
Peroxisomal ABC Transporters: An Update



Abstract
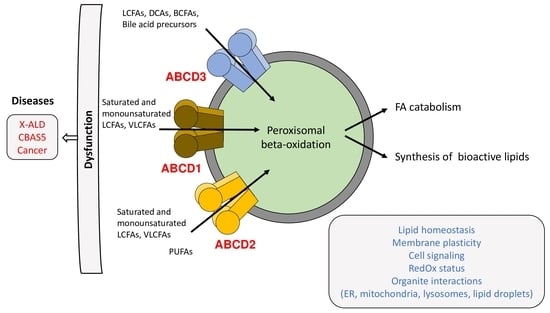
:
1. Introduction
2. Structure, Function, and Mechanism of Transport
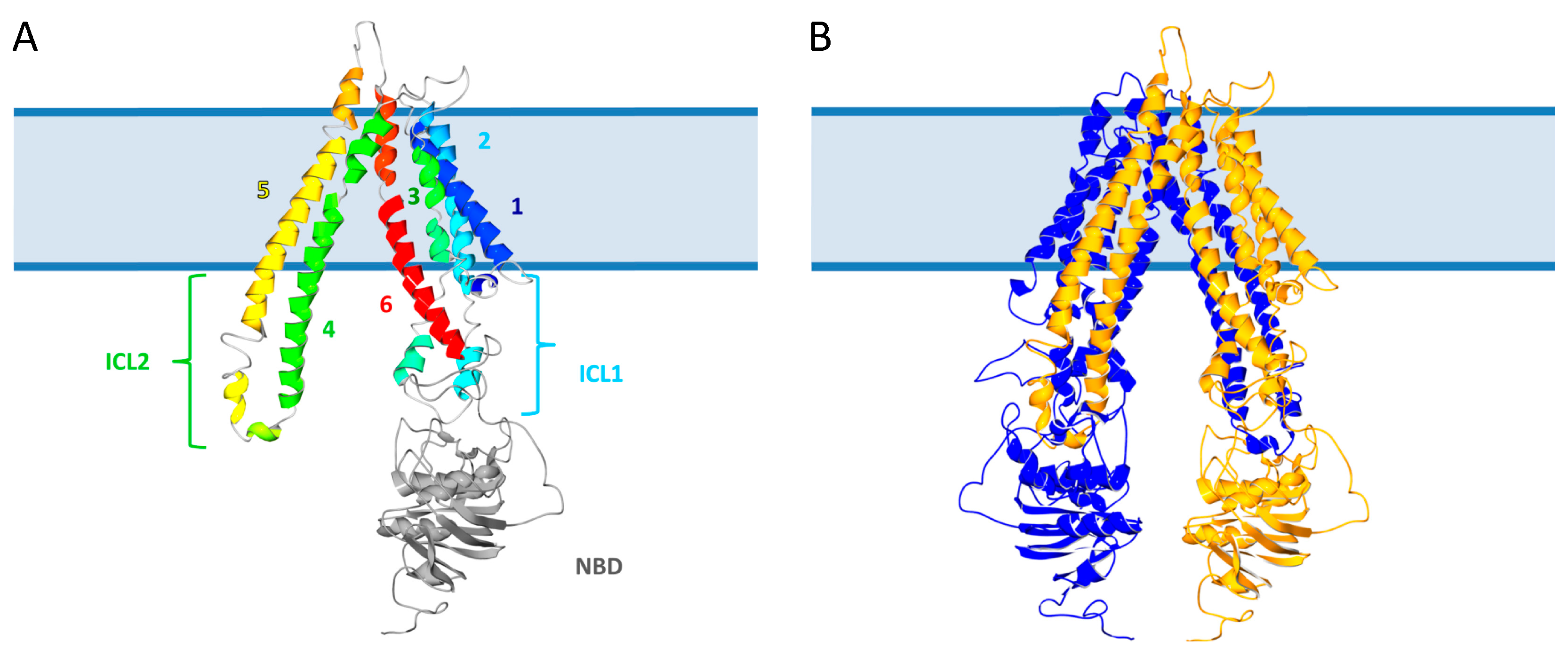
2.1. Structure
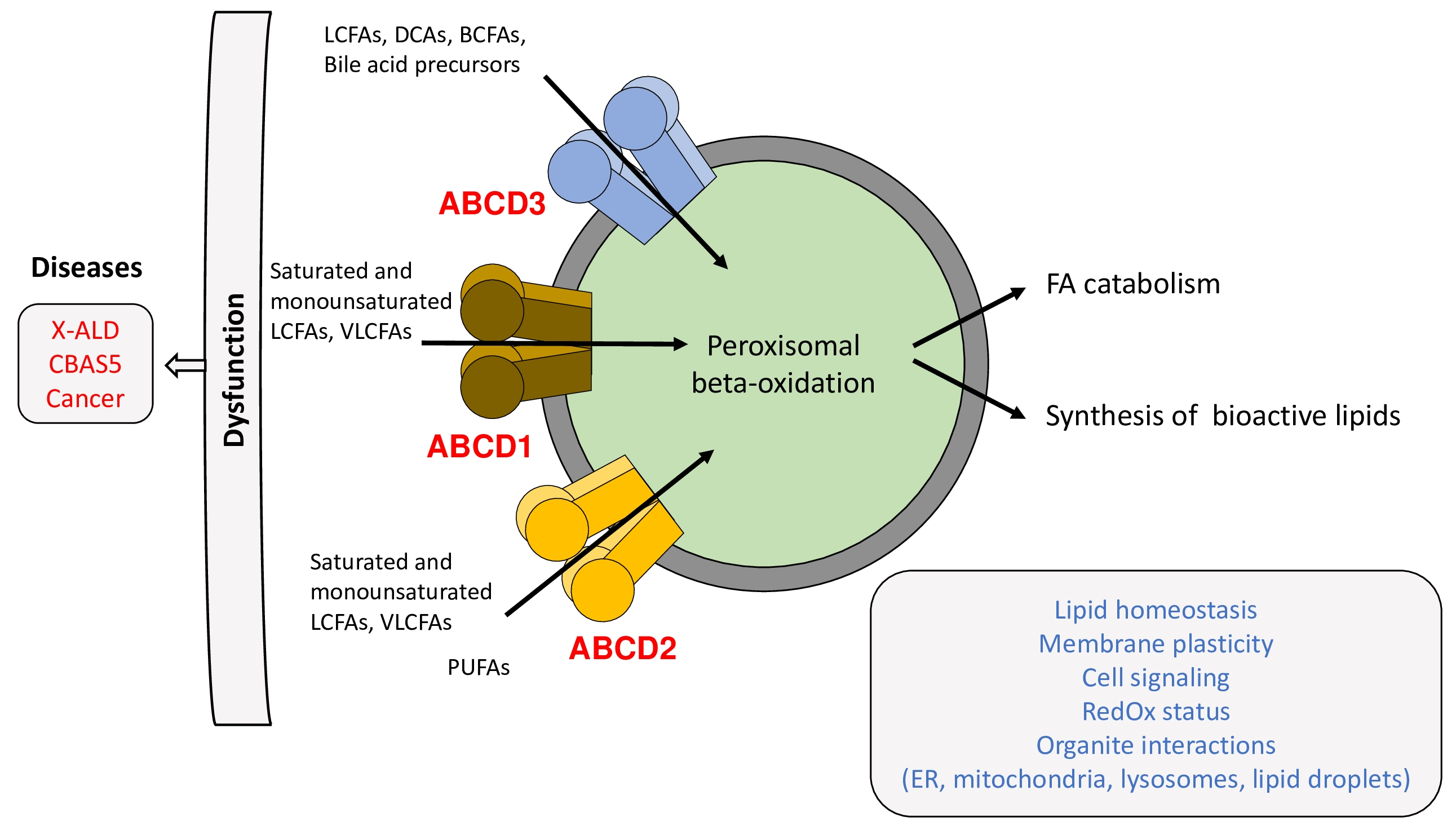
2.2. Substrate Specificity
2.3. Mechanism
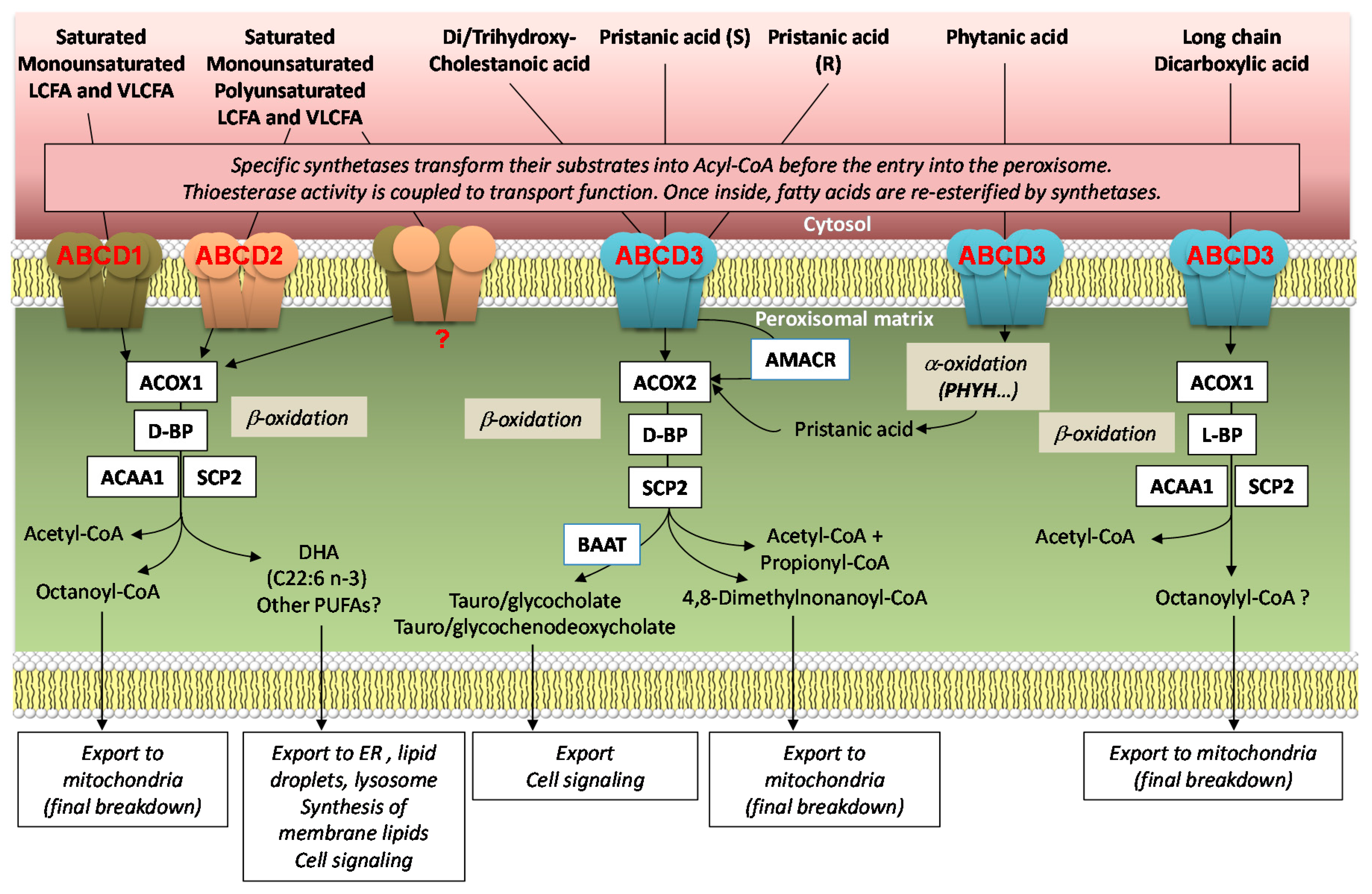
3. Human Diseases
3.1. X-Linked Adrenoleukodystrophy
3.2. Congenital Bile Acid Synthesis Defect Type 5
3.3. Peroxisomal ABC Transporters and Cancer
4. Cell, Plant, and Animal Models
4.1. Yeast
4.2. Plant
4.3. Nematode
4.4. Insect
4.5. Fish
4.6. Rat and Mouse
4.7. Human
5. Protein Interactions and Unexpected Roles
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| ACOX1 | Acyl-coenzyme A oxidase 1 |
| AMN | Adrenomyeloneuropathy |
| BBB | Blood-brain barrier |
| cALD | Cerebral adrenoleukodystrophy |
| CBAS | Congenital bile acid synthesis defect |
| CNS | Central nervous system |
| CoA | Coenzyme A |
| CTS | Comatose |
| DHA | Docosahexaenoic acid, C22:6 n-3 |
| ER | Endoplasmic reticulum |
| FA | Fatty acid |
| FC | Fold change |
| HSCT | Hematopoietic stem cell transplantation |
| iPSC | Induced pluripotent stem cell |
| LCFA | Long-chain fatty acid |
| LD | Lipid droplet |
| MCFA | Medium-chain fatty acid |
| MUFA | Monounsaturated fatty acid |
| NBD | Nucleotide binding domain |
| PBMC | Peripheral blood mononuclear cell |
| PMP | Peroxisomal membrane protein |
| PUFA | Polyunsaturated fatty acid |
| TMD | Transmembrane domain |
| VLCFA | Very long-chain fatty acid |
| X-ALD | X-linked adrenoleukodystrophy |
References
- Thomas, C.; Tampé, R. Structural and Mechanistic Principles of ABC Transporters. Annu. Rev. Biochem. 2020, 89, 605–636. [Google Scholar] [CrossRef]
- Dean, M.; Annilo, T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genom. Hum. Genet. 2005, 6, 123–142. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Morita, M. ABC Transporter Subfamily D: Distinct Differences in Behavior between ABCD1-3 and ABCD4 in Subcellular Localization, Function, and Human Disease. BioMed Res. Int. 2016, 2016, 6786245. [Google Scholar] [CrossRef] [Green Version]
- Shani, N.; Jimenez-Sanchez, G.; Steel, G.; Dean, M.; Valle, D. Identification of a fourth half ABC transporter in the human peroxisomal membrane. Hum. Mol. Genet. 1997, 6, 1925–1931. [Google Scholar] [CrossRef]
