
Protein Aggregation Landscape in Neurodegenerative Diseases: Clinical Relevance and Future Applications

 ,
, 

Abstract
:1. Introduction
2. Intrinsic Disorder
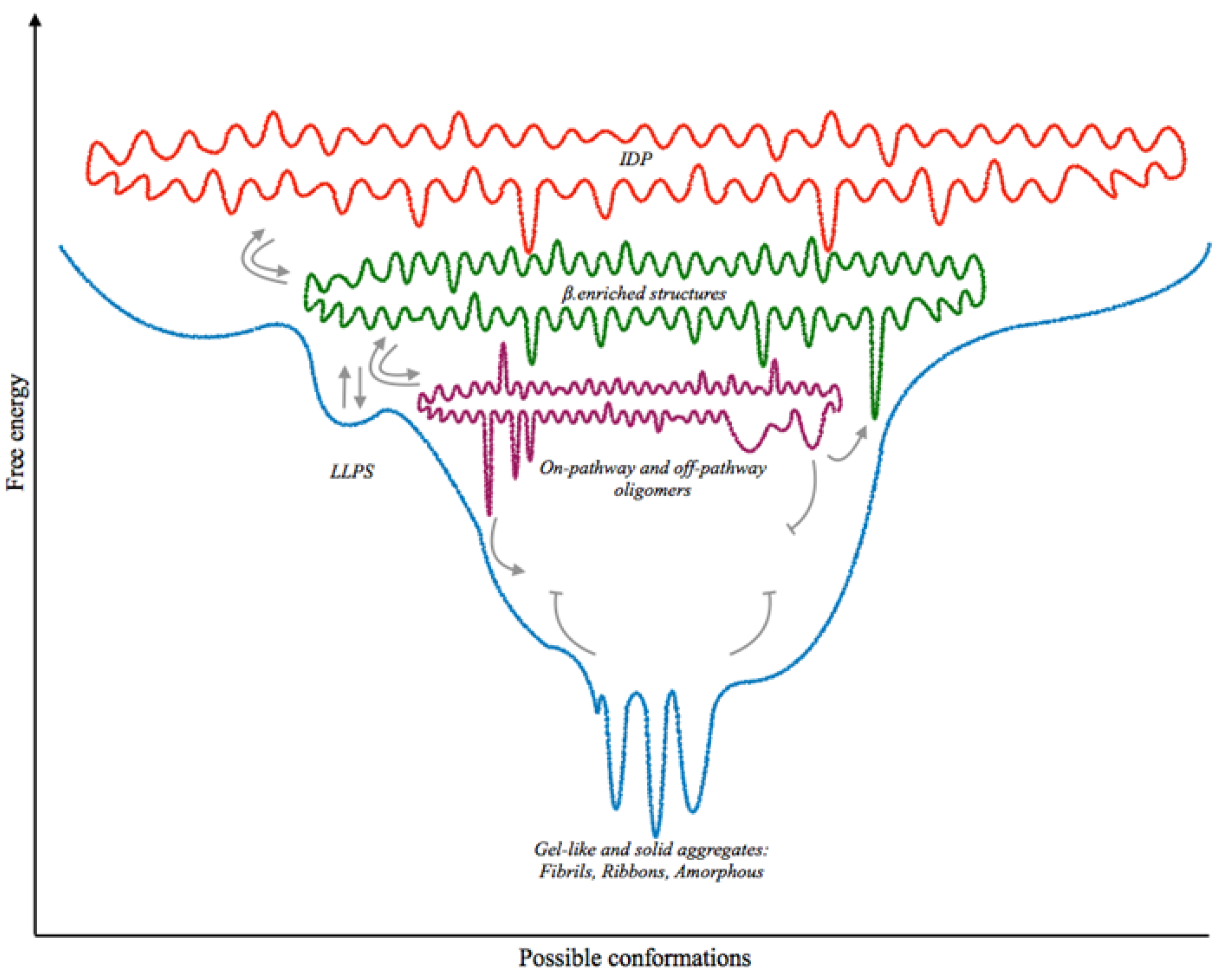
3. Phase Transition
4. The Paths towards Toxicity
5. PrP: The Protein That Started It All
6. α-Synuclein and Synucleinopathies
7. TDP-43
8. Tau and Tauopathies
9. The Curious Case of Aβ
10. Protein Quality Control
11. Clinical Outlook and Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IDR | Intrinsically disordered region |
| IDP | Intrinsically disordered protein |
| LCD | Low complexity domain |
| PrP | Prion protein |
| Aβ | Amyloid β |
| TDP-43 | TAR DNA binding protein 43 |
| LLPS | Liquid-liquid phase separation |
| MLO | Membraneless Organelle |
| SG | Stress Granules |
| PTM | Post-translational modification |
| CJD | Creutzfeldt-Jakob disease |
| PrP-C | Cellular prion protein |
| AD | Alzheimer’s Disease |
| PD | Parkinson’s Disease |
| NAC | Non- Amyloid β Component |
| PDD | Parkinson’s Disease Dementia |
| DLB | Dementia with Lewy Bodies |
| MSA | Multiple System Atrophy |
| ALS | Amyotrophic Lateral Sclerosis |
| FTD | Frontotemporal Dementia |
| APP | Amyloid Precursor Protein |
| FFI | Fatal Familial Insomnia |
| GSS | Gerstmann-Sträussler-Scheinker |
| vPSPr | variably Protease-Sensitive Prionopathy |
| PiD | Pick’s Disease |
| CBD | Corticobasal Degeneration |
| PSP | Progressive Supranuclear Palsy |
| PART | Primary Age-Related Tauopathy |
| CAA | Cerebral Amyloid Angiopathy |
| PQC | Protein Quality Control |
| UPS | Ubiquitin-proteasome system |
| CMA | Chaperone-mediated autophagy |
| MAM | Mitochondria Associated Membrane |
References
- Soto, C. Protein misfolding and disease; protein refolding and therapy. FEBS Lett. 2001, 498, 204–207. [Google Scholar] [CrossRef] [Green Version]
- Soto, C. Unfolding protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Foit, L.; Morgan, G.J.; Kern, M.J.; Steimer, L.R.; von Hacht, A.A.; Titchmarsh, J.; Warriner, S.L.; Radford, S.E.; Bradwell, J.C.A. Optimizing protein stability in vivo. Mol. Cell. 2009, 36, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.J.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic Disorder, Protein-Protein Interactions, and Disease. Adv. Protein. Chem. Struct. Biol. 2018, 110, 85–121. [Google Scholar] [CrossRef]
- Hegyi, H.; Tompa, P. Intrinsically Disordered Proteins Display No Preference for Chaperone Binding In Vivo. PLoS Comput. Biol. 2008, 4, e1000017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- March, Z.M.; King, O.D.; Shorter, J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016, 1647, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Wiseman, R.L.; Matteson, J.; Hammarström, P.; Miller, S.R.; Sawkar, A.R.; Balch, W.E.; Kelly, J.W. The biological and chemical basis for tissue-selective amyloid disease. Cell 2005, 121, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, A.F.; Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef] [Green Version]
- Wallman, A.; Kesten, C. Common Functions of Disordered Proteins across Evolutionary Distant Organisms. Int. J. Mol. Sci. 2020, 21, 2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef] [Green Version]
- Vymetal, J.; Vondrášek, J.; Hlouchová, K. Sequence Versus Composition: What Prescribes IDP Biophysical Properties? Entropy 2019, 21, 654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: Comprehensive characterization of intrinsic disorder in all domains of life. Cell Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically Disordered Proteins in Cellular Signaling and Regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Chavali, S.; Gunnarsson, A.; Babu, M.M. Intrinsically Disordered Proteins Adaptively Reorganize Cellular Matter During Stress. Trends Biochem. Sci. 2017, 42, 410–412. [Google Scholar] [CrossRef]
- Uversky, V.N. The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett. 2015, 589, 2498–2506. [Google Scholar] [CrossRef] [Green Version]
- Calabretta, S.; Richard, S. Emerging Roles of Disordered Sequences in RNA-Binding Proteins. Trends Biochem. Sci. 2015, 40, 662–672. [Google Scholar] [CrossRef]
- Basu, S.; Bahadur, R.P. A structural perspective of RNA recognition by intrinsically disordered proteins. Cell Mol. Life Sci. 2016, 73, 4075–4084. [Google Scholar] [CrossRef]
- Zagorvic, B.; Bartonek, L.; Polyansky, A.A. RNA-protein interactions in an unstructured context. FEBS Lett. 2018, 592, 2901–2916. [Google Scholar] [CrossRef]
- Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. What Macromolecular Crowding Can Do to a Protein. Int. J. Mol. Sci. 2014, 15, 23090–23140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crick, S.L.; Ruff, K.M.; Garai, K.; Frieden, C.; Pappu, R.V. Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA 2013, 110, 20075–20080. [Google Scholar] [CrossRef] [Green Version]
- Bergeron-Sandoval, L.P.; Safaee, N.; Michnick, S.W. Mechanisms and Consequences of Macromolecular Phase Separation. Cell 2016, 165, 1067–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieplak, M.; Chwastyk, M.; Mioduszewski, L.; de Aquino, B.R.H. Transient knots in intrinsically disordered proteins and neurodegeneration. Prog. Mol. Biol. Transl. Sci. 2020, 174, 79–103. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Pal, U.; Sad, S.; Bagga, K.; Roy, A.; Mrigwani, A.; Maiti, N.C. Sequence Complexity of Amyloidogenic Regions in Intrinsically Disordered Human Proteins. PLoS ONE 2014, 9, e89781. [Google Scholar] [CrossRef]
- Kumari, B.; Kumar, R.; Kumar, M. Low complexity and disordered regions of proteins have different structural and amino acid preferences. Mol. Biosyst. 2015, 11, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.W.; Mittag, T. Relationship of Sequence and Phase Separation in Protein Low-Complexity Regions. Biochemistry. 2018, 57, 2478–2487. [Google Scholar] [CrossRef]
- Pak, C.W.; Kosno, M.; Holehouse, A.S.; Padrick, S.B.; Mittal, A.; Ali, R.; Yunus, A.A.; Liu, D.R.; Pappu, R.V.; Rosen, M.K. Sequence Determinants of Intracellular Phase Separation by Complex Coacervation of a Disordered Protein. Mol. Cell. 2016, 63, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Zarin, T.; Strome, B.; Nguyen Ba, A.N.; Alberti, S.; Forman-Kay, J.D.; Moses, A.M. Proteome-wide signatures of function in highly diverged intrinsically disordered regions. eLife 2019, 8, e46883. [Google Scholar] [CrossRef]
- Uversky, V.N. Paradoxes and wonders of intrinsic disorder: Complexity of simplicity. Intrinsically Disord. Proteins 2016, 4, e1135015. [Google Scholar] [CrossRef] [Green Version]
