
Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis


Abstract
:1. Introduction
2. Neurofibromatosis Type 1
2.1. Clinical Characteristics
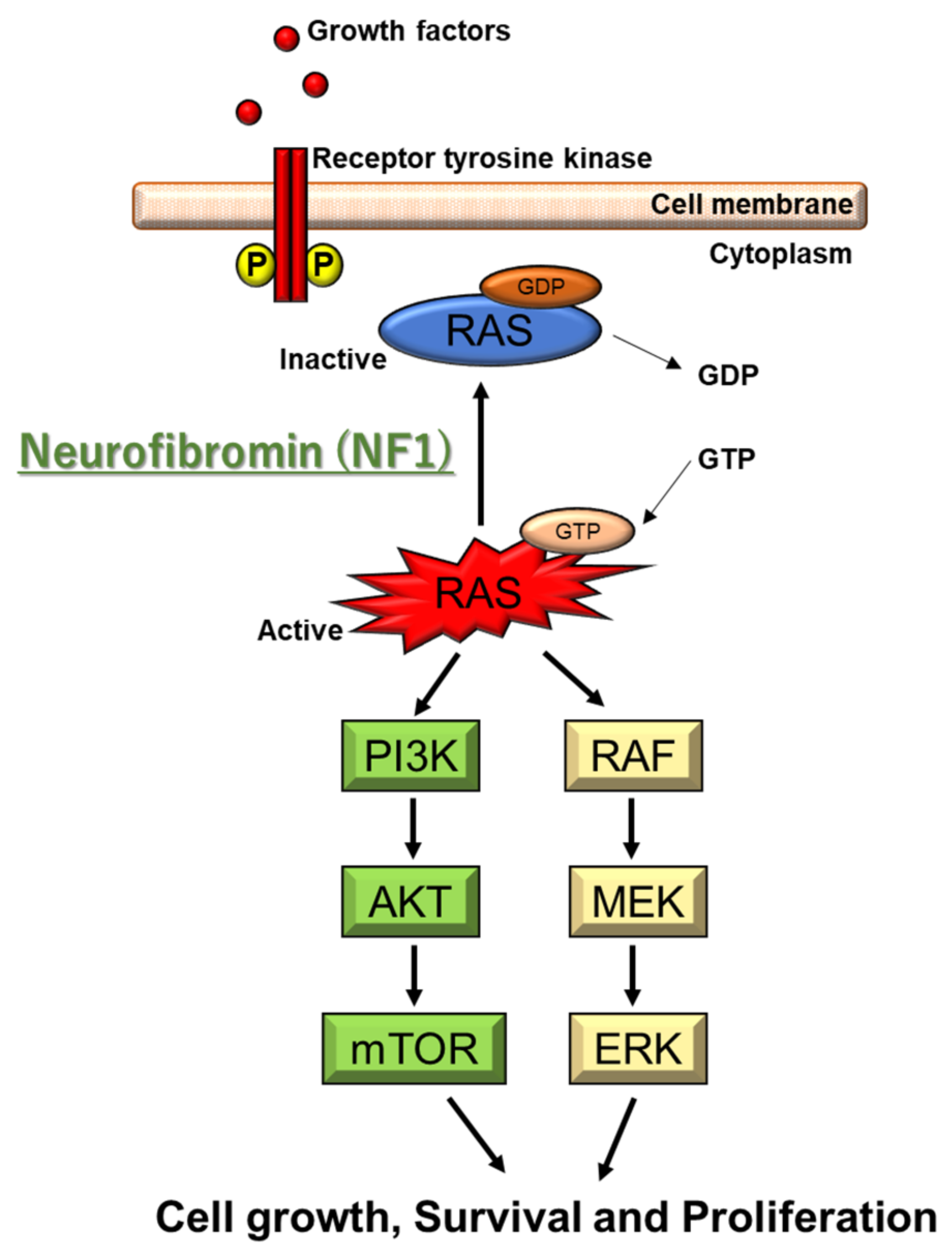
2.2. Genetic and Molecular Characteristics
2.3. Therapeutic Strategies
2.4. Ongoing Clinical Trials
2.5. Animal Models
3. Neurofibromatosis Type 2
3.1. Clinical Characteristics
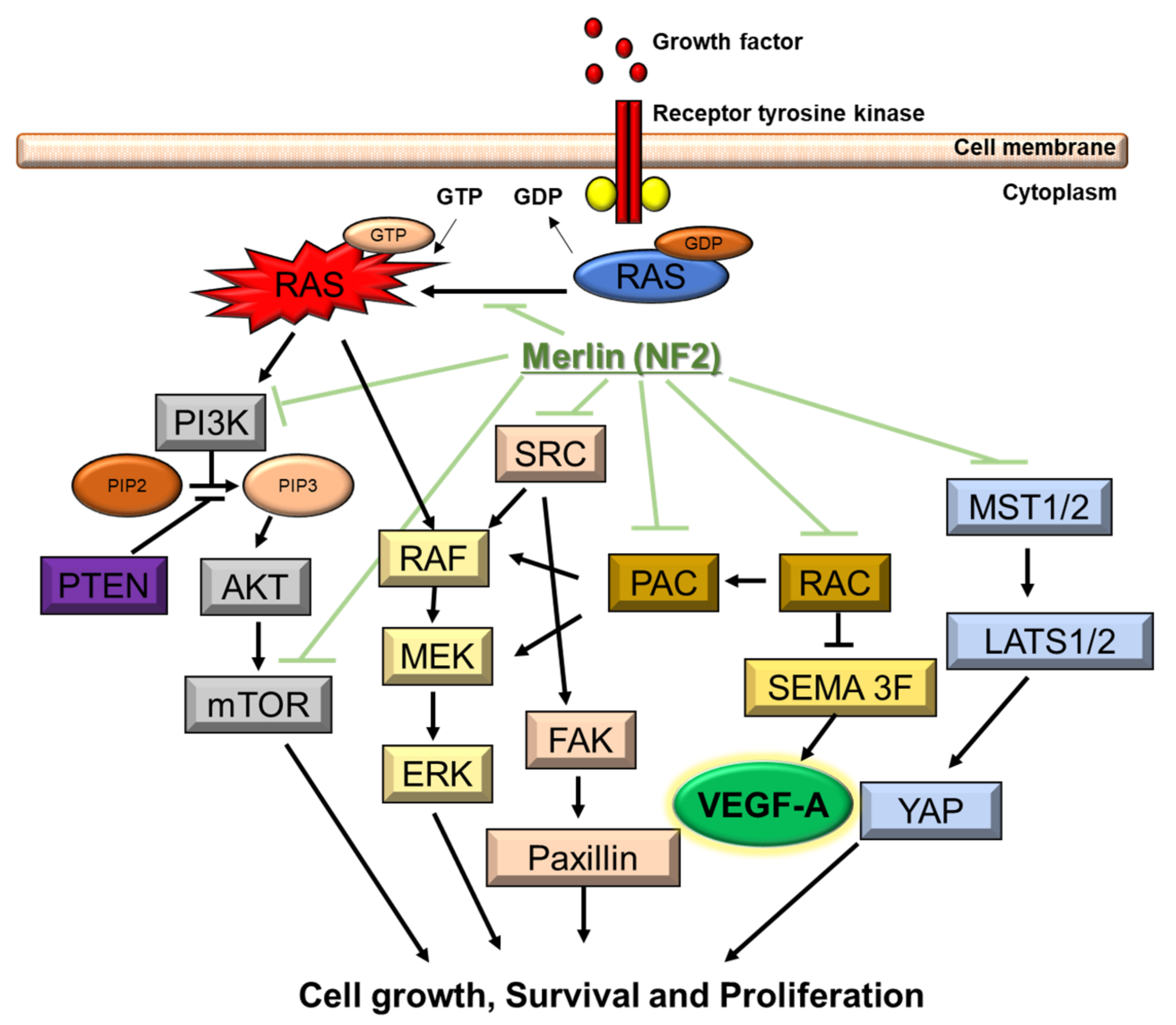
3.2. Genetic and Molecular Characteristics
3.3. Therapeutic Strategies
3.4. Ongoing Clinical Trials
3.5. Animal Models
4. Schwannomatosis
4.1. Clinical Characteristics
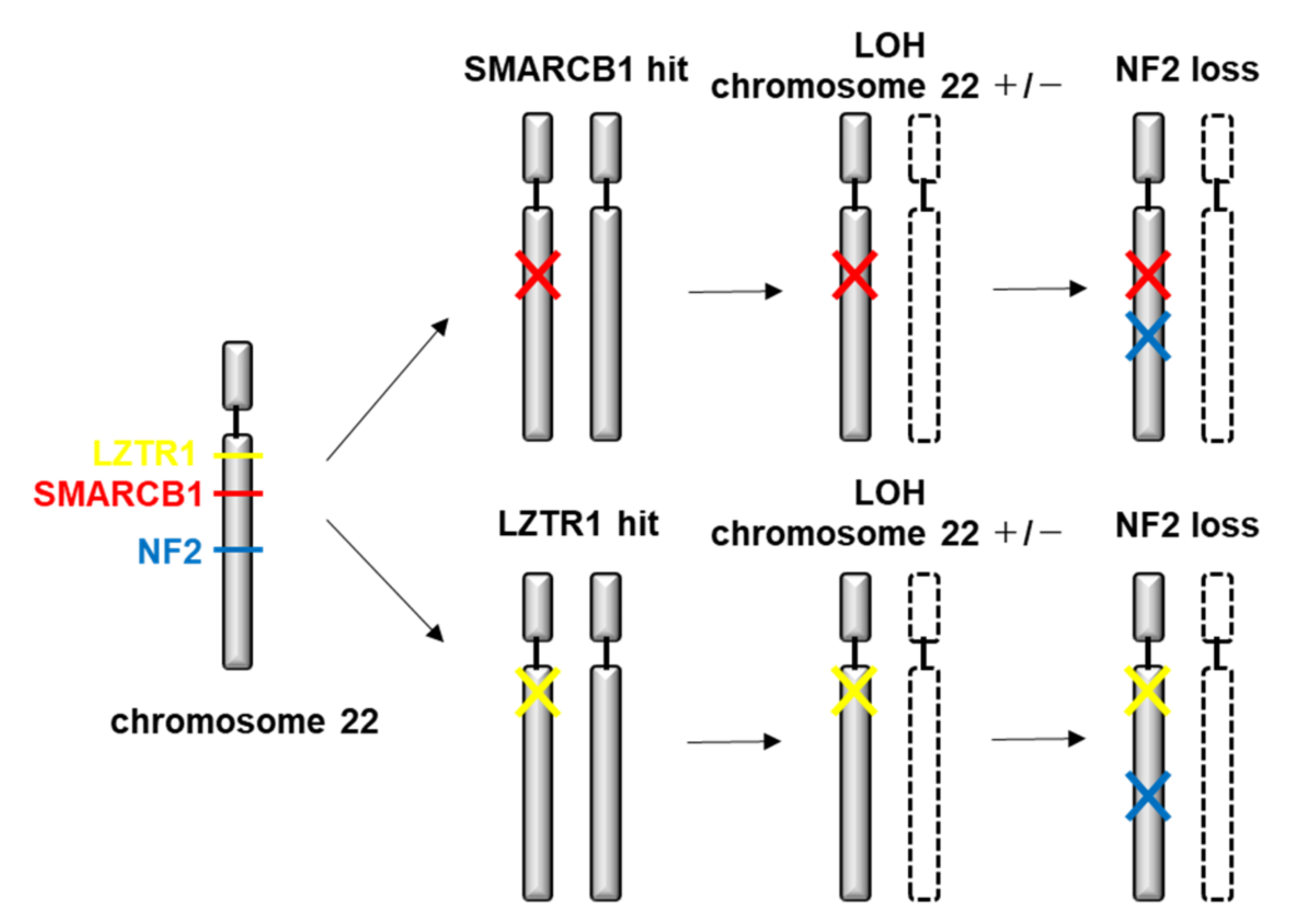
4.2. Genetic and Molecular Characteristics
4.3. Therapeutic Strategies
4.4. Ongoing Clinical Trials
5. Future Direction
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coy, S.; Rashid, R.; Stemmer-Rachamimov, A.; Santagata, S. An update on the CNS manifestations of neurofibromatosis type 2. Acta Neuropathol. 2020, 139, 643–665. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.G.; Bowers, N.L.; Tobi, S.; Hartley, C.; Wallace, A.J.; King, A.T.; Lloyd, S.K.W.; Rutherford, S.A.; Hammerbeck-Ward, C.; Pathmanaban, O.N.; et al. Schwannomatosis: A genetic and epidemiological study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1215. [Google Scholar] [CrossRef]
- Nix, J.S.; Blakeley, J.; Rodriguez, F.J. An update on the central nervous system manifestations of neurofibromatosis type 1. Acta Neuropathol. 2020, 139, 625–641. [Google Scholar] [CrossRef]
- Kresak, J.L. Neurofibromatosis: A Review of NF1, NF2, and Schwannomatosis. J. Pediatr. Genet. 2016, 5, 98–104. [Google Scholar]
- Blakeley, J.O.; Plotkin, S.R. Therapeutic advances for the tumors associated with neurofibromatosis type 1, type 2, and schwannomatosis. Neuro. Oncol. 2016, 18, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Campian, J.; Gutmann, D.H. CNS Tumors in Neurofibromatosis. J. Clin. Oncol. 2017, 35, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.M.; Lallo, F. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am. J. Med. Genet. A 2010, 152A, 327. [Google Scholar] [CrossRef]
- Kallionpää, R.A.; Uusitalo, E.; Leppävirta, J.; Pöyhönen, M.; Peltonen, S.; Peltonen, J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet. Med. 2018, 20, 1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggieri, M.; Huson, S.M. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology 2001, 56, 1433. [Google Scholar] [CrossRef] [PubMed]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet. Med. 2021, 19. [Google Scholar] [CrossRef]
- Stephens, K.; Kayes, L.; Riccardi, V.M.; Rising, M.; Sybert, V.P.; Pagon, R.A. Preferential mutation of the neurofibromatosis type 1 gene in paternally derived chromosomes. Hum. Genet. 1992, 88, 279. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Yang, Q.; Friedman, J.M. Mortality in neurofibromatosis 1: An analysis using U.S. death certificates. Am. J. Hum. Genet. 2001, 68, 1110–1118. [Google Scholar] [CrossRef] [Green Version]
- Landry, J.P.; Schertz, K.L.; Chiang, Y.J.; Bhalla, A.D.; Yi, M.; Keung, E.Z.; Scally, C.P.; Feig, B.W.; Hunt, K.K.; Roland, C.L.; et al. Comparison of Cancer Prevalence in Patients With Neurofibromatosis Type 1 at an Academic Cancer Center vs in the General Population From 1985 to 2020. JAMA Netw. Open. 2021, 4, e210945. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Sabbagh, A.; Hanna, N.; Masliah-Planchon, J.; Jolly, E.; Goussard, P.; Ballerini, P.; Cartault, F.; Barbarot, S.; Landman-Parker, J.; et al. SPRED1 germline mutations caused a neurofibromatosis type 1 overlapping phenotype. J. Med. Genet. 2009, 46, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffzek, K.; Ahmadian, M.R.; Wiesmüller, L.; Kabsch, W.; Stege, P.; Schmitz, F.; Wittinghofer, A. Structural analysis of the GAP-related domain from neurofibromin and its implications. EMBO J. 1998, 17, 4313–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltonen, S.; Kallionpää, R.A.; Peltonen, J. Neurofibromatosis type 1 (NF1) gene: Beyond café au lait spots and dermal neurofibromas. Exp. Dermatol. 2017, 26, 645–648. [Google Scholar] [CrossRef] [Green Version]
- Trovó-Marqui, A.B.; Tajara, E.H. Neurofibromin: A general outlook. Clin. Genet. 2006, 70, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Messiaen, L.; Wimmer, K. NF1 mutational spectrum. Monogr. Hum. Genet. 2008, 16, 63. [Google Scholar]
- Kluwe, L.; Friedrich, R.E.; Peiper, M.; Friedman, J.; Mautner, V.F. Constitutional NF1 mutations in neurofibromatosis 1 patients with malignant peripheral nerve sheath tumors. Hum. Mutat. 2003, 22, 420. [Google Scholar] [CrossRef]