- Coelho, D.; Kim, J.C.; Miousse, I.R.; Fung, S.; du Moulin, M.; Buers, I.; Suormala, T.; Burda, P.; Frapolli, M.; Stucki, M.; et al. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat. Genet. 2012, 44, 1152–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwayama, Y.; Seki, M.; Yasui, A.; Murasaki, Y.; Morita, M.; Yamashita, Y.; Sakaguchi, M.; Tanaka, Y.; Imanaka, T. 70-kDa peroxisomal membrane protein related protein (P70R/ABCD4) localizes to endoplasmic reticulum not peroxisomes, and NH2-terminal hydrophobic property determines the subcellular localization of ABC subfamily D proteins. Exp. Cell Res. 2009, 315, 190–205. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef] [PubMed]
- Trompier, D.; Vejux, A.; Zarrouk, A.; Gondcaille, C.; Geillon, F.; Nury, T.; Savary, S.; Lizard, G. Brain peroxisomes. Biochimie 2014, 98, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandusse, S.; Denis, S.; Mooijer, P.A.; Zhang, Z.; Reddy, J.K.; Spector, A.A.; Wanders, R.J. Identification of the peroxisomal beta-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J. Lipid Res. 2001, 42, 1987–1995. [Google Scholar] [CrossRef]
- Chapkin, R.S.; Kim, W.; Lupton, J.R.; McMurray, D.N. Dietary docosahexaenoic and eicosapentaenoic acid: Emerging mediators of inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2009, 81, 187–191. [Google Scholar] [CrossRef] [Green Version]
- Lodhi, I.J.; Semenkovich, C.F. Peroxisomes: A nexus for lipid metabolism and cellular signaling. Cell. Metab. 2014, 19, 380–392. [Google Scholar] [CrossRef] [Green Version]
- Di Cara, F.; Andreoletti, P.; Trompier, D.; Vejux, A.; Bulow, M.H.; Sellin, J.; Lizard, G.; Cherkaoui-Malki, M.; Savary, S. Peroxisomes in Immune Response and Inflammation. Int. J. Mol. Sci. 2019, 20, 3877. [Google Scholar] [CrossRef] [Green Version]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef]
- Lismont, C.; Revenco, I.; Fransen, M. Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. Int. J. Mol. Sci. 2019, 20, 3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, M.; Sengupta, T.K.; Sheikh, F.; Aubourg, P.; Singh, I. Topology of ATP-binding domain of adrenoleukodystrophy gene product in peroxisomes. Arch. Biochem. Biophys. 1996, 334, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Andreoletti, P.; Raas, Q.; Gondcaille, C.; Cherkaoui-Malki, M.; Trompier, D.; Savary, S. Predictive Structure and Topology of Peroxisomal ATP-Binding Cassette (ABC) Transporters. Int. J. Mol. Sci. 2017, 18, 1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, A.; Carrier, D.J.; Schaedler, T.; Waterham, H.R.; van Roermund, C.W.; Theodoulou, F.L. Peroxisomal ABC transporters: Functions and mechanism. Biochem. Soc. Trans. 