- Disfani, F.M.; Hsu, W.L.; Mizianty, M.J.; Oldfield, C.J.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. MoRFpred, a Computational Tool for Sequence-based Prediction and Characterization of Short Disorder-to-order Transitioning Binding Regions in Proteins. Bioinformatics 2012, 28, i75–i83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuxreiter, M.; Tompa, P.; Simon, I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics 2007, 23, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.J.; Davey, N.E.; Shields, D.C. SLiMFinder: A probabilistic method for identifying over-represented, convergently evolved, short linear motifs in proteins. PLoS ONE 2007, 2, e967. [Google Scholar] [CrossRef] [PubMed]
- Yruela, I.; Oldfield, C.J.; Niklas, K.J.; Keith Dunker, A. Evidence for a Strong Correlation Between Transcription Factor Protein Disorder and Organismic Complexity. Genome Biol. Evol. 2017, 9, 1248–1265. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Babinchak, W.M.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.K.; Surewicz, W.K. The role of liquid–liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 2019, 294, 6306–6317. [Google Scholar] [CrossRef] [Green Version]
- Šarić, A.; Michaels, T.C.T.; Zaccone, A.; Knowles, T.P.J.; Frenkel, D. Kinetics of spontaneous filament nucleation via oligomers: Insights from theory and simulation. J. Chem. Phys. 2016, 145, 211926. [Google Scholar] [CrossRef]
- Fomicheva, A.; Ross, E. From Prions to Stress Granules: Defining the Compositional Features of Prion-Like Domains That Promote Different Types of Assemblies. Int. J. Mol. Sci. 2021, 22, 1251. [Google Scholar] [CrossRef]
- Halfmann, R.; Alberti, S.; Krishnan, R.; Lyle, N.; O’Donnell, C.W.; King, O.D.; Berger, B.; Pappu, R.V.; Lindquist, S. Opposing Effects of Glutamine and Asparagine Govern Prion Formation by Intrinsically Disordered Proteins. Mol. Cell. 2011, 43, 72–84. [Google Scholar] [CrossRef]
- Bousset, L.; Savistchenko, J.; Melki, R. Assembly of the asparagine- and glutamine-rich yeast prions into protein fibrils. Curr. Alzheimer Res. 2008, 5, 251–259. [Google Scholar] [CrossRef]
- Jayaraman, M.; Kodali, R.; Sahoo, B.; Thakur, A.K.; Mayasundari, A.; Mishra, R.; Peterson, C.B.; Wetzel, R. Slow amyloid nucleation via α-helix-rich oligomeric intermediates in short polyglutamine-containing huntingtin fragments. J. Mol. Biol. 2012, 415, 881–899. [Google Scholar] [CrossRef] [Green Version]
- Elden, A.C.; Kim, H.J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Krishnan, R.; Lindquist, S.L. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 2005, 435, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inayathullah, M.; Tan, A.; Jeyaraj, R.; Lam, J.; Cho, N.J.; Liu, C.W.; Manoukian, M.A.; Ashkan, K.; Mahmoudi, M.; Rajadas, J. Self-assembly and sequence length dependence on nanofibrils of polyglutamine peptides. Neuropeptides. 2016, 57, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.W.; Huang, M. Protein Aggregation and Polyasparagine-Mediated Cellular Toxicity in Saccharomyces cerevisiae. Prion 2007, 1, 144–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crick, F.H.; Rich, A. Structure of polyglycine II. Nature 1955, 176, 780–781. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.B.; Riekel, C.; Guinea, G.V.; Madurga, R.; Daza, R.; Burghammer, M.; Hayashi, C.; Elices, M.; Plaza, G.R.; Pérez-Rigueiro, J. Identification and dynamics of polyglycine II nanocrystals in Argiope trifasciata flagelliform silk. Sci. Rep. 2013, 3, 3061. [Google Scholar] [CrossRef] [PubMed]
- Ozdilek, B.A.; Thompson, V.F.; Ahmed, N.S.; White, C.I.; Batey, R.T.; Schwartz, J.C. Intrinsically disordered RGG/RG domains mediate degenerate specificity in RNA binding. Nucleic Acids Res. 2017, 45, 7984–7996. [Google Scholar] [CrossRef] [PubMed]
- McGuffee, S.R.; Elcock, A.H. Diffusion, crowding & protein stability in a dynamic molecular model of the bacterial cytoplasm. PLoS Comput. Biol. 2010, 6, e1000694. [Google Scholar] [CrossRef]
- André, A.A.M.; Spruijt, E. Liquid–Liquid Phase Separation in Crowded Environments. Int. J. Mol. Sci. 2020, 21, 5908. [Google Scholar] [CrossRef]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Hu, G.; Wu, Z.; Uversky, V.N.; Kurgan, L. Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions. Int. J. Mol. Sci. 2017, 18, 2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumdar, A.; Dogra, P.; Maity, S.; Mukhopadhyay, S. Liquid−Liquid Phase Separation Is Driven by Large-Scale Conformational Unwinding and Fluctuations of Intrinsically Disordered Protein Molecules. J. Phys. Chem. Lett. 2019, 10, 3929–3936. [Google Scholar] [CrossRef]
- Aumiller, W.M., Jr.; Keating, C.D. Experimental models for dynamic compartmentalization of biomolecules in liquid organelles: Reversible formation and partitioning in aqueous biphasic systems. Adv. Colloid Interface Sci. 2017, 239, 75–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Signal 2016, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, Y.; Ford, L.K.; Fioriti, L.; McGurk, L.; Zhang, M. Liquid-Liquid Phase Separation in Physiology and Pathophysiology of the Nervous System. J. Neurosci. 2021, 41, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Protein intrinsic disorder-based liquid-liquid phase transitions in biological systems: Complex coacervates and membrane-less organelles. Adv. Colloid. Interface Sci. 2017, 239, 97–114. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Cika, J.A.; Guy, C.S.; Ban, D.; Banerjee, P.R.; Stanley, C.B.; Nourse, A.; Deniz, A.A.; Kriwacki, R.W. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. eLife 2016, 5, e13571. [Google Scholar] [CrossRef]
- Falahati, H.; Wieschaus, E. Independent active and thermodynamic processes govern the nucleolus assembly in vivo. Proc. Natl Acad. Sci. USA 2017, 114, 1335–1340. [Google Scholar] [CrossRef] [Green Version]
- Dundr, M. Nuclear bodies: Multifunctional companions of the genome. Curr. Opin. Cell Biol. 2012, 24, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.R.; Matheny, T.; Jain, S.; Abrisch, R.; Parker, R. Distinct stages in stress granule assembly and disassembly. eLife 2016, 5, e18413. [Google Scholar] [CrossRef]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting liquid phases underlie nucleolar sub-compartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Choi, J.M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riback, J.A.; Katanski, C.D.; Kear-Scott, J.L.; Pilipenko, E.V.; Rojek, A.E.; Sosnick, T.R.; Drummond, D.A. Stress-Triggered Phase Separation Is an Adaptive, Evolutionarily Tuned Response. Cell 2017, 168, 1028–1040.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, I.; Shewmaker, F. The Role of Post-Translational Modifications in the Phase Transitions of Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2019, 20, 5501. [Google Scholar] [CrossRef] [Green Version]