- Farid, M.; Demicco, E.G.; Garcia, R.; Ahn, L.; Merola, P.R.; Cioffi, A.; Maki, R.G. Malignant peripheral nerve sheath tumors. Oncologist 2014, 19, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, M.; Spurlock, G.; Majounie, E.; Griffiths, S.; Forrester, N.; Baser, M.; Huson, S.M.; Gareth, E.D.; Ferner, R. The heterogeneous nature of germline mutations in NF1 patients with malignant peripheral serve sheath tumours (MPNSTs). Hum. Mutat. 2006, 27, 716. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Callens, T.; Gomes, A.; Sharp, A.; Chen, Y.; Hicks, A.D.; Aylsworth, A.S.; Azizi, A.A.; Basel, D.G.; Bellus, G.; et al. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): An update of genotype-phenotype correlation. Genet. Med. 2019, 21, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844-848. Am. J. Hum. Genet. 2018, 102, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczkowska, M.; Callens, T.; Chen, Y.; Gomes, A.; Hicks, A.D.; Sharp, A.; Johns, E.; Uhas, K.A.; Armstrong, L.; Bosanko, K.A.; et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: Genotype-phenotype study in neurofibromatosis type 1. Hum. Mutat. 2020, 41, 299. [Google Scholar] [CrossRef] [PubMed]
- Quesnel, B.; Preudhomme, C.; Vanrumbeke, M.; Vachee, A.; Lai, J.L.; Fenaux, P. Absence of rearrangement of the neurofibromatosis 1 (NF1) gene in myelodysplastic syndromes and acute myeloid leukemia. Leukemia 1994, 8, 878–880. [Google Scholar]
- Messiaen, L.M.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; Roy, N.V.; Paepe, A.D. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum. Mutat. 2000, 15, 541. [Google Scholar] [CrossRef]
- Castle, B.; Baser, M.E.; Huson, S.M.; Cooper, D.N.; Upadhyaya, M. Evaluation of genotype-phenotype correlations in neurofibromatosis type 1. J. Med. Genet. 2003, 40, e109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardecchia, E.; Perfetti, L.; Castiglioni, M.; Di Natale, D.; Imperatori, A.; Rotolo, N. Bullous lung disease and neurofibromatosis type-1. Monaldi Arch. Chest Dis. 2012, 77, 105. [Google Scholar] [CrossRef] [Green Version]
- Katz, D.; Lazar, A.; Lev, D. Malignant peripheral nerve sheath tumour (MPNST): The clinical implications of cellular signalling pathways. Expert Rev. Mol. Med. 2009, 11, e30. [Google Scholar] [CrossRef]
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Maitra, A.; Choudhury, S. Selumetinib: The first ever approved drug for neurofibromatosis-1 related inoperable plexiform neurofibroma. Curr. Med. Res. Opin. 2021, 8, 1. [Google Scholar]
- DeBella, K.; Szudek, J.; Friedman, J.M. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000, 105, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, L.M.; Doroftei, O. Optic glioma warranting treatment in children. Eye 2006, 20, 1149–1164. [Google Scholar] [CrossRef] [PubMed]
- Brannan, C.I.; Perkins, A.S.; Vogel, K.S.; Ratner, N.; Nordlund, M.L.; Reid, S.W.; Buchberg, A.M.; Jenkins, N.A.; Parada, L.F.; Copeland, N.G. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994, 8, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Jacks, T.; Shih, T.S.; Schmitt, E.M.; Bronson, R.T.; Bernards, A.; Weinberg, R.A. Tumor predisposition in mice heterozygous for a targeted mutation in NF1. Nat. Genet. 1994, 7, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Lakkis, M.M.; Epstein, J.A. Neurofibromin modulation of ras activity is required for normal endocardial-mesenchymal transformation in the developing heart. Development 1998, 125, 4359–4367. [Google Scholar] [CrossRef] [PubMed]
- Lakkis, M.M.; Golden, J.A.; O’Shea, S.; Epstein, J.A. Neurofibromin deficiency in mice causes exencephaly and is a modifier for Splotch neural tube defects. Dev. Biol. 1999, 212, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Bajenaru, M.L.; Donahoe, J.; Corral, T.; Keilly, K.M.; Brophy, S.; Pellicer, A.; Gutmann, D.H. Neurofibromatosis 1 (NF1) heterozygosity results in a cell—Autonomous growth advantage for astrocytes. Glia 2001, 33, 314–323. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Loehr, A.; Zhang, Y.; Kim, J.; Henkemeyer, M.; Cashen, A. Haploinsufficiency for the neurofibromatosis 1 (NF1) tumor suppressor results in increased astrocyte proliferation. Oncogene 1999, 18, 4450–4459. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Romero, M.I.; Ghosh, P.; Ye, Z.; Charnay, P.; Rushing, E.J.; Marth, J.D.; Parada, L.F. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001, 15, 859–876. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.M.; Yang, T.; Huynh, D.P.; Pulst, S.M.; Viskochil, D.H.; Silva, A.J.; Brannan, C.I. Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nat. Genet. 2001, 27, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, M.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.; Tschida, B.; Moriarity, B.; et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat. Commun. 2015, 6, 7391. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.H.; Zhao, D.; Chen, J.; Alcantara, S.; Li, Y.; Burns, D.K.; Mason, R.P.; Lee, E.Y.; Wu, H.; Parada, L.F. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 2008, 68, 3286–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, K.M.; Loisel, D.A.; Bronson, R.T.; McLaughlin, M.E.; Jacks, T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat. Genet. 2000, 26, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Marumoto, T.; Tashiro, A.; Friedmann-Morvinski, D.; Scadeng, M.; Soda, Y.; Gage, F.H.; Verma, I.M. Development of a novel mouse glioma model using lentiviral vectors. Nat. Med. 2009, 15, 110–116. [Google Scholar] [CrossRef]