2015, 43, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Geillon, F.; Gondcaille, C.; Charbonnier, S.; Van Roermund, C.W.; Lopez, T.E.; Dias, A.M.M.; de Barros, J.-P.P.; Arnould, C.; Wanders, R.J.; Trompier, D.; et al. Structure-function analysis of peroxisomal ATP-binding cassette transporters using chimeric dimers. J. Biol. Chem. 2014, 289, 24511–24520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genin, E.; Geillon, F.; Gondcaille, C.; Athias, A.; Gambert, P.; Trompier, D.; Savary, S. Substrate specificity overlap and interaction between adrenoleukodystrophy protein (ALDP/ABCD1) and adrenoleukodystrophy-related protein (ALDRP/ABCD2). J. Biol. Chem. 2011, 286, 8075–8084. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.D.; Kemp, S.; Braiterman, L.T.; Lu, J.F.; Wei, H.M.; Geraghty, M.; Stetten, G.; Bergin, J.S.; Pevsner, J.; Watkins, P.A. X-linked adrenoleukodystrophy: Genes, mutations, and phenotypes. Neurochem. Res. 1999, 24, 521–535. [Google Scholar] [CrossRef]
- Tanaka, A.R.; Tanabe, K.; Morita, M.; Kurisu, M.; Kasiwayama, Y.; Matsuo, M.; Kioka, N.; Amachi, T.; Imanaka, T.; Ueda, K. ATP binding/hydrolysis by and phosphorylation of peroxisomal ATP-binding cassette proteins PMP70 (ABCD3) and adrenoleukodystrophy protein (ABCD1). J. Biol. Chem. 2002, 277, 40142–40147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillebrand, M.; Verrier, S.E.; Ohlenbusch, A.; Schafer, A.; Soling, H.D.; Wouters, F.S.; Gartner, J. Live cell FRET microscopy: Homo- and heterodimerization of two human peroxisomal ABC transporters, the adrenoleukodystrophy protein (ALDP, ABCD1) and PMP70 (ABCD3). J. Biol. Chem. 2007, 282, 26997–27005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimaraes, C.P.; Domingues, P.; Aubourg, P.; Fouquet, F.; Pujol, A.; Jimenez-Sanchez, G.; Sa-Miranda, C.; Azevedo, J.E. Mouse liver PMP70 and ALDP: Homomeric interactions prevail in vivo. Biochim. Biophys. Acta 2004, 1689, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.X.; Janvier, K.; Berteaux-Lecellier, V.; Cartier, N.; Benarous, R.; Aubourg, P. Homo- and heterodimerization of peroxisomal ATP-binding cassette half-transporters. J. Biol. Chem. 1999, 274, 32738–32743. [Google Scholar] [CrossRef] [Green Version]
- Van Roermund, C.W.; Visser, W.F.; Ijlst, L.; Waterham, H.R.; Wanders, R.J. Differential substrate specificities of human ABCD1 and ABCD2 in peroxisomal fatty acid beta-oxidation. Biochim. Biophys. Acta 2011, 1811, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Van Roermund, C.W.; Visser, W.F.; Ijlst, L.; van Cruchten, A.; Boek, M.; Kulik, W.; Waterham, H.R.; Wanders, R.J. The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl-CoA esters. FASEB J. 2008, 22, 4201–4208. [Google Scholar] [CrossRef] [Green Version]