- Yao, R.W.; Xu, G.; Wang, Y.; Shan, L.; Luan, P.F.; Wang, Y.; Wu, M.; Yang, L.Z.; Xing, Y.H.; Yang, L.; et al. Nascent Pre-rRNA Sorting via Phase Separation Drives the Assembly of Dense Fibrillar Components in the Human Nucleolus. Mol. Cell. 2019, 76, 767–783.e11. [Google Scholar] [CrossRef]
- Hughes, M.P.; Sawaya, M.R.; Boyer, D.R.; Goldschmidt, L.; Rodriguez, J.A.; Cascio, D.; Chong, L.; Gonen, T.; Eisenberg, D.S. Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science 2018, 359, 698–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.K.; Sindi, S.S. A study in nucleated polymerization models of protein aggregation. Appl. Math. Lett. 2016, 72, 1555–1578. [Google Scholar] [CrossRef] [Green Version]
- Hernańdez-Vega, A.; Braun, M.; Scharrel, L.; Jahnel, M.; Wegmann, S.; Hyman, B.T.; Alberti, S.; Diez, S.; Hyman, A.A. Local Nucleation of Microtubule Bundles through Tubulin Concentration into a Condensed Tau Phase. Cell Rep. 2017, 20, 2304–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Carrell, R.W.; Gooptu, B. Conformational changes and disease—Serpins, prions and Alzheimer’s. Curr. Opin. Struct. Biol. 1999, 8, 799–809. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Reik, R. The Three-Dimensional Structures of Amyloids. Cold Spring Harb. Perspect. Biol. 2017, 9, a023572. [Google Scholar] [CrossRef] [Green Version]
- Seuring, C.; Verasdonck, J.; Gath, J.; Ghosh, D.; Nespovitaya, N.; Wälti, M.A.; Maji, S.K.; Cadalbert, R.; Güntert, P.; Meier, B.H.; et al. The three-dimensional structure of human β-endorphin amyloid fibrils. Nat. Struct. Mol. Biol. 2020, 27, 1178–1184. [Google Scholar] [CrossRef]
- Shi, Y.; Murzin, A.G.; Falcon, B.; Epstein, A.; Machin, J.; Tempest, P.; Newell, K.L.; Vidal, R.; Garringer, H.J.; Sahara, N.; et al. Cryo-EM structures of tau filaments from Alzheimer’s disease with PET ligand APN-1607. Acta Neuropathol. 2021, 141, 697–708. [Google Scholar] [CrossRef]
- Moran, S.D.; Zhang, T.O.; Decatur, S.M.; Zanni, M.T. Amyloid fiber formation in human γD-Crystallin induced by UV-B photodamage. Biochemistry 2013, 52, 6169–6181. [Google Scholar] [CrossRef] [Green Version]
- Daskalov, A.; Martinez, D.; Coustou, V.; El Mammeri, N.; Berbon, M.; Andreas, L.B.; Bardiaux, B.; Stanek, J.; Noubhani, A.; Kauffmann, B.; et al. Structural and molecular basis of cross-seeding barriers in amyloids. Proc. Natl. Acad. Sci. USA 2021, 118, e2014085118. [Google Scholar] [CrossRef]
- Scialò, C.; De Cecco, E.; Manganotti, P.; Legname, G. Prion and Prion-Like Protein Strains: Deciphering the Molecular Basis of Heterogeneity in Neurodegeneration. Viruses 2019, 11, 261. [Google Scholar] [CrossRef] [Green Version]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular Mechanisms of Amyloid Oligomers Toxicity. J. Alzheimer’s Dis. 2012, 33, S67–S78. [Google Scholar] [CrossRef] [Green Version]
- Lemarre, P.; Pujo-Menjouet, L.; Sindi, S.S. Generalizing a mathematical model of prion aggregation allows strain coexistence and co-stability by including a novel misfolded species. J. Math. Biol. 2019, 78, 465–495. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.C.; Condello, C.; Stöhr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial propagation of distinct strains of Aβ prions from Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zaer, S.; Drori, P.; Zamel, J.; Joron, K.; Kalisman, N.; Lerner, E.; Dokholyan, N.V. The structural heterogeneity of α-synuclein is governed by several distinct subpopulations with interconversion times slower than milliseconds. Structure 2021, 29, 1–17. [Google Scholar] [CrossRef]
- Gyparaki, M.T.; Arab, A.; Sorokina, E.M.; Santiago-Ruiz, A.N.; Bohrer, C.H.; Xiao, J.; Lakadamyali, M. Tau forms oligomeric complexes on microtubules that are distinct from tau aggregates. Proc. Natl. Acad. Sci. USA 2021, 118, e2021461118. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Laferrière, F.; Maniecka, Z.; Pérez-Berlanga, M.; Hruska-Plochan, M.; Gilhespy, L.; Hock, E.M.; Wagner, U.; Afroz, T.; Boersema, P.J.; Barmettler, G.; et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci. 2019, 22, 65–77. [Google Scholar] [CrossRef]
- Karamanos, T.K.; Jackson, M.P.; Calabrese, A.N.; Goodchild, S.C.; Cawood, E.E.; Thompson, G.S.; Kalverda, A.P.; Hewitt, E.W.; Radford, S.E. Structural mapping of oligomeric intermediates in an amyloid assembly pathway. eLife 2019, 8, e46574. [Google Scholar] [CrossRef]
- Dear, A.J.; Meisl, G.; Šarić, A.; Michaels, T.C.T.; Kjaergaard, M.; Linse, S.; Knowles, T.P.J. Identification of on- and off-pathway oligomers in amyloid fibril formation. Chem. Sci. 2020, 11, 6236–6247. [Google Scholar] [CrossRef]
- Dear, A.J.; Michaels, T.C.T.; Meisl, G.; Klenerman, D.; Wu, S.; Perrett, S.; Linse, S.; Dobson, C.M.; Knowles, T.P.J. Kinetic diversity of amyloid oligomers. Proc. Natl. Acad. Sci. USA 2020, 117, 12087–12094. [Google Scholar] [CrossRef]
- Collins, S.R.; Douglass, A.; Vale, R.D.; Weissman, J.S. Mechanism of Prion Propagation: Amyloid Growth Occurs by Monomer Addition. PLoS Biol. 2004, 2, e321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Close, W.; Neumann, M.; Schmidt, A.; Hora, M.; Annamalai, K.; Schmidt, M.; Reif, B.; Schmidt, V.; Grigorieff, N.; Fändrich, M. Physical basis of amyloid fibril polymorphism. Nat. Commun. 2018, 9, 699. [Google Scholar] [CrossRef] [PubMed]
- Makowski, L. The Structural Basis of Amyloid Strains in Alzheimer’s Disease. ACS Biomater. Sci. Eng. 2020, 6, 2498–2505. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 36–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajdusek, D.C. Unconventional viruses and the origin and disappearance of kuru. Science 1977, 197, 943–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimberlin, R.H.; Walker, C.A. Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains from a mixture. J. Gen. Virol. 1978, 39, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; McKinley, M.P.; Groth, D.F.; Bowman, K.A.; Mock, N.I.; Cochran, S.P.; Masiarz, F.R. Scrapie agent contains a hydrophobic protein. Proc. Natl. Acad. Sci. USA 1981, 78, 6675–6679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinley, M.P.; Braunfeld, M.B.; Bellinger, C.G.; Prusiner, S.B. Molecular Characteristics of Prion Rods Purified from Scrapie-Infected Hamster Brains. J. Infect. Dis. 1986, 54, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.T.; Inouye, H.; Baldwin, M.A.; Fletterick, R.J.; Cohen, F.E.; Prusiner, S.B.; Kirschner, D.A. X-ray diffraction of scrapie prion rods and PrP peptides. J. Mol. Biol. 1995, 252, 412–422. [Google Scholar] [CrossRef]