- Bajenaru, M.L.; Hernandez, M.R.; Perry, A.; Zhu, Y.; Parada, L.F.; Garbow, J.R.; Gutmann, D.H. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003, 63, 8573–8577. [Google Scholar] [PubMed]
- Isakson, S.H.; Rizzardi, A.E.; Coutts, A.W.; Carlson, D.F.; Kirstein, M.N.; Fisher, J.; Vitte, J.; Williams, K.B.; Pluhar, G.E.; Dahiya, S.; et al. Genetically engineered minipigs model the major clinical features of human neurofibromatosis type 1. Commun Biol. 2018, 1, 158. [Google Scholar] [CrossRef] [Green Version]
- White, K.A.; Swier, V.J.; Cain, J.T.; Kohlmeyer, J.L.; Meyerholz, D.K.; Tanas, M.R.; Uthoff, J.; Hammond, E.; Li, H.; Rohret, F.A.; et al. A porcine model of neurofibromatosis type 1 that mimics the human disease. JCI Insight. 2018, 3, e120402. [Google Scholar] [CrossRef]
- Evans, D.G.; Hartley, C.L.; Smith, P.T.; King, A.T.; Bowers, N.L.; Tobi, S.; Wallace, A.J.; Perry, M.; Anup, R.; Lloyd, S.K.W.; et al. Incidence of mosaicism in 1055 de novo NF2 cases: Much higher than previous estimates with high utility of next-generation sequencing. Genet. Med. 2020, 22, 53. [Google Scholar] [CrossRef]
- Evans, D.G.; Ramsden, R.T.; Shenton, A.; Gokhale, C.; Bowers, N.L.; Huson, S.M.; Pichert, G.; Wallace, A. Mosaicism in neurofibromatosis type 2: An update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including multiple ligation-dependent probe amplification. J. Med. Genet. 2007, 44, 424. [Google Scholar] [CrossRef]
- Evans, D.G.; Moran, A.; King, A.; Saeed, S.; Gurusinghe, N.; Ramsden, R. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: Higher incidence than previously thought. Otol. Neurotol. 2005, 26, 93. [Google Scholar] [CrossRef]
- Godel, T.; Bäumer, P.; Farschtschi, S.; Gugel, I.; Kronlage, M.; Hofstadler, B.; Heiland, S.; Gelderblom, M.; Bendszus, M.; Mautner, V.F. Peripheral nervous system alterations in infant and adult neurofibromatosis type 2. Neurology 2019, 93, e590. [Google Scholar] [CrossRef] [PubMed]
- Picry, A.; Bonne, N.X.; Ding, J.; Aboukais, R.; Lejeune, J.P.; Baroncini, M.; Dubrulle, F.; Vincent, C. Long-term growth rate of vestibular schwannoma in neurofibromatosis 2: A volumetric consideration. Laryngoscope 2016, 126, 2358–2362. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J. Rare Dis. 2009, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Sperfeld, A.D.; Hein, C.; Schröder, J.M.; Ludolph, A.C.; Hanemann, C.O. Occurrence and characterization of peripheral nerve involvement in neurofibromatosis type 2. Brain 2002, 125, 996. [Google Scholar] [CrossRef] [PubMed]
- Gaudioso, C.; Listernick, R.; Fisher, M.J.; Campen, C.J.; Paz, A.; Gutmann, D.H. Neurofibromatosis 2 in children presenting during the first decade of life. Neurology 2019, 93, e964. [Google Scholar] [CrossRef]
- McLaughlin, M.E.; Pepin, S.M.; Maccollin, M.; Choopong, P.; Lessell, S. Ocular pathologic findings of neurofibromatosis type 2. Arch. Ophthalmol. 2007, 125, 389. [Google Scholar] [CrossRef] [Green Version]
- Painter, S.L.; Sipkova, Z.; Emmanouil, B.; Halliday, D.; Parry, A.; Elston, J.S. Neurofibromatosis Type 2-Related Eye Disease Correlated with Genetic Severity Type. J. Neuroophthalmol. 2019, 39, 44. [Google Scholar] [CrossRef]
- Castellanos, E.; Plana, A.; Carrato, C.; Carrió, M.; Rosas, I.; Amilibia, E.; Roca-Ribas, F.; Hostalot, C.; Castillo, A.; Ros, A.; et al. Early Genetic Diagnosis of Neurofibromatosis Type 2 from Skin Plaque Plexiform Schwannomas in Childhood. JAMA Dermatol. 2018, 154, 341. [Google Scholar] [CrossRef]
- Kluwe, L.; Mautner, V.; Heinrich, B.; Dezube, R.; Jacoby, L.B.; Friedrich, R.E.; MacCollin, M. Molecular study of frequency of mosaicism in neurofibromatosis 2 patients with bilateral vestibular schwannomas. J. Med. Genet. 2003, 40, 109. [Google Scholar] [CrossRef] [Green Version]
- Moyhuddin, A.; Baser, M.E.; Watson, C.; Purcell, S.; Ramsden, R.T.; Heiberg, A.; Wallace, A.J.; Evans, D.G. Somatic mosaicism in neurofibromatosis 2: Prevalence and risk of disease transmission to offspring. J. Med. Genet. 2003, 40, 459. [Google Scholar] [CrossRef] [Green Version]
- Selvanathan, S.K.; Shenton, A.; Ferner, R.; Wallace, A.J.; Huson, S.M.; Ramsden, R.T.; Evans, D.G. Further genotype—Phenotype correlations in neurofibromatosis 2. Clin. Genet. 2010, 77, 163. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Higgs, J.E.; Bowers, N.L.; Halliday, D.; Paterson, J.; Gillespie, J.; Huson, S.M.; Freeman, S.R.; Lloyd, S.; Rutherford, S.A.; et al. Cranial meningiomas in 411 neurofibromatosis type 2 (NF2) patients with proven gene mutations: Clear positional effect of mutations, but absence of female severity effect on age at onset. J. Med. Genet. 2011, 48, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hexter, A.; Jones, A.; Joe, H.; Heap, L.; Smith, M.J.; Wallace, A.J.; Halliday, D.; Parry, A.; Taylor, A.; Raymond, L.; et al. Clinical and molecular predictors of mortality in neurofibromatosis 2: A UK national analysis of 1192 patients. J. Med. Genet. 2015, 52, 699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.G.; Ramsden, R.T.; Gokhale, C.; Bowers, N.; Huson, S.M.; Wallace, A. Should NF2 mutation screening be undertaken in patients with an apparently isolated vestibular schwannoma? Clin. Genet. 