- Geillon, F.; Gondcaille, C.; Raas, Q.; Dias, A.M.M.; Pecqueur, D.; Truntzer, C.; Lucchi, G.; Ducoroy, P.; Falson, P.; Savary, S.; et al. Peroxisomal ATP-binding cassette transporters form mainly tetramers. J. Biol. Chem. 2017, 292, 6965–6977. [Google Scholar] [CrossRef] [Green Version]
- Woudenberg, J.; Rembacz, K.P.; Hoekstra, M.; Pellicoro, A.; van den Heuvel, F.A.; Heegsma, J.; van Ijzendoorn, S.C.; Holzinger, A.; Imanaka, T.; Moshage, H.; et al. Lipid rafts are essential for peroxisome biogenesis in HepG2 cells. Hepatology 2010, 52, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Mosser, J.; Douar, A.M.; Sarde, C.O.; Kioschis, P.; Feil, R.; Moser, H.; Poustka, A.M.; Mandel, J.L.; Aubourg, P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 1993, 361, 726–730. [Google Scholar] [CrossRef]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.; Aubourg, P.; Poll-The, B.T. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis. 2012, 7, 51. [Google Scholar] [CrossRef]
- Rattay, T.W.; Rautenberg, M.; Söhn, A.S.; Hengel, H.; Traschütz, A.; Röben, B.; Hayer, S.N.; Schüle, R.; Wiethoff, S.; Zeltner, L.; et al. Defining diagnostic cutoffs in neurological patients for serum very long chain fatty acids (VLCFA) in genetically confirmed X-Adrenoleukodystrophy. Sci. Rep. 2020, 10, 15093. [Google Scholar] [CrossRef]
- Forss-Petter, S.; Werner, H.; Berger, J.; Lassmann, H.; Molzer, B.; Schwab, M.H.; Bernheimer, H.; Zimmermann, F.; Nave, K.A. Targeted inactivation of the X-linked adrenoleukodystrophy gene in mice. J. Neurosci. Res. 1997, 50, 829–843. [Google Scholar] [CrossRef]
- Kobayashi, T.; Shinnoh, N.; Kondo, A.; Yamada, T. Adrenoleukodystrophy protein-deficient mice represent abnormality of very long chain fatty acid metabolism. Biochem. Biophys. Res. Commun. 1997, 232, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.F.; Lawler, A.M.; Watkins, P.A.; Powers, J.M.; Moser, A.B.; Moser, H.W.; Smith, K.D. A mouse model for X-linked adrenoleukodystrophy. Proc. Natl. Acad. Sci. USA 1997, 94, 9366–9371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartier, N.; Lopez, J.; Moullier, P.; Rocchiccioli, F.; Rolland, M.O.; Jorge, P.; Mosser, J.; Mandel, J.L.; Bougneres, P.F.; Danos, O.; et al. Retroviral-mediated gene transfer corrects very-long-chain fatty acid metabolism in adrenoleukodystrophy fibroblasts. Proc. Natl. Acad. Sci. USA 1995, 92, 1674–1678. [Google Scholar] [CrossRef] [Green Version]