- Caughey, B.; Race, R.E. Potent inhibition of scrapie-associated PrP accumulation by Congo red. J. Neurochem. 1992, 59, 768–771. [Google Scholar] [CrossRef]
- Griffith, J.S. Self-replication and scrapie. Nature 1967, 215, 1043–1044. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Scott, M.; Foster, D.; Pan, K.M.; Groth, D.; Mirenda, C.; Torchia, M.; Yang, S.L.; Serban, D.; Carlson, G.A.; et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 1990, 63, 673–686. [Google Scholar] [CrossRef]
- Flechsig, E.; Shmerling, D.; Hegyi, I.; Raeber, A.J.; Fischer, M.; Cozzio, A.; von Mering, C.; Aguzzi, A.; Weissmann, C. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron 2000, 27, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.H.; Huang, M.Y.; Lee, Y.R.; Lin, Y.K.; Chen, H.R.; Lee, C.I. The Effect of Octapeptide Repeats on Prion Folding and Misfolding. Int. J. Mol. Sci. 2021, 22, 1800. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.F.; Lawson, V.A.; Coleman, B.M.; Kim, Y.S.; Masters, C.L.; Cappai, R.; Barnham, K.J.; Hill, A.F. Conservation of a glycine-rich region in the prion protein is required for uptake of prion infectivity. J. Biol. Chem. 2010, 285, 20213–20223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahn, R.; Liu, T.; Lührs, T.; Riek, R.; von Schroetter, F.; López García, F.; Billeter, M.; Calzolai, G.; Wider, K.; Wüthrich, K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baral, P.K.; Yin, J.; Aguzzi, A.; James, M.N.G. Transition of the prion protein from a structured cellular form (PrP C) to the infectious scrapie agent (PrP Sc). Protein Sci. 2019, 28, 2055–2063. [Google Scholar] [CrossRef]
- Moore, R.A.; Herzog, C.; Errett, J.; Kocisko, D.A.; Arnold, K.M.; Hayes, S.F.; Priola, S.A. Octapeptide repeat insertions increase the rate of protease-resistant prion protein formation. Protein Sci. 2006, 15, 609–619. [Google Scholar] [CrossRef]
- Schmitz, M.; Dittmar, K.; Llorens, F.; Gelpi, E.; Ferrer, I.; Schulz-Schaeffer, W.J.; Zerr, I. Hereditary Human Prion Diseases: An Update. Mol. Neurobiol. 2017, 54, 4138–4149. [Google Scholar] [CrossRef]
- Brandel, J.P.; Knight, R. Variant Creutzfeldt-Jakob Disease. Handb. Clin. Neurol. 2018, 153, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Cracco, L.; Xiao, X.; Nemani, S.K.; Lavrich, J.; Cali, I.; Ghetti, B.; Notari, S.; Surewicz, W.K.; Gambetti, P. Gerstmann-Sträussler-Scheinker disease revisited: Accumulation of covalently-linked multimers of internal prion protein fragments. Acta Neuropathol. Commun. 2019, 7, 85. [Google Scholar] [CrossRef] [Green Version]
- Llorens, F.; Zarranz, J.J.; Fischer, A.; Zerr, I.; Ferrer, I. Fatal Familial Insomnia: Clinical Aspects and Molecular Alterations. Curr. Neurol. Neurosci. Rep. 2017, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Notari, S.; Appleby, B.S.; Gambetti, P. Variably protease-sensitive prionopathy. Handb. Clin. Neurol. 2018, 153, 175–190. [Google Scholar] [CrossRef]
- Pattison, I.H.; Millson, G.C. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J. Comp. Pathol. 1961, 71, 101–109. [Google Scholar] [CrossRef]
- Zanusso, G.; Monaco, S.; Pocchiari, M.; Caughey, B. Advanced tests for early and accurate diagnosis of Creutzfeldt-Jakob disease. Nat. Rev. Neurol. 2016, 12, 325–333. [Google Scholar] [CrossRef]
- Nemani, S.K.; Xiao, X.; Cali, I.; Cracco, L.; Puoti, G.; Nigro, M.; Lavrich, J.; Bharara Singh, A.; Appleby, B.S.; Sim, V.L.; et al. A novel mechanism of phenotypic heterogeneity in Creutzfeldt-Jakob disease. Acta Neuropathol. Commun. 2020, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Saverioni, D.; Di Bari, M.; Baiardi, S.; Lemstra, A.W.; Pirisinu, L.; Capellari, S.; Rozemuller, A.; Nonno, R.; Parchi, P. Atypical Creutzfeldt-Jakob disease with PrP-amyloid plaques in white matter: Molecular characterization and transmission to bank voles show the M1 strain signature. Acta Neuropathol. Commun. 2017, 5, 87. [Google Scholar] [CrossRef] [Green Version]
- Cassard, H.; Huor, A.; Espinosa, J.C.; Douet, J.Y.; Lugan, S.; Aron, N.; Vilette, D.; Delisle, M.B.; Marín-Moreno, A.; Peran, P.; et al. Prions from Sporadic Creutzfeldt-Jakob Disease Patients Propagate as Strain Mixtures. mBio 2020, 11, e00393-20. [Google Scholar] [CrossRef]
- Cali, I.; Puoti, G.; Smucny, J.; Curtiss, P.M.; Cracco, L.; Kitamoto, T.; Occhipinti, R.; Cohen, M.L.; Appleby, B.S.; Gambetti, P. Co-existence of PrPD types 1 and 2 in sporadic Creutzfeldt-Jakob disease of the VV subgroup: Phenotypic and prion protein characteristics. Sci. Rep. 2020, 10, 1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cali, I.; Espinosa, J.C.; Nemani, S.K.; Marin-Moreno, A.; Camacho, M.V.; Aslam, R.; Kitamoto, T.; Appleby, B.S.; Torres, J.M.; Gambetti, P. Two distinct conformers of PrPD type 1 of sporadic Creutzfeldt–Jakob disease with codon 129VV genotype faithfully propagate in vivo. Acta Neuropathol. Commun. 2021, 9, 55. [Google Scholar] [CrossRef]
- Combet, S.; Cousin, F.; Rezaei, H.; Noinville, S. Membrane interaction of off-pathway prion oligomers and lipid-induced on-pathway intermediates during prion conversion: A clue for neurotoxicity. Biochim. Biophys. Acta Biomembr. 2019, 1861, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Tange, H.; Ishibashi, D.; Nakagaki, T.; Taguchi, Y.; Kamatari, Y.O.; Ozawa, H.; Nishida, N. Liquid-liquid phase separation of full-length prion protein initiates conformational conversion in vitro. J. Biol. Chem. 2021, 2, 100367. [Google Scholar] [CrossRef] [PubMed]
- Uéda, K.; Fukushima, K.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T., Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; von Saucken, V.E.; Bartels, T.; Selkoe, D. KTKEGV repeat motifs are key mediators of normal α-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 9596–9601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jao, C.C.; Der-Sarkissian, A.; Chen, J.; Langen, R. Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. USA 2004, 101, 8331–8336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tofaris, G.K.; Spillantini, M.G. Physiological and pathological properties of α-synuclein. Cell. Mol. Life Sci. 2007, 64, 2194–2201. [Google Scholar] [CrossRef]