2007, 71, 354. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Wallace, A.J.; Hartley, C.; Freeman, S.R.; Lloyd, S.K.; Thomas, O.; Axon, P.; Hammerbeck-Ward, C.L.; Pathmanaban, O.; Rutherford, S.A.; et al. Familial unilateral vestibular schwannoma is rarely caused by inherited variants in the NF2 gene. Laryngoscope 2019, 129, 967. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.G.; Baser, M.E.; O’Reilly, B.; Rowe, J.; Gleeson, M.; Saeed, S.; King, A.; Huson, S.M.; Kerr, R.; Thomas, N.; et al. Management of the patient and family with neurofibromatosis 2: A consensus conference statement. Br. J. Neurosurg. 2005, 19, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Dewan, R.; Pemov, A.; Kim, H.J.; Morgan, K.L.; Vasquez, R.A.; Chittiboina, P.; Wang, X.; Chandrasekharappa, S.C.; Ray-Chaudhury, A.; Butman, J.A.; et al. Evidence of polyclonality in neurofibromatosis type 2-associated multilobulated vestibular schwannomas. Neuro. Oncol. 2015, 17, 566. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, D.; Kondziolka, D.; Flickinger, J.C.; Niranjan, A.; Williamson, R.; Martin, J.J.; Lunsford, L.D. Stereotactic radiosurgery for vestibular schwannomas in patients with neurofibromatosis type 2: An analysis of tumor control, complications, and hearing preservation rates. Neurosurgery 2007, 60, 460. [Google Scholar] [CrossRef]
- Sharma, M.S.; Singh, R.; Kale, S.S.; Agrawal, D.; Sharma, B.S.; Mahapatra, A.K. Tumor control and hearing preservation after Gamma Knife radiosurgery for vestibular schwannomas in neurofibromatosis type 2. J. Neurooncol. 2010, 98, 265–270. [Google Scholar] [CrossRef]
- McClelland, S., 3rd; Gerbi, B.J.; Cho, K.H.; Hall, W.A. The treatment of a large acoustic tumor with fractionated stereotactic radiotherapy. J. Robot. Surg. 2007, 1, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Seferis, C.; Torrens, M.; Paraskevopoulou, C.; Psichidis, G. Malignant transformation in vestibular schwannoma: Report of a single case, literature search, and debate. J. Neurosurg. 2014, 121, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Balasubramaniam, A.; Shannon, P.; Hodaie, M.; Laperriere, N.; Michaels, H.; Guha, A. Glioblastoma multiforme after stereotactic radiotherapy for acoustic neuroma: Case report and review of the literature. Neuro Oncol. 2007, 9, 447. [Google Scholar] [CrossRef] [PubMed]
- Cayé-Thomasen, P. VEGF and VEGF receptor-1 concentration in vestibular schwannoma homogenates correlates to tumor growth rate. Otol. Neurotol. 2005, 26, 98–101. [Google Scholar] [CrossRef]
- Komotar, R.J.; Starke, R.M.; Sisti, M.B.; Connolly, E.S. The role of bevacizumab in hearing preservation and tumor volume control in patients with vestibular schwannomas. Neurosurgery 2009, 65, N12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alanin, M.C.; Klausen, C.; Caye-Thomasen, P.; Thomsen, C.; Fugleholm, K.; Poulsgaard, L.; Lassen, U.; Mau-Sorensen, M.; Hofland, K.F. The effect of bevacizumab on vestibular schwannoma tumour size and hearing in patients with neurofibromatosis type 2. Eur. Arch. Otorhinolaryngol. 2015, 272, 3627–3633. [Google Scholar] [CrossRef] [PubMed]
- Blakeley, J.; Schreck, K.C.; Evans, D.G.; Korf, B.R.; Zagzag, D.; Karajannis, M.A.; Bergner, A.L.; Belzberg, A.J. Clinical response to bevacizumab in schwannomatosis. Neurology 2014, 83, 1986–1987. [Google Scholar] [CrossRef] [Green Version]
- Eminowicz, G.K.; Raman, R.; Conibear, J.; Plowman, P.N. Bevacizumab treatment for vestibular schwannomas in neurofibromatosis type two: Report of two cases, including responses after prior gamma knife and vascular endothelial growth factor inhibition therapy. J. Laryngol. Otol. 2012, 126, 79–82. [Google Scholar] [CrossRef]
- Liu, P.; Yao, Q.; Li, N.A.; Liu, Y.; Wang, Y.; Li, M.; Li, Z.; Li, J.; Li, G. Low-dose bevacizumab induces radiographic regression of vestibular schwannomas in neurofibromatosis type 2: A case report and literature review. Oncol. Lett. 2016, 11, 2981–2986. [Google Scholar] [CrossRef] [Green Version]
- Mautner, V.F.; Nguyen, R.; Kutta, H.; Fuensterer, C.; Bokemeyer, C.; Hagel, C.; Friedrich, R.E.; Panse, J. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro. Oncol. 2010, 12, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, S.R.; Stemmer-Rachamimov, A.O.; Barker, F.G., 2nd; Halpin, C.; Padera, T.P.; Tyrrell, A.; Sorensen, A.G.; Jain, R.K.; di Tomaso, E. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N. Engl. J. Med. 2009, 361, 358–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, S.R.; Merker, V.L.; Halpin, C.; Jennings, D.; McKenna, M.J.; Harris, G.J.; Barker, F.G., 2nd. Bevacizumab for progressive vestibular schwannoma in neurofibromatosis type 2: A retrospective review of 31 patients. Otol. Neurotol. 2012, 33, 1046–1052. [Google Scholar] [CrossRef]
- Subbiah, V.; Slopis, J.; Hong, D.S.; Ketonen, L.M.; Hamilton, J.; McCutcheon, I.E.; Kurzrock, R. Treatment of patients with advanced neurofibromatosis type 2 with novel molecularly targeted therapies: From bench to bedside. J. Clin. Oncol. 2012, 30, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Versleijen, M.W.; Verbist, B.M.; Mulder, J.J.; de Geus-Oei, L.F.; van Herpen, C.M. Avastin scintigraphy in surveillance of bevacizumab treatment in a patient with neurofibromatosis type 2: A case report. Clin. Nucl. Med. 2014, 39, 277–280. [Google Scholar] [CrossRef]