- Braiterman, L.T.; Zheng, S.; Watkins, P.A.; Geraghty, M.T.; Johnson, G.; McGuinness, M.C.; Moser, A.B.; Smith, K.D. Suppression of peroxisomal membrane protein defects by peroxisomal ATP binding cassette (ABC) proteins. Hum. Mol. Genet. 1998, 7, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Lombard-Platet, G.; Savary, S.; Sarde, C.O.; Mandel, J.L.; Chimini, G. A close relative of the adrenoleukodystrophy (ALD) gene codes for a peroxisomal protein with a specific expression pattern. Proc. Natl. Acad. Sci. USA 1996, 93, 1265–1269. [Google Scholar] [CrossRef] [Green Version]
- Netik, A.; Forss-Petter, S.; Holzinger, A.; Molzer, B.; Unterrainer, G.; Berger, J. Adrenoleukodystrophy-related protein can compensate functionally for adrenoleukodystrophy protein deficiency (X-ALD): Implications for therapy. Hum. Mol. Genet. 1999, 8, 907–913. [Google Scholar] [CrossRef] [Green Version]
- Pujol, A.; Ferrer, I.; Camps, C.; Metzger, E.; Hindelang, C.; Callizot, N.; Ruiz, M.; Pampols, T.; Giros, M.; Mandel, J.L. Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: A therapeutic target for X-adrenoleukodystrophy. Hum. Mol. Genet. 2004, 13, 2997–3006. [Google Scholar] [CrossRef] [Green Version]
- McGuinness, M.C.; Zhang, H.P.; Smith, K.D. Evaluation of Pharmacological Induction of Fatty Acid beta-Oxidation in X-Linked Adrenoleukodystrophy. Mol. Genet. Metab. 2001, 74, 256–263. [Google Scholar] [CrossRef]
- Bugaut, M.; Fourcade, S.; Gondcaille, C.; Gueugnon, F.; Depreter, M.; Roels, F.; Netik, A.; Berger, J.; Martin, P.; Pineau, T.; et al. Pharmacological induction of redundant genes for a therapy of X-ALD: Phenylbutyrate and other compounds. Adv. Exp. Med. Biol. 2003, 544, 281–291. [Google Scholar]
- Kemp, S.; Wei, H.M.; Lu, J.F.; Braiterman, L.T.; McGuinness, M.C.; Moser, A.B.; Watkins, P.A.; Smith, K.D. Gene redundancy and pharmacological gene therapy: Implications for X-linked adrenoleukodystrophy. Nat. Med. 1998, 4, 1261–1268. [Google Scholar] [CrossRef]
- Weber, F.D.; Weinhofer, I.; Einwich, A.; Forss-Petter, S.; Muneer, Z.; Maier, H.; Weber, W.H.; Berger, J. Evaluation of retinoids for induction of the redundant gene ABCD2 as an alternative treatment option in X-linked adrenoleukodystrophy. PLoS ONE 2014, 9, e103742. [Google Scholar] [CrossRef]