- Apetri, M.M.; Maiti, N.C.; Zagorski, M.G.; Carey, P.R.; Anderson, V.E. Secondary Structure of a-Synuclein Oligomers: Characterization by Raman and Atomic Force Microscopy. J. Mol. Biol. 2006, 6, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Fagerqvist, T.; Näsström, T.; Ihse, E.; Lindström, V.; Sahlin, C.; Tucker, S.M.; Kasaryan, A.; Karlsson, M.; Nikolajeff, F.; Schell, H.; et al. Off-pathway α-synuclein oligomers seem to alter α-synuclein turnover in a cell model but lack seeding capability in vivo. Amyloid 2013, 20, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein aggregation nucleates through liquid–liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellinger, K.A.; Korczyn, A.D. Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med. 2018, 16, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2013, 24, 197–211. [Google Scholar] [CrossRef]
- Alafuzoff, I.; Ince, P.G.; Arzberger, T.; Al-Sarraj, S.; Bell, J.; Bodi, I.; Bogdanovic, N.; Bugiani, O.; Ferrer, I.; Gelpi, E.; et al. Staging/typing of Lewy body related alpha-synuclein pathology: A study of the BrainNet Europe Consortium. Acta Neuropathol. 2009, 117, 635–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulzer, D.; Surmeier, D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 2013, 28, 715–724. [Google Scholar] [CrossRef]
- Peelaerts, W.; Baekelandt, V. ɑ-Synuclein strains and the variable pathologies of synucleinopathies. J. Neurochem. 2016, 139 (Suppl. 1), 256–274. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.H.; Wu, F.; García-Martínez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Tziortzouda, P.; Van Den Bosch, L.; Hirth, F. Triad of TDP43 control in neurodegeneration: Autoregulation, localization and aggregation. Nat. Ver. Neurosci. 2021, 22, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Afroz, T.; Hock, E.M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakravan, D.; Michiels, E.; Bratek-Skicki, A.; De Decker, M.; Van Lindt, J.; Alsteens, D.; Derclaye, S.; Van Damme, P.; Schymkowitz, J.; Rousseau, F.; et al. Liquid-Liquid Phase Separation Enhances TDP-43 LCD Aggregation but Delays Seeded Aggregation. Biomolecules 2021, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droppelmann, C.A.; Campos-Melo, D.; Moszczynski, A.J.; Amzil, H.; Strong, M.J. TDP-43 aggregation inside micronuclei reveals a potential mechanism for protein inclusion formation in ALS. Sci. Rep. 2019, 9, 19928. [Google Scholar] [CrossRef]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.; Guo, L.; Shorter, J.; Da Cruz, S.; Cleveland, D.W. Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 2019, 102, 339–357.e7. [Google Scholar] [CrossRef] [Green Version]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- Bozzo, F.; Salvatori, I.; Iacovelli, F.; Mirra, A.; Rossi, S.; Cozzolino, M.; Falconi, M.; Valle, C.; Carrì, M.T. Structural insights into the multi-determinant aggregation of TDP-43 in motor neuron-like cells. Neurobiol Dis. 2016, 94, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.B.; Porta, S.; Michael Baer, G.; Xu, Y.; Suh, E.; Kwong, L.K.; Elman, L.; Grossman, M.; Lee, V.M.; Irwin, D.J.; et al. Expansion of the classification of FTLD-TDP: Distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 2017, 134, 65–78. [Google Scholar] [CrossRef]
- Ayers, J.I.; Borchelt, D.R. Phenotypic diversity in ALS and the role of poly-conformational protein misfolding. Acta Neuropathol. 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Smethurst, P.; Newcombe, J.; Troakes, C.; Simone, R.; Chen, Y.R.; Patani, R.; Sidle, K. In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol. Dis. 2016, 96, 236–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C. Skeletal Muscle in ALS: An Unappreciated Therapeutic Opportunity? Cells 2021, 10, 525. [Google Scholar] [CrossRef]
- Picchiarelli, G.; Dupuis, L. Role of RNA Binding Proteins with prion-like domains in muscle and neuromuscular diseases. Cell Stress. 2020, 4, 76–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimer‘s Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [Green Version]
- Derreumaux, P.; Man, V.H.; Wang, J.; Nguyen, P.H. Tau R3-R4 Domain Dimer of the Wild Type and Phosphorylated Ser356 Sequences. I. In Solution by Atomistic Simulations. J. Phys. Chem. B 2020, 124, 2975–2983. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225–4230. [Google Scholar] [CrossRef]
- Tuerde, D.; Kimura, T.; Miyasaka, T.; Furusawa, K.; Shimozawa, A.; Hasegawa, M.; Ando, K.; Hisanaga, S.I. Isoform-independent and -dependent phosphorylation of microtubule-associated protein tau in mouse brain during postnatal development. J. Biol. Chem. 2018, 293, 1781–1793. [Google Scholar] [CrossRef] [Green Version]
- Couchie, D.; Mavilia, C.; Georgieff, I.S.; Liem, R.K.; Shelanski, M.L.; Nunez, J. Primary structure of high molecular weight tau present in the peripheral nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 4378–4381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.C.; Haggarty, S.J. Tauopathies: Deciphering Disease Mechanisms to Develop Effective Therapies. Int. J. Mol. Sci. 2020, 21, 8948. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Derreumaux, P. Structures of the intrinsically disordered Aβ, tau and α-synuclein proteins in aqueous solution from computer simulations. Biophys. Chem. 2020, 264, 106421. [Google Scholar] [CrossRef]
- Zhu, S.; Shala, A.; Bezginov, A.; Sljoka, A.; Audette, G.; Wilson, D.J. Hyperphosphorylation of intrinsically disordered tau protein induces an amyloidogenic shift in its conformational ensemble. PLoS ONE 2015, 10, e0120416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Yang, J.; Zhang, B.; Gao, M.; Su, Z.; Huang, Y. The structure and phase of tau: From monomer to amyloid filament. Cell Mol. Life Sci. 2021, 78, 1873–1886. [Google Scholar] [CrossRef]
- Jeganathan, S.; von Bergen, M.; Brutlach, H.; Steinhoff, H.J.; Mandelkow, E. Global hairpin folding of tau in solution. Biochemistry 2006, 45, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Agnihotri, M.V.; Huseby, C.J.; Kuret, J.; Singer, S.J. A theoretical study of polymorphism in VQIVYK fibrils. Biophys. J. 2021, 120, 1396–1416. [Google Scholar] [CrossRef] [PubMed]
- Foote, A.K.; Manger, L.H.; Holden, M.R.; Margittai, M.; Goldsmith, R.H. Time-resolved multirotational dynamics of single solution-phase tau proteins reveals details of conformational variation. Phys. Chem. Chem. Phys. 2019, 21, 1863–1871. [Google Scholar] [CrossRef]
- Narasimhan, S.; Guo, J.L.; Changolkar, L.; Stieber, A.; McBride, J.D.; Silva, L.V.; He, Z.; Zhang, B.; Gathagan, R.J.; Trojanowski, J.Q.; et al. Pathological Tau Strains from Human Brains Recapitulate the Diversity of Tauopathies in Nontransgenic Mouse Brain. J. Neurosci. 2017, 37, 11406–11423. [Google Scholar] [CrossRef]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid–liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef]