- Morris, K.A.; Golding, J.F.; Axon, P.R.; Afridi, S.; Blesing, C.; Ferner, R.E.; Halliday, D.; Jena, R.; Pretorius, P.M. Bevacizumab in Neurofibromatosis type 2 (NF2) related vestibular schwannomas: A nationally coordinated approach to delivery and prospective evaluation. Neuro-Oncol. Pract. 2016, 3, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Lu, V.M.; Ravindran, K.; Graffeo, C.S.; Perry, A.; Van Gompel, J.J.; Daniels, D.J.; Link, M.J. Efficacy and safety of bevacizumab for vestibular schwannoma in neurofibromatosis type 2: A systematic review and meta-analysis of treatment outcomes. J. Neurooncol. 2019, 144, 239. [Google Scholar] [CrossRef]
- Plotkin, S.R.; Duda, D.G.; Muzikansky, A.; Allen, J.; Blakeley, J.; Rosser, T.; Campian, J.L.; Clapp, D.W.; Fisher, M.J.; Tonsgard, J.; et al. Multicenter, Prospective, Phase II and Biomarker Study of High-Dose Bevacizumab as Induction Therapy in Patients with Neurofibromatosis Type 2 and Progressive Vestibular Schwannoma. J. Clin. Oncol. 2019, 37, 3446. [Google Scholar] [CrossRef]
- Ouerdani, A.; Goutagny, S.; Kalamarides, M.; Trocóniz, I.F.; Ribba, B. Mechanism-based modeling of the clinical effects of bevacizumab and everolimus on vestibular schwannomas of patients with neurofibromatosis type 2. Cancer Chemother. Pharmacol. 2016, 77, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Fujioka, M.; Morimoto, Y.; Ohara, K.; Kosugi, K.; Oishi, Y.; Sato, M.; Ueda, R.; Fujiwara, H.; Hikichi, T.; et al. A VEGF receptor vaccine demonstrates preliminary efficacy in neurofibromatosis type 2. Nat. Commun. 2020, 11, 2028. [Google Scholar] [CrossRef]
- Goutagny, S.; Raymond, E.; Esposito-Farese, M.; Trunet, S.; Mawrin, C.; Bernardeschi, D.; Larroque, B.; Sterkers, O.; Giovannini, M.; Kalamarides, M. Phase II study of mTORC1 inhibition by everolimus in neurofibromatosis type 2 patients with growing vestibular schwannomas. J. Neurooncol. 2015, 122, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karajannis, M.A.; Legault, G.; Hagiwara, M.; Giancotti, F.G.; Filatov, A.; Derman, A.; Hochman, T.; Goldberg, J.D.; Vega, E.; Wisoff, J.H.; et al. Phase II study of everolimus in children and adults with neurofibromatosis type 2 and progressive vestibular schwannomas. Neuro. Oncol. 2014, 16, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karajannis, M.A.; Legault, G.; Hagiwara, M.; Ballas, M.S.; Brown, K.; Nusbaum, A.O.; Hochman, T.; Goldberg, J.D.; Koch, K.M.; Golfinos, J.G.; et al. Phase II trial of lapatinib in adult and pediatric patients with neurofibromatosis type 2 and progressive vestibular schwannomas. Neuro. Oncol. 2012, 14, 1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, S.R.; Halpin, C.; McKenna, M.J.; Loeffler, J.S.; Batchelor, T.T.; Barker, F.G., 2nd. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol. Neurotol. 2010, 31, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, R.; Morimoto, Y.; Sato, M.; Kuranari, Y.; Oishi, Y.; Kosugi, K.; Yoshida, K.; Toda, M. Difference in the hypoxic immunosuppressive microenvironment of patients with neurofibromatosis type 2 schwannomas and sporadic schwannomas. J. Neurooncol. 2020, 146, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Deep, N.L.; Patel, E.J.; Shapiro, W.H.; Waltzman, S.B.; Jethanamest, D.; McMenomey, S.O.; Roland, J.T., Jr.; Friedmann, D.R. Cochlear Implant Outcomes in Neurofibromatosis Type 2: Implications for Management. Otol. Neurotol. 2021, 42, 540–548. [Google Scholar] [CrossRef]
- Neff, B.A.; Wiet, R.M.; Lasak, J.M.; Cohen, N.L.; Pillsbury, H.C.; Ramsden, R.T.; Welling, D.B. Cochlear implantation in the neurofibromatosis type 2 patient: Long-term follow-up. Laryngoscope 2007, 117, 1069. [Google Scholar] [CrossRef]
- Funes, C.J.; Mace, R.A.; Macklin, E.A.; Plotkin, S.R.; Jordan, J.T.; Vranceanu, A.M. First report of quality of life in adults with neurofibromatosis 2 who are deafened or have significant hearing loss: Results of a live-video randomized control trial. J. Neurooncol. 2019, 143, 505. [Google Scholar] [CrossRef]
- Evans, D.G.; Watson, C.; King, A.; Wallace, A.J.; Baser, M.E. Multiple meningiomas: Differential involvement of the NF2 gene in children and adults. J. Med. Genet. 2005, 42, 45. [Google Scholar] [CrossRef]
- Larson, J.J.; van Loveren, H.R.; Balko, M.G.; Tew, J.M., Jr. Evidence of meningioma infiltration into cranial nerves: Clinical implications for cavernous sinus meningiomas. J. Neurosurg. 1995, 83, 596. [Google Scholar] [CrossRef]
- Wentworth, S.; Pinn, M.; Bourland, J.D.; Deguzman, A.F.; Ekstrand, K.; Ellis, T.L.; Glazier, S.S.; McMullen, K.P.; Munley, M.; Stieber, V.W.; et al. Clinical experience with radiation therapy in the management of neurofibromatosis-associated central nervous system tumors. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 208. [Google Scholar] [CrossRef]