- Rampler, H.; Weinhofer, I.; Netik, A.; Forss-Petter, S.; Brown, P.J.; Oplinger, J.A.; Bugaut, M.; Berger, J. Evaluation of the therapeutic potential of PPARalpha agonists for X-linked adrenoleukodystrophy. Mol. Genet. Metab. 2003, 80, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, S.; Savary, S.; Albet, S.; Gauthe, D.; Gondcaille, C.; Pineau, T.; Bellenger, J.; Bentejac, M.; Holzinger, A.; Berger, J.; et al. Fibrate induction of the adrenoleukodystrophy-related gene (ABCD2)—Promoter analysis and role of the peroxisome proliferator-activated receptor PPAR alpha. Eur. J. Biochem. 2001, 268, 3490–3500. [Google Scholar] [CrossRef]
- Fourcade, S.; Savary, S.; Gondcaille, C.; Berger, J.; Netik, A.; Cadepond, F.; El Etr, M.; Molzer, B.; Bugaut, M. Thyroid hormone induction of the adrenoleukodystrophy-related gene (ABCD2). Mol. Pharmacol. 2003, 63, 1296–1303. [Google Scholar] [CrossRef] [Green Version]
- Gondcaille, C.; Depreter, M.; Fourcade, S.; Lecca, M.; Leclercq, S.; Martin, P.; Pineau, T.; Cadepond, F.; El-Etr, M.; Bertrand, N.; et al. Phenylbutyrate up-regulates the adrenoleukodystrophy-related gene as a nonclassical peroxisome proliferator. J. Cell Biol. 2005, 169, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, S.; Skrzypski, J.; Courvoisier, A.; Gondcaille, C.; Bonnetain, F.; Andre, A.; Chardigny, J.; Bellenger, S.; Bellenger, J.; Narce, M.; et al. Effect of dietary polyunsaturated fatty acids on the expression of peroxisomal ABC transporters. Biochimie 2008, 90, 1602–1607. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.; Gondcaille, C.; Trompier, D.; Savary, S. Induction of the adrenoleukodystrophy-related gene (ABCD2) by thyromimetics. J. Steroid Biochem. Mol. Biol. 2009, 116, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Gondcaille, C.; Genin, E.C.; Lopez, T.E.; Dias, A.M.M.; Geillon, F.; Andreoletti, P.; Cherkaoui-Malki, M.; Nury, T.; Lizard, G.; Weinhofer, I.; et al. LXR antagonists induce ABCD2 expression. Biochim. Biophys. Acta 2014, 1841, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trompier, D.; Gondcaille, C.; Lizard, G.; Savary, S. Regulation of the adrenoleukodystrophy-related gene (ABCD2): Focus on oxysterols and LXR antagonists. Biochem. Biophys. Res. Commun. 2014, 446, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Khan, M.; Singh, I. HDAC inhibitor SAHA normalizes the levels of VLCFAs in human skin fibroblasts from X-ALD patients and downregulates the expression of proinflammatory cytokines in Abcd1/2-silenced mouse astrocytes. J. Lipid Res. 2011, 52, 2056–2069. [Google Scholar] [CrossRef] [Green Version]
- Hartley, M.D.; Kirkemo, L.L.; Banerji, T.; Scanlan, T.S. A Thyroid Hormone-Based Strategy for Correcting the Biochemical Abnormality in X-Linked Adrenoleukodystrophy. Endocrinology 2017, 158, 1328–1338. [Google Scholar] [CrossRef] [Green Version]
- Hartley, M.D.; Shokat, M.D.; DeBell, M.J.; Banerji, T.; Kirkemo, L.L.; Scanlan, T.S. Pharmacological Complementation Remedies an Inborn Error of Lipid Metabolism. Cell Chem. Biol. 2020, 27, 551–559.e4. [Google Scholar] [CrossRef]
- Weinhofer, I.; Kunze, M.; Rampler, H.; Bookout, A.L.; Forss-Petter, S.; Berger, J. Liver X receptor alpha interferes with SREBP1c-mediated Abcd2 expression. Novel cross-talk in gene regulation. J. Biol. Chem. 2005, 280, 41243–41251. [Google Scholar] [CrossRef] [Green Version]
- Weinhofer, I.; Forss-Petter, S.; Zigman, M.; Berger, J. Cholesterol regulates ABCD2 expression: Implications for the therapy of X-linked adrenoleukodystrophy. Hum. Mol. Genet. 2002, 11, 2701–2708. [Google Scholar] [CrossRef] [Green Version]
- Zierfuss, B.; Weinhofer, I.; Kühl, J.S.; Köhler, W.; Bley, A.; Zauner, K.; Binder, J.; Martinović, K.; Seiser, C.; Hertzberg, C.; et al. Vorinostat in the acute neuroinflammatory form of X-linked adrenoleukodystrophy. Ann. Clin. Transl. Neurol. 2020, 7, 639–652. [Google Scholar] [CrossRef]
- Liu, J.; Liang, S.; Liu, X.; Brown, J.A.; Newman, K.E.; Sunkara, M.; Morris, A.J.; Bhatnagar, S.; Li, X.; Pujol, A.; et al. The absence of ABCD2 sensitizes mice to disruptions in lipid metabolism by dietary erucic acid. J. Lipid Res. 2012, 53, 1071–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Sabeva, N.S.; Bhatnagar, S.; Li, X.A.; Pujol, A.; Graf, G.A. ABCD2 is abundant in adipose tissue and opposes the accumulation of dietary erucic acid (C22:1) in fat. J. Lipid Res. 2010, 51, 162–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourcade, S.; Ruiz, M.; Camps, C.; Schluter, A.; Houten, S.M.; Mooyer, P.A.; Pampols, T.; Dacremont, G.; Wanders, R.J.; Giros, M.; et al. A key role for the peroxisomal ABCD2 transporter in fatty acid homeostasis. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E211–E221. [Google Scholar] [CrossRef] [Green Version]
- Imanaka, T.; Aihara, K.; Takano, T.; Yamashita, A.; Sato, R.; Suzuki, Y.; Yokota, S.; Osumi, T. Characterization of the 70-kDa peroxisomal membrane protein, an ATP binding cassette transporter. J. Biol. Chem. 1999, 274, 11968–11976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamijo, K.; Taketani, S.; Yokota, S.; Osumi, T.; Hashimoto, T. The 70-kDa peroxisomal membrane protein is a member of the Mdr (P-glycoprotein)-related ATP-binding protein superfamily. J. Biol. Chem. 1990, 265, 4534–4540. [Google Scholar] [CrossRef]
- Gartner, J.; Moser, H.; Valle, D. Mutations in the 70K peroxisomal membrane protein gene in Zellweger syndrome. Nat. Genet. 1992, 1, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Van Roermund, C.W.; Ijlst, L.; Wagemans, T.; Wanders, R.J.; Waterham, H.R. A role for the human peroxisomal half-transporter ABCD3 in the oxidation of dicarboxylic acids. Biochim. Biophys. Acta 2014, 1841, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, G.; Silva-Zolezzi, I.; Hebron, K.J.; Mihalik, S.; Watkins, P.; Moser, A.; Thomas, G.; Wood, P.A.; Valle, D. Defective phytanic and pristanic acids metabolism in PMP70 deficient mice results in defective nonshivering thermogenesis and dicarboxylic aciduria. J. Inherit. Metab. Dis. 2000, 23, 256. [Google Scholar]
- Ferdinandusse, S.; Jimenez-Sanchez, G.; Koster, J.; Denis, S.; Van Roermund, C.W.; Silva-Zolezzi, I.; Moser, A.B.; Visser, W.F.; Gulluoglu, M.; Durmaz, O.; et al. A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum. Mol. Genet. 2015, 24, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Violante, S.; Achetib, N.; van Roermund, C.W.T.; Hagen, J.; Dodatko, T.; Vaz, F.M.; Waterham, H.R.; Chen, H.; Baes, M.; Yu, C.; et al. Peroxisomes can oxidize medium- and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4. FASEB J. 2019, 33, 4355–4364. [Google Scholar] [CrossRef]
- Watkins, P.A.; Ellis, J.M. Peroxisomal acyl-CoA synthetases. Biochim. Biophys. Acta Mol. Bas. Dis. 2012, 1822, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Guimaraes, C.P.; Sa-Miranda, C.; Azevedo, J.E. Probing substrate-induced conformational alterations in adrenoleukodystrophy protein by proteolysis. J. Hum. Genet. 2005, 50, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Roerig, P.; Mayerhofer, P.; Holzinger, A.; Gartner, J. Characterization and functional analysis of the nucleotide binding fold in human peroxisomal ATP binding cassette transporters. FEBS Lett. 2001, 492, 66–72. [Google Scholar] [CrossRef] [Green Version]
- van Roermund, C.W.; Ijlst, L.; Majczak, W.; Waterham, H.R.; Folkerts, H.; Wanders, R.J.; Hellingwerf, K.J. Peroxisomal fatty acid uptake mechanism in Saccharomyces cerevisiae. J. Biol. Chem. 2012, 287, 20144–20153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marcos Lousa, C.; van Roermund, C.W.; Postis, V.L.; Dietrich, D.; Kerr, I.D.; Wanders, R.J.; Baldwin, S