- Boyko, S.; Qi, X.; Chen, T.H.; Surewicz, K.; Surewicz, W.K. Liquid-liquid phase separation of tau protein: The crucial role of electrostatic interactions. J. Biol. Chem. 2019, 294, 11054–11059. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Fichou, Y.; Longhini, A.P.; Llanes, L.C.; Yin, P.; Bazan, G.C.; Kosik, K.S.; Han, S. Electrostatically driven complex coacervation and amyloid aggregation of tau are independent processes with overlapping conditions. J. Mol. Biol. 2021, 433, 166731. [Google Scholar] [CrossRef] [PubMed]
- Lester, E.; Ooi, F.K.; Bakkar, N.; Ayers, J.; Woerman, A.L.; Wheeler, J.; Bowser, R.; Carlson, G.A.; Prusiner, S.B.; Parker, R. Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron 2021, 7. [Google Scholar] [CrossRef]
- Yu, H.; Lu, S.; Gasior, K.; Singh, D.; Vazquez-Sanchez, S.; Tapia, O.; Toprani, D.; Beccari, M.S.; Yates, J.R., 3rd; Da Cruz, S.; et al. HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science 2021, 371, eabb4309. [Google Scholar] [CrossRef] [PubMed]
- Ercan-Herbst, E.; Ehrig, J.; Schöndorf, D.C.; Behrendt, A.; Klaus, B.; Gomez Ramos, B.; Prat Oriol, N.; Weber, C.; Ehrnhoefer, D.E. A post-translational modification signature defines changes in soluble tau correlating with oligomerization in early stage Alzheimer’s disease brain. Acta Neuropathol. Commun. 2019, 7, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzeciakiewicz, H.; Tseng, J.H.; Wander, C.M.; Madden, V.; Tripathy, A.; Yuan, C.X.; Cohen, T.J. A Dual Pathogenic Mechanism Links Tau Acetylation to Sporadic Tauopathy. Sci. Rep. 2017, 7, 44102. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, J.C.; Jain, A.; Choi, K.J.; Tsoi, P.S.; MacKenzie, K.R.; Jung, S.Y.; Ferreon, A.C. Acetylation Disfavors Tau Phase Separation. Int. J. Mol. Sci. 2018, 19, 1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef]
- Biernat, J.; Mandelkow, E.M. The development of cell processes induced by tau protein requires phosphorylation of serine 262 and 356 in the repeat domain and is inhibited by phosphorylation in the proline-rich domains. Mol. Biol. Cell. 1999, 10, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Patterson, K.R.; Remmers, C.; Fu, Y.; Brooker, S.; Kanaan, N.M.; Vana, L.; Ward, S.; Reyes, J.F.; Philibert, K.; Glucksman, M.J.; et al. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 2011, 286, 23063–23076. [Google Scholar] [CrossRef] [Green Version]
- Clavaguera, F.; Duyckaerts, C.; Haïk, S. Prion-like properties of Tau assemblies. Curr. Opin. Neurobiol. 2020, 61, 49–57. [Google Scholar] [CrossRef]
- Popov, K.I.; Makepeace, K.A.T.; Petrotchenko, E.V.; Dokholyan, N.V.; Borchers, C.H. Insight into the Structure of the ‘‘Unstructured’’ Tau Protein. Structure 2019, 27, 1710–1715.e4. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef]
- Zhang, W.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Schenk, D. Alzheimer’s disease: Molecular understanding predicts amyloid-based therapeutics. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 545–584. [Google Scholar] [CrossRef]
- Haass, C.; Lemere, C.A.; Capell, A.; Citron, M.; Seubert, P.; Schenk, D.; Lannfelt, L.; Selkoe, D.J. The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat. Med. 1995, 1, 1291–1296. [Google Scholar] [CrossRef]
- Carter, D.B.; Dunn, E.; Pauley, A.M.; McKinley, D.D.; Fleck, T.J.; Ellerbrook, B.R.; Stratman, N.C.; Zhou, X.; Himes, C.S.; Nye, J.S.; et al. Changes in gamma-secretase activity and specificity caused by the introduction of consensus aspartyl protease active motif in Presenilin 1. Mol. Neurodegener. 2008, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Muvva, C.; Murugan, N.A.; Subramanian, V. Assessment of Amyloid Forming Tendency of Peptide Sequences from Amyloid Beta and Tau Proteins Using Force-Field, Semi-Empirical, and Density Functional Theory Calculations. Int. J. Mol. Sci. 2021, 22, 3244. [Google Scholar] [CrossRef] [PubMed]
- Eker, F.; Griebenow, K.; Schweitzer-Stenner, R. Abeta(1-28) fragment of the amyloid peptide predominantly adopts a polyproline II conformation in an acidic solution. Biochemistry 2004, 43, 6893–6898. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimer‘s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Eom, K.; Kwon, T. Different Aggregation Pathways and Structures for A 40 and A 42 Peptides. Biomolecules 2021, 11, 198. [Google Scholar] [CrossRef]
- Sgourakis, N.G.; Yan, Y.; McCallum, S.A.; Wang, C.; Garcia, A.E. The Alzheimer’s peptides Abeta40 and 42 adopt distinct conformations in water: A combined MD/NMR study. J. Mol. Biol. 2007, 368, 1448–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coles, M.; Bicknell, W.; Watson, A.A.; Fairlie, D.P.; Craik, D.J. Solution structure of amyloid beta-peptide(1-40) in a water-micelle environment. Is the membrane-spanning domain where we think it is? Biochemistry 1998, 37, 11064–11077. [Google Scholar] [CrossRef]
- Eisenberg, D.S.; Sawaya, M.R. Implications for Alzheimer’s disease of an atomic resolution structure of amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA 2016, 113, 9398–9400. [Google Scholar] [CrossRef] [Green Version]
- Wildburger, N.C.; Esparza, T.J.; LeDuc, R.D.; Fellers, R.T.; Thomas, P.M.; Cairns, N.J.; Kelleher, N.L.; Bateman, R.J.; Brody, D.L. Diversity of Amyloid-beta Proteoforms in the Alzheimer’s Disease Brain. Sci. Rep. 2017, 7, 9520. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.L.; Kim, C.; Haldiman, T.; ElHag, M.; Mehndiratta, P.; Pichet, T.; Lissemore, F.; Shea, M.; Cohen, Y.; Chen, W.; et al. Rapidly progressive Alzheimer’s disease features distinct structures of amyloid-β. Brain 2015, 138, 1009–1022. [Google Scholar] [CrossRef] [Green Version]
- Tay, W.M.; Huang, D.; Rosenberry, T.L.; Paravastu, A.K. The Alzheimer’s amyloid-β(1-42) peptide forms off-pathway oligomers and fibrils that are distinguished structurally by intermolecular organization. J. Mol. Biol. 2013, 425, 2494–2508. [Google Scholar] [CrossRef]
- Hayden, E.Y.; Teplow, D.B. Amyloid β-protein oligomers and Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 60. [Google Scholar] [CrossRef] [Green Version]
- Lau, H.H.C.; Ingelsson, M.; Watts, J.C. The existence of Aβ strains and their potential for driving phenotypic heterogeneity in Alzheimer’s disease. Acta Neuropathol. 2020. [Google Scholar] [CrossRef]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 2017, 140, 1829–1850. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Fromholt, S.E.; Chakrabarty, P.; Zhu, F.; Liu, X.; Pace, M.C.; Koh, J.; Golde, T.E.; Levites, Y.; Lewis, J.; et al. Diversity in Aβ deposit morphology and secondary proteome insolubility across models of Alzheimer-type amyloidosis. Acta Neuropathol. Commun. 2020, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Jagust, W. Is amyloid-β harmful to the brain? Insights from human imaging studies. Brain 2016, 139, 23–30. [Google Scholar] [CrossRef] [Green Version]