- Osorio, D.S.; Hu, J.; Mitchell, C.; Allen, J.C.; Stanek, J.; Hagiwara, M.; Karajannis, M.A. Effect of lapatinib on meningioma growth in adults with neurofibromatosis type 2. J. Neurooncol. 2018, 139, 749. [Google Scholar] [CrossRef]
- Nunes, F.P.; Merker, V.L.; Jennings, D.; Caruso, P.A.; di Tomaso, E.; Muzikansky, A.; Barker, F.G., 2nd; Stemmer-Rachamimov, A.; Plotkin, S.R. Bevacizumab treatment for meningiomas in NF2: A retrospective analysis of 15 patients. PLoS ONE 2013, 8, e59941. [Google Scholar] [CrossRef] [Green Version]
- Fujii, M.; Ichikawa, M.; Iwatate, K.; Bakhit, M.; Yamada, M.; Kuromi, Y.; Sato, T.; Sakuma, J.; Saito, K. Bevacizumab Therapy of Neurofibromatosis Type 2 Associated Vestibular Schwannoma in Japanese Patients. Neurol. Med. Chir. 2020, 60, 75–82. [Google Scholar] [CrossRef] [Green Version]
- McClatchey, A.I.; Saotome, I.; Ramesh, V.; Gusella, J.F.; Jacks, T. The NF2 tumor suppressor gene product is essential for extraembryonic development immediately prior to gastrulation. Genes Dev. 1997, 11, 1253–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannini, M.; Robanus-Maandag, E.; Niwa-Kawakita, M.; van der Valk, M.; Woodruff, J.M.; Goutebroze, L.; Merel, P.; Berns, A.; Thomas, G. Schwann cell hyperplasia and tumors in transgenic mice expressing a naturally occurring mutant NF2 protein. Genes Dev. 1999, 13, 978–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalamarides, M.; Niwa-Kawakita, M.; Leblois, H.; Abramowski, V.; Perricaudet, M.; Janin, A.; Thomas, G.; Gutmann, D.H.; Giovanni, M. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev. 2002, 16, 1060–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Landegger, L.D.; Sun, Y.; Ren, J.; Maimon, N.; Wu, L.; Ng, M.R.; Chen, J.W.; Zhang, N.; Zhao, Y.; et al. A cerebellopontine angle mouse model for the investigation of tumor biology, hearing, and neurological function in NF2-related vestibular schwannoma. Nat. Protoc. 2019, 14, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Kalamarides, M.; Stemmer-Rachamimov, A.O.; Takahashi, M.; Han, Z.Y.; Chareyre, F.; Niwa-Kawakita, M.; Black, P.M.; Carroll, R.S.; Giovannini, M. Natural history of meningioma development in mice reveals: A synergy of Nf2 and p16(Ink4a) mutations. Brain Pathol. 2008, 18, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, S.S.; Chang, L.S. Generation of noninvasive, quantifiable, orthotopic animal models for NF2-associated schwannoma and meningioma. Methods Mol. Biol. 2016, 1427, 59–72. [Google Scholar]
- MacCollin, M.; Woodfin, W.; Kronn, D.; Short, M.P. Schwannomatosis: A clinical and pathologic study. Neurology 1996, 46, 1072. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.R.; Bredella, M.A.; Cai, W.; Kassarjian, A.; Harris, G.J.; Esparza, S.; Merker, V.L.; Munn, L.L.; Muzikansky, A.; Askenazi, M.; et al. Quantitative assessment of whole-body tumor burden in adult patients with neurofibromatosis. PLoS ONE 2012, 7, e35711. [Google Scholar] [CrossRef] [Green Version]
- Merker, V.L.; Esparza, S.; Smith, M.J.; Stemmer-Rachamimov, A.; Plotkin, S.R. Clinical features of schwannomatosis: A retrospective analysis of 87 patients. Oncologist 2012, 17, 1317. [Google Scholar] [CrossRef] [Green Version]
- MacCollin, M.; Chiocca, E.A.; Evans, D.G.; Friedman, J.M.; Horvitz, R.; Jaramillo, D.; Lev, M.; Mautner, V.F.; Niimura, M.; Plotkin, S.R.; et al. Diagnostic criteria for schwannomatosis. Neurology 2005, 64, 1838. [Google Scholar] [CrossRef]
- Carter, J.M.; O’Hara, C.; Dundas, G.; Gilchrist, D.; Collins, M.S.; Eaton, K.; Judkins, A.R.; Biegel, J.A.; Folpe, A.L. Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with “neuroblastoma-like” schwannomatosis and a novel germline SMARCB1 mutation. Am. J. Surg. Pathol. 2012, 36, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacoby, L.B.; MacCollin, M.; Parry, D.M.; Kluwe, L.; Lynch, J.; Jones, D.; Gusella, J.F. Allelic expression of the NF2 gene in neurofibromatosis 2 and schwannomatosis. Neurogenetics 1999, 2, 101. [Google Scholar] [CrossRef] [PubMed]
- Van den Munckhof, P.; Christiaans, I.; Kenter, S.B.; Baas, F.; Hulsebos, T.J. Germline SMARCB1 mutation predisposes to multiple meningiomas and schwannomas with preferential location of cranial meningiomas at the falx cerebri. Neurogenetics 2012, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, K.D.; Newman, W.G.; Bowers, N.L.; Wallace, A.; Bolger, C.; Colley, A.; McCann, E.; Trump, D.; Prescott, T.; Evans, D.G. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J. Med. Genet. 2008, 45, 332. [Google Scholar] [CrossRef]
- Boyd, C.; Smith, M.J.; Kluwe, L.; Balogh, A.; Maccollin, M.; Plotkin, S.R. Alterations in the SMARCB1 (INI1) tumor suppressor gene in familial schwannomatosis. Clin. Genet. 2008, 74, 358. [Google Scholar] [CrossRef]
- Hadfield, K.D.; Smith, M.J.; Urquhart, J.E.; Wallace, A.J.; Bowers, N.L.; King, A.T.; Rutherford, S.A.; Trump, D.; Newman, W.G.; Evans, D.G. Rates of loss of heterozygosity and mitotic recombination in NF2 schwannomas, sporadic vestibular schwannomas and schwannomatosis schwannomas. Oncogene 2010, 29, 6216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsebos, T.J.; Kenter, S.B.; Jakobs, M.E.; Baas, F.; Chong, B.; Delatycki, M.B. SMARCB1/INI1 maternal germ line mosaicism in schwannomatosis. Clin. Genet. 2010, 77, 86. [Google Scholar] [CrossRef]
- Sestini, R.; Bacci, C.; Provenzano, A.; Genuardi, M.; Papi, L. Evidence of a four-hit mechanism involving SMARCB1 and NF2 in schwannomatosis-associated schwannomas. Hum. Mutat. 2008, 29, 227. [Google Scholar] [CrossRef]
- Rousseau, G.; Noguchi, T.; Bourdon, V.; Sobol, H.; Olschwang, S. SMARCB1/INI1 germline mutations contribute to 10% of sporadic schwannomatosis. BMC Neurol. 2011, 11, 9. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.J.; Wallace, A.J.; Bowers, N.L.; Rustad, C.F.; Woods, C.G.; Leschziner, G.D.; Ferner, R.E.; Evans, D.G. Frequency of SMARCB1 mutations in familial and sporadic schwannomatosis. Neurogenetics 2012, 13, 141. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.R.; Blakeley, J.O.; Evans, D.G.; Hanemann, C.O.; Hulsebos, T.J.; Hunter-Schaedle, K.; Kalpana, G.V.; Korf, B.; Messiaen, L.; Papi, L.; et al. Update from the 2011 International Schwannomatosis Workshop: From genetics to diagnostic criteria. Am. J. Med. Genet. A 2013, 161A, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versteege, I.; Sévenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203. [Google Scholar] [CrossRef] [PubMed]
- Eaton, K.W.; Tooke, L.S.; Wainwright, L.M.; Judkins, A.R.; Biegel, J.A. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr. Blood Cancer 2011, 56, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swensen, J.J.; Keyser, J.; Coffin, C.M.; Biegel, J.A.; Viskochil, D.H.; Williams, M.S. Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J. Med. Genet. 2009, 46, 68. [Google Scholar] [CrossRef] [Green Version]
- Caltabiano, R.; Magro, G.; Polizzi, A.; Praticò, A.D.; Ortensi, A.; D’Orazi, V.; Panunzi, A.; Milone, P.; Maiolino, L.; Nicita, F.; et al. A mosaic pattern of INI1/SMARCB1 protein expression distinguishes Schwannomatosis and NF2-associated peripheral schwannomas from solitary peripheral schwannomas and NF2-associated vestibular schwannomas. Childs Nerv. Syst. 2017, 33, 933. [Google Scholar] [CrossRef]
- Piotrowski, A.; Xie, J.; Liu, Y.F.; Poplawski, A.B.; Gomes, A.R.; Madanecki, P.; Fu, C.; Crowley, M.R.; Crossman, D.K.; Armstrong, L.; et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat. Genet. 2014, 46, 182. [Google Scholar] [CrossRef]
- Smith, M.J.; Isidor, B.; Beetz, C.; Williams, S.G.; Bhaskar, S.S.; Richer, W.; O’Sullivan, J.; Anderson, B.; Daly, S.B.; Urquhart, J.E.; et al. Mutations in LZTR1 add to the complex heterogeneity of schwannomatosis. Neurology 2015, 84, 141. [Google Scholar] [CrossRef] [Green Version]
- Frattini, V.; Trifonov, V.; Chan, J.M.; Castano, A.; Lia, M.; Abate, F.; Keir, S.T.; Ji, A.X.; Zoppoli, P.; Niola, F.; et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013, 5, 1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945. [Google Scholar] [CrossRef] [Green Version]
- Algar, E.M.; Muscat, A.; Dagar, V.; Rickert, C.; Chow, C.W.; Biegel, J.A.; Ekert, P.G.; Saffery, R.; Craig, J.; Johnstone, R.W.; et al. Imprinted CDKN1C is a tumor suppressor in rhabdoid tumor and activated by restoration of SMARCB1 and histone deacetylase inhibitors. PLoS ONE 2009, 4, e4482. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, M.J.; Elia, A.E.; Xu, Q.; Thoma, C.R.; Izhar, L.; Leng, Y.; Guo, A.; Chen, Y.N.; Rush, J.; Hsu, P.W.; et al. Global identification of modular cullin-RING ligase substrates. Cell 2011, 147, 459. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| A: The Diagnostic Criteria for NF1 Are Met in an Individual Who Does Not Have a Parent Diagnosed with NF1 if Two or More of the Following Are Present: |
| At least six café-au-lait macules (>5 mm diameter in prepubertal individuals and >15 mm in postpubertal individuals) |
| Freckling in axillary or inguinal regions #1 |
| Optic glioma |
| At least two Lisch nodules identified by slit lamp examination or two or more choroidal abnormalities—defined as bright, patchy nodules imaged by optical coherence tomography/near-infrared reflectance imaging |
| At least two neurofibromas of any type, or one plexiform neurofibroma |
| A distinctive osseous lesion such as sphenoid dysplasia, #2 anterolateral bowing of the tibia, or pseudarthrosis of a long bone |
| A heterozygous pathogenic NF1 variant with a variant allele fraction of 50% in apparently normal tissue such as white blood cells |
| B: A child of a parent who meets the diagnostic criteria specified in A merits a diagnosis of NF1 if one or more of the criteria in A are present |
| ID | Initiation Date | Phase | Nation | N | Disease | Treatment | Primary Outcome |
|---|---|---|---|---|---|---|---|
| NCT04495127 | 8, 2020 | 1 | Japan | 12 | NF1 | Selumetinib | Toxicity |
| NCT01968590 | 8, 2017 | 2 | USA | 320 | NF1 | Cholecalciferol | Bone mineral density |
| NCT03962543 | 9, 2019 | 2 | USA | 100 | NF1 Plexiform Neurofibroma | Mirdametinib (PD-0325901) oral capsule | Complete or partial response rate compared to baseline. |
| NCT03231306 | 11, 2017 | 2 | USA | 40 | NF1 Plexiform Neurofibroma | Binimetinib | Change from Baseline Target Tumor Volume at 12 months |
| NCT02839720 | 4, 2017 | 2 | USA | 24 | Cutaneous Neurofibroma NF1 Optic Nerve Glioma | Selumetinib | Change in the size |
| NCT02407405 | 1, 2016 | 2 | USA | 60 | NF1 Plexiform Neurofibromas | Selumetinib | Determine objective response rate |
| NCT04461886 | 7, 2020 | 3 | Japan | 100 | NF | NPC-12G gel | Discontinuation rate associated with adverse events |
| NCT03871257 | 10, 2019 | 3 | USA | 290 | Low Grade Glioma NF1 Visual Pathway Glioma | Carboplatin Selumetinib Sulfate Vincristine Sulfate | Event-free survival |
| NCT02101736 | 6, 2014 | 2 | USA | 48 | NF1 Neurofibromatosis Plexiform Neurofibromas | Cabozantinib | The change in tumor size based on radiographic assessment |
| NCT03326388 | 9, 2019 | 1/2 | USA | 30 | NF1 Plexiform Neurofibroma Optic Nerve Glioma | Selumetinib | To evaluate the Maximum Tolerated Dose Objective response rate |
| NCT03741101 | 6, 2019 | 2 | Sweden | 15 | NF1 Plexiform Neurofibromas | Trametinib | Remission of tumor volume ≥20% |
| NCT02728388 | 8, 2016 | 2 | USA | 30 | NF1 | aminolevulinic acid | Time to disease progression |
| NCT04435665 | 8, 2020 | 2 | USA | 48 | NF1 Cutaneous Neurofibroma | NFX-179 Gel | Phospho-erk (p-ERK) levels of Target cNF Tumors Toxicity |
| NCT02390752 | 4, 2015 | 1/2 | USA | 81 | Neurofibroma, Plexiform | PLX3397 | Toxicity Objective response rate |
| NCT03688568 | 9, 2018 | 2 | USA | 20 | Neurofibroma, Plexiform | Imatinib Mesylate | Quantitative Functional Airway Response |
| NCT03433183 | 10, 2019 | 2 | USA | 21 | Malignant Peripheral Nerve Sheath Tumors NF1 | Selumetinib Sirolimus | Clinical benefit rate of selumetinib in combination with sirolimus |
| NCT04085159 | 9, 2019 | 1/2 | China | 100 | Neurofibromatosis Schwannomatosis | Antigen-specific T cells CART/CTL and DCvac | Percentage of adverse effects |
| Bilateral vestibular schwannomas or |
| First-degree relative with neurofibromatosis type 2 plus |
| 1. Unilateral vestibular schwannomas or |
| 2. Any two of the following: Meningioma, glioma, schwannoma, or juvenile PLO |
| ID | Initiation Date | Phase | Nation | N | Disease | Treatment | Primary Outcome |
|---|---|---|---|---|---|---|---|
| NCT02934256 | 7, 2016 | 2 | China | 20 | NF2 | Icotinib | Change from Baseline in volume of tumor |
| NCT02129647 | 4, 2014 | 2 | USA | 12 | NF2 Progressive VS | Axitinib | volumetric response rates |
| NCT01345136 | 7, 2015 | 2 | USA | 4 | NF2 | RAD001, everolimus | Vestibular schwannoma volume |
| NCT01767792 | 5, 2013 | 2 | USA | 22 | NF2 Progressive VS | Bevacizumab | Hearing |
| NCT04283669 | 2, 2020 | 2 | USA | 19 | NF2 Progressive VS | Crizotinib | Volumetric response rate |
| NCT02831257 | 8, 2016 | 2 | USA | 18 | NF2 Meningioma | AZD2014 | Volumetric response rate |
| NCT04374305 | 6, 2020 | 2 | USA | 80 | NF2 Vestibular Schwannoma Non-vestibular Schwannoma Meningioma Ependymoma | Brigatinib | Volumetric response rate |
| NCT03095248 | 5, 2017 | 2 | USA | 34 | NF2 Vestibular Schwannoma Meningioma Ependymoma Glioma | Selumetinib | Hearing response Volumetric response rate |
| NCT03079999 | 6, 2018 | 2 | USA | 300 | NF2 Vestibular schwannoma | Aspirin | Progression-free survival |
| Definite Schwannomatosis |
| A. Age >30 years and two or more schwannomas (not intradermal), at least one with histologic confirmation with no evidence of vestibular tumor on brain MRI scan and no known NF mutation |
| B. Vestibular schwannoma (pathologically confirmed) plus first-degree relative who meets the criteria of schwannomatosis |
| Possible schwannomatosis |
| A. Age <30 years plus two or more schwannomas (not intradermal), at least one with histologic confirmation with no evidence of vestibular tumor on brain MRI scan and no known NF mutation |
| B. Age >45 years plus two or more schwannomas (not dermal), at least one with histologic confirmation and no symptoms of 8th nerve dysfunction and NF type 2 |
| C. Evidence of a non-vestibular schwannoma and first-degree relative meeting criteria for definite schwannomatosis |
| ID | Initiation Date | Phase | Nation | N | Disease | Treatment | Primary Outcome |
|---|---|---|---|---|---|---|---|
| NCT04163419 | 4, 2020 | 2 | USA | 46 | Schwannomatosis | Tanezumab | Change in pain level |
| NCT04085159 | 9, 2019 | 1/2 | China | 100 | Neurofibromatosis Schwannomatosis | Antigen-specific T cells CART/CTL and DCvac | Percentage of adverse effects |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamura, R. Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis. Int. J. Mol. Sci. 2021, 22, 5850. https://doi.org/10.3390/ijms22115850
Tamura R. Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis. International Journal of Molecular Sciences. 2021; 22(11):5850. https://doi.org/10.3390/ijms22115850
Chicago/Turabian StyleTamura, Ryota. 2021. "Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis" International Journal of Molecular Sciences 22, no. 11: 5850. https://doi.org/10.3390/ijms22115850
APA StyleTamura, R. (2021). Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis. International Journal of Molecular Sciences, 22(11), 5850. https://doi.org/10.3390/ijms22115850