosharp, BS-15-GJM, Hefei, China) overnight and treated with 30 μM CVB-D for 24 h. The morphology of mitochondria was determined by staining 100 nM Mito-Tracker green (Beyotime, C1048) for 30 min at 37 °C. Cells were washed twice, and images were visualized under confocal microscopy (Leica TCS SP8 microscope, Wetzlar, Germany). To evaluate autophagic flux, GFP-LC3-transfected cells were treated with 30 μM CVB-D for 24 h, followed by fixation with 4% paraformaldehyde, then cells were washed twice and visualized under confocal microscopy (Leica TCS SP8 microscope). To detect the endogenous LC3, cells were treated with 30 μM CVB-D for 24 h, fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and then this process was followed by blocking with 10% goat serum. Then cells were incubated with the anti-LC3 antibody and Alexa Fluor 555 goat anti-mouse IgG (H+L) antibody, and images were visualized under confocal microscopy (Leica TCS SP8 microscope). To evaluate the colocalization of mitochondria and lysosomes, cells were treated with 30 μM CVB-D for 24 h, labeled with 100 nM Mito-Tracker green (Beyotime, C1048) and 50 nM Lyso-Tracker Red (Beyotime, C1046) for 30 min at 37 °C and visualized under confocal microscopy (Leica TCS SP8 microscope) immediately. To evaluate the change of MMP (mitochondrial membrane potential), we treated cells with 30 μM CVB-D for 24 h. Then cells were labeled with a JC-1 probe using the mitochondrial membrane potential assay kit with JC-1 (Beyotime, C2005) according to the corresponding manufacturer’s instructions, incubated for 20 min at 37 °C, washed twice and visualized under confocal microscopy (Leica TCS SP8 microscope) immediately.
4.9. Transmission Electron Microscopy
Briefly, cells were treated with 30 μM CVB-D for 24 h, fixed with 2.5% glutaraldehyde buffered in a 0.1 mM phosphate buffer (pH 7.4) overnight at 4 °C, washed with phosphate buffer, followed by being embedded in 1% osmium tetroxide and dehydrated in a series concentration of ethanol. Next, cell pellets were embedded in epon resin; ultrathin sections were cut and stained with 2% uranyl acetate, and images were captured using TEM (JEM-1400plus).
4.10. RNA Extraction, RNA-Seq Library Preparation and Sequencing
A549 cells were treated with 30 μM CVB-D for 24 h. Total RNA was extracted using the TRIZOL reagent (Takara, Kyoto, Japan), and genome DNA was digested with DNase I (Takara). The RNA library was constructed by using a TruSeqTM RNA sample preparation kit (Illumina), according to the corresponding manufacturer’s instructions, using 1 μg of total RNA. Briefly, mRNA was isolated, fragmented, and double-stranded cDNA was synthesized with random hexamer primers (Illumina, San Diego, CA, USA) using a SuperScript double-stranded cDNA synthesis kit (Invitrogen, Carlsbad, CA, USA). Subsequently, the synthesized cDNA was subjected to phosphorylation, end-repair and ‘A’ base addition according to Illumina’s library construction instructions. cDNA target fragments of 300 bp were selected and amplified. The library was subjected to sequencing with the Illumina NovaSeq 6000 sequencer (2 × 150 bp read length).
4.11. Immunohistochemistry Analysis
Tumors obtained from A549 and H446 xenograft mice were fixed in 4% paraformaldehyde, then were paraffin-embedded and sectioned. Tumor slides were incubated with primary antibodies at 4 °C overnight, incubated with a secondary antibody, and then the chromogenic reaction was conducted with 3, 3-diaminobenzidine (Solarbio, DA1015) and counterstained with hematoxylin (Solarbio, G1080, Beijing China) according to the corresponding manufacturer’s instructions. Images were captured using the Nikon microscope.
4.12. Animal Study
All animal experiments were approved by the institutional animal care and use committee of Chongqing University. Briefly, 5 × 106 of A549 and H446 cells were suspended in 100 μL of PBS and subcutaneously implanted into the flank of nude mice (male, 6 weeks). When the tumor volume reached ~50 mm3, mice were randomly divided into different groups and were intraperitoneally injected with CVB-D (20 mg/kg) or a vehicle every two days. Tumor volume was measured every five days. The mice were sacrificed after 20 days of drug treatment, and tumors were harvested and analyzed.
4.13. Statistical Analysis
The Student’s t test and a one-way ANOVA were used to compare the differences of two groups or multiple groups. Data were presented as means ± SD of three independent experiments. A value of p < 0.05 was considered statistically significant.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