- McAleese, K.E.; Colloby, S.J.; Thomas, A.J.; Al-Sarraj, S.; Ansorge, O.; Neal, J.; Roncaroli, F.; Love, S.; Francis, P.T.; Attems, J. Concomitant neurodegenerative pathologies contribute to the transition from mild cognitive impairment to dementia. Alzheimers Dement. 2021. [Google Scholar] [CrossRef]
- Shafiq, M.; Zafar, S.; Younas, N.; Noor, A.; Puig, B.; Altmeppen, H.C.; Schmitz, M.; Matschke, J.; Ferrer, I.; Glatzel, M.; et al. Prion protein oligomers cause neuronal cytoskeletal damage in rapidly progressive Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 11. [Google Scholar] [CrossRef]
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A.; Kwon, Y.T. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Maldonado, M.A. The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol. Immunol. 2006, 3, 255–261. [Google Scholar] [PubMed]
- Andre, R.; Tabrizi, S.J. Misfolded PrP and a novel mechanism of proteasome inhibition. Prion 2012, 6, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, A.N.; Upadhya, S.C. Role of ubiquitin-proteasome-mediated proteolysis in nervous system disease. Biochim. Biophys. Acta 2011, 1809, 128–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Dice, J.F. Traffic. Regulation of lamp2a levels in the lysosomal membrane. Traffic 2000, 1, 570–583. [Google Scholar] [CrossRef] [Green Version]
- Bourdenx, M.; Martín-Segura, A.; Scrivo, A.; Rodriguez-Navarro, J.A.; Kaushik, S.; Tasset, I.; Diaz, A.; Storm, N.J.; Xin, Q.; Juste, Y.R.; et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 2021, 184, 2696–2714. [Google Scholar] [CrossRef]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef]
- Garofalo, T.; Ferri, A.; Sorice, M.; Azmoon, P.; Grasso, M.; Mattei, V.; Capozzi, A.; Manganelli, V.; Misasi, R. Neuroglobin overexpression plays a pivotal role in neuroprotection through mitochondrial raft-like microdomains in neuroblastoma SK-N-BE2 cells. Mol. Cell. Neurosci. 2018, 88, 167–176. [Google Scholar] [CrossRef]
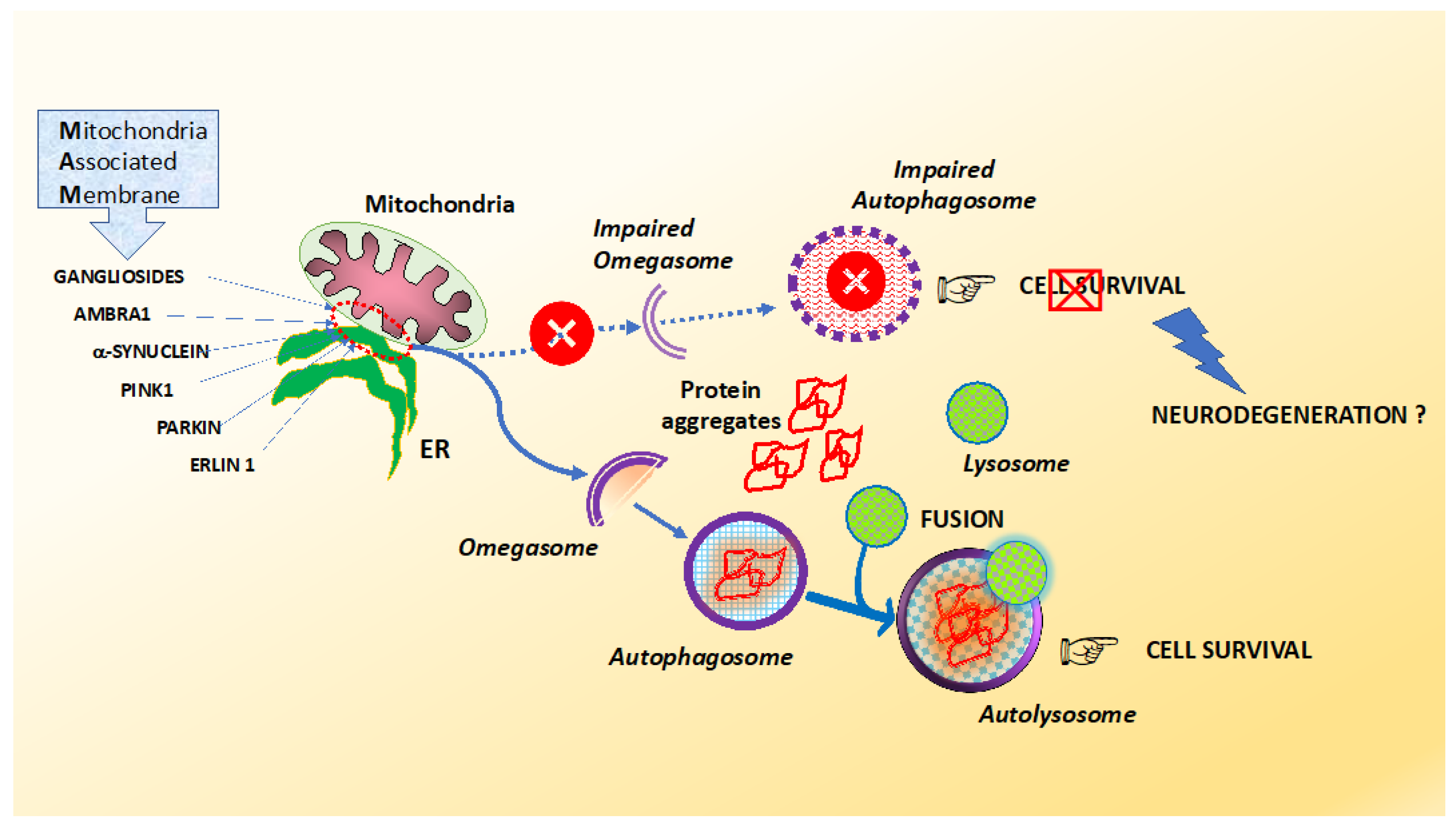
- Area-Gomez, E.; de Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 19, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciarlo, L.; Manganelli, V.; Matarrese, P.; Garofalo, T.; Tinari, A.; Gambardella, L.; Marconi, M.; Grasso, M.; Misasi, R.; Sorice, M.; et al. Raft-like microdomains play a key role in mitochondrial impairment in lymphoid cells from patients with Huntington’s disease. J. Lipid Res. 2012, 53, 2057–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, T.; Matarrese, P.; Manganelli, V.; Marconi, M.; Tinari, A.; Gambardella, L.; Faggioni, A.; Misasi, R.; Sorice, M.; Malorni, W. Evidence for the involvement of lipid rafts localized at the ER-mitochondria associated membranes in autophagosome formation. Autophagy 2016, 12, 917–935. [Google Scholar] [CrossRef] [Green Version]
- Matarrese, P.; Garofalo, T.; Manganelli, V.; Gambardella, L.; Marconi, M.; Grasso, M.; Tinari, A.; Misasi, R.; Malorni, W.; Sorice, M. Evidence for the involvement of GD3 ganglioside in autophagosome formation and maturation. Autophagy 2014, 10, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, V.; Matarrese, P.; Antonioli, M.; Gambardella, L.; Vescovo, T.; Gretzmeier, C.; Longo, A.; Capozzi, A.; Recalchi, S.; Riitano, G.; et al. Raft-like lipid microdomains drive autophagy initiation via AMBRA1-ERLIN1 molecular association within MAMs. Autophagy 2020. [Google Scholar] [CrossRef]
- Cha-Molstad, H.; Sung, K.S.; Hwang, J.; Kim, K.A.; Yu, J.E.; Yoo, Y.D.; Jang, J.M.; Han, D.H.; Molstad, M.; Kim, J.G.; et al. Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat. Cell Biol. 2015, 17, 917–929. [Google Scholar] [CrossRef]
- Stephan, J.S.; Fioriti, L.; Lamba, N.; Colnaghi, L.; Karl, K.; Derkatch, I.L.; Kandel, E.R. The CPEB3 Protein Is a Functional Prion that Interacts with the Actin Cytoskeleton. Cell Rep. 2015, 11, 1772–1785. [Google Scholar] [CrossRef]
- Mattei, V.; Martellucci, S.; Santilli, F.; Manganelli, V.; Garofalo, T.; Candelise, N.; Caruso, A.; Sorice, M.; Scaccianoce, S.; Misasi, R. Morphine Withdrawal Modifies Prion Protein Expression in Rat Hippocampus. PLoS ONE 2017, 12, e0169571. [Google Scholar] [CrossRef] [Green Version]
- Arendt, T.; Bullmann, T. Neuronal plasticity in hibernation and the proposed role of the microtubule-associated protein tau as a “master switch” regulating synaptic gain in neuronal networks. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R478–R489. [Google Scholar] [CrossRef] [Green Version]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochem. Biophy. Acta 2010, 1804, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- Atarashi, R.; Satoh, K.; Sano, K.; Fuse, T.; Yamaguchi, N.; Ishibashi, D.; Matsubara, T.; Nakagaki, T.; Yamanaka, H.; Shirabe, S.; et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat. Med. 2011, 17, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Orrú, C.D.; Groveman, B.R.; Hughson, A.G.; Zanusso, G.; Coulthart, M.B.; Caughey, B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. mBio 2015, 6, e02451-14. [Google Scholar] [CrossRef] [Green Version]
- Fairfoul, G.; McGuire, L.I.; Pal, S.; Ironside, J.W.; Neumann, J.; Christie, S.; Joachim, C.; Esiri, M.; Evetts, S.G.; Rolinski, M.; et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 2016, 3, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Candelise, N.; Baiardi, S.; Capellari, S.; Giannini, G.; Orrù, C.D.; Antelmi, E.; Mammana, A.; Hughson, A.G.; Calandra-Buonaura, G.; et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol. 2020, 140, 49–62. [Google Scholar] [CrossRef]
- Saijo, E.; Ghetti, B.; Zanusso, G.; Oblak, A.; Furman, J.L.; Diamond, M.I.; Kraus, A.; Caughey, B. Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol. 2017, 133, 751–765. [Google Scholar] [CrossRef]
- Saijo, E.; Metrick, M.A., 2nd; Koga, S.; Parchi, P.; Litvan, I.; Spina, S.; Boxer, A.; Rojas, J.C.; Galasko, D.; Kraus, A.; et al. 4-Repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol. 2020, 139, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Saijo, E.; Metrick, M.A., 2nd; Newell, K.; Sigurdson, C.J.; Zanusso, G.; Ghetti, B.; Caughey, B. Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol. 2019, 137, 585–598. [Google Scholar] [CrossRef] [Green Version]
- Scialò, C.; Tran, T.H.; Salzano, G.; Novi, G.; Caponnetto, C.; Chiò, A.; Calvo, A.; Canosa, A.; Moda, F.; Caroppo, P.; et al. TDP-43 real-time quaking induced conversion reaction optimization and detection of seeding activity in CSF of amyotrophic lateral sclerosis and frontotemporal dementia patients. Brain Commun. 2020, 2, fcaa142. [Google Scholar] [CrossRef] [PubMed]
- Orrú, C.D.; Groveman, B.R.; Raymond, L.D.; Hughson, A.G.; Nonno, R.; Zou, W.; Ghetti, B.; Gambetti, P.; Caughey, B. Bank Vole Prion Protein As an Apparently Universal Substrate for RT-QuIC-Based Detection and Discrimination of Prion Strains. PLoS Pathog. 2015, 11, e1004983. [Google Scholar] [CrossRef]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Candelise, N.; Schmitz, M.; Llorens, F.; Villar-Piqué, A.; Cramm, M.; Thom, T.; da Silva Correia, S.M.; da Cunha, J.E.G.; Möbius, W.; Outeiro, T.F.; et al. Seeding variability of different alpha synuclein strains in synucleinopathies. Ann. Neurol. 2019, 85, 691–703. [Google Scholar] [CrossRef]
- Hermann, P.; Appleby, B.; Brandel, J.P.; Caughey, B.; Collins, S.; Geschwind, M.D.; Green, A.; Haïk, S.; Kovacs, G.G.; Ladogana, A.; et al. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Lancet Neurol. 2021, 20, 235–246. [Google Scholar] [CrossRef]
- Llorens, F.; Villar-Piqué, A.; Schmitz, M.; Diaz-Lucena, D.; Wohlhage, M.; Hermann, P.; Goebel, S.; Schmidt, I.; Glatzel, M.; Hauw, J.J.; et al. Plasma total prion protein as a potential biomarker for neurodegenerative dementia: Diagnostic accuracy in the spectrum of prion diseases. Neuropathol. Appl. Neurobiol. 2020, 46, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA 2020, 324, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Kabir, M.T.; Niaz, K.; Jeandet, P.; Clément, C.; Mathew, B.; Rauf, A.; Rengasamy, K.R.R.; Sobarzo-Sánchez, E.; Ashraf, G.M.; et al. Molecular Insight into the Therapeutic Promise of Flavonoids against Alzheimer’s Disease. Molecules 2020, 25, 1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, M.; Candelise, N.; Kanata, E.; Llorens, F.; Thüne, K.; Villar-Piqué, A.; da Silva Correia, S.M.; Dafou, D.; Sklaviadis, T.; Appelhans, D.; et al. Validation of Poly(Propylene Imine) Glycodendrimers Towards Their Anti-prion Conversion Efficiency. Mol. Neurobiol. 2020, 57, 1863–1874. [Google Scholar] [CrossRef]
- Peña-Díaz, S.; Pujols, J.; Pinheiro, F.; Santos, J.; Pallarés, I.; Navarro, S.; Conde-Gimenez, M.; García, J.; Salvatella, X.; Dalfó, E.; et al. Inhibition of alpha-Synuclein Aggregation and Mature Fibril Disassembling With a Minimalistic Compound, ZPDm. Front. Bioeng. Biotechnol. 2020, 8, 588947. [Google Scholar] [CrossRef]
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering 2021, 8, 30. [Google Scholar] [CrossRef]
- Salman, M.M.; Al-Obaidi, Z.; Kitchen, P.; Loreto, A.; Bill, R.M.; Wade-Martins, R. Advances in Applying Computer-Aided Drug Design for Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4688. [Google Scholar] [CrossRef]
- Brunden, K.R.; Ballatore, C.; Crowe, A.; Smith, A.B., III; Lee, V.M.Y.; Trojanowski, J.Q. Tau-directed drug discovery for Alzheimer’s disease and related tauopathies: A focus on tau assembly inhibitors. Exp. Neurol. 2010, 223, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.I.; Song, E. Lab-on-a-Chip Systems for Aptamer-Based Biosensing. Micromachines 2020, 11, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| LCD Features | Type of Interaction | String Example | Protein Example | References |
|---|---|---|---|---|
| Hydrophobic-enriched (G, A, I, L, V, F, Y) | Hydrophobic | VTNVGGAVVTGVTAVA | α-synuclein | [26] |
| Poly-Q | weak side-by-side | ≥8 Q residues | Huntingtin | [42] |
| Q/N-stretches | weak side-by-side, intra- and inter-molecular | P/QQGGYQQ/SYN repeats | [PSI+] (S.cerevisiae) | [44] |
| Homopolymers | H-bonds | GPG/GGX motifs | Flag (Silk protein) | [48] |
| Poly-basic | Net surface charge | RGRGG repeats | FUS | [49] |
| Protein | Strains | Off Pathway | LLPS | Associated Neurodegenerative Disease | References |
|---|---|---|---|---|---|
| Prion protein | Drowsy and scratching in sheeps; Type 120, Type 121, Type 2 in humans | V | V | Kuru, CJD, FFI, GSS, vPSPr | [111,112,113,114,116,121,122,123] |
| α-synuclein | MSA associated; PD/DLB associated; Fibrils and Ribbons | V | V | PD, DLB, MSA, AD * | [134,138,140] |
| TDP-43 | Type A to Type E | / | V | ALS, FTD, AD * | [144,149,152,153] |
| Tau | CBD associated; AD associated | V | V | PiD, CBD, PSP, FTD, PART, AD | [157,170] |
| Aβ | Possibly associated to AD heterogeneity | V | / | AD, CAA, PDD *, DLB * | [201,204] |
| Protein | Sequence(s) Features | LCD Sequence(s) | References |
|---|---|---|---|
| Prion protein | N-terminal octapeptide repeats | 51PQGGTWGQ58 | [105] |
| C-terminal hydrophobic stretch | 112MAGAAAAGAVVGGLGGYVLGSAM134 | [106] | |
| α-synuclein | N-terminal imperfectly repeated LCD | consensus sequence: KTKEGV | [127] |
| C-terminal hydrophobic NAC domain | 61EQVTNVGGAVVTGVTAVAQK TVEGAGSIAAATGFVKKDQL GKNEE105 | [126] | |
| TDP-43 | Long C-terminal glycine-rich LCD | IDR1: 216RAFAFVTFADDQIAQSLCGEDLIIKGISVHISNAEPKHNSNRQLERSGRFGGNPGGFGNQGGFGNSRGGGAGLGNNQGSNMGGGM310 | [144,145] |
| Amyloidogenic core: 311NFGAFSINPAMMAAAQAALQSSWGMMGMLASQQNQSGPSGNNQNQGNMQ360 | |||
| IDR2: 361REPNQAFGSGNNSYSGSNSGAAIGWGSASNAGSGSGFNGGFGSSMDSKSSGWGM414 | |||
| Tau | C-terminal hydrophobic PHF6 and PHF6* motifs | 275VQIINK280; 306VQIVYK311 | [166,168] |
| Aβ | N-terminal Hydrophobic-aromatic | 16KLVFFA21 | [75] |
| C-terminal Hydrophobic | 29GAIIGL34; 29GGVVIA34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Candelise, N.; Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C.; Manganelli, V.; Garofalo, T.; Sorice, M.; Misasi, R. Protein Aggregation Landscape in Neurodegenerative Diseases: Clinical Relevance and Future Applications. Int. J. Mol. Sci. 2021, 22, 6016. https://doi.org/10.3390/ijms22116016
Candelise N, Scaricamazza S, Salvatori I, Ferri A, Valle C, Manganelli V, Garofalo T, Sorice M, Misasi R. Protein Aggregation Landscape in Neurodegenerative Diseases: Clinical Relevance and Future Applications. International Journal of Molecular Sciences. 2021; 22(11):6016. https://doi.org/10.3390/ijms22116016
Chicago/Turabian StyleCandelise, Niccolò, Silvia Scaricamazza, Illari Salvatori, Alberto Ferri, Cristiana Valle, Valeria Manganelli, Tina Garofalo, Maurizio Sorice, and Roberta Misasi. 2021. "Protein Aggregation Landscape in Neurodegenerative Diseases: Clinical Relevance and Future Applications" International Journal of Molecular Sciences 22, no. 11: 6016. https://doi.org/10.3390/ijms22116016
APA StyleCandelise, N., Scaricamazza, S., Salvatori, I., Ferri, A., Valle, C., Manganelli, V., Garofalo, T., Sorice, M., & Misasi, R. (2021). Protein Aggregation Landscape in Neurodegenerative Diseases: Clinical Relevance and Future Applications. International Journal of Molecular Sciences, 22(11), 6016. https://doi.org/10.3390/ijms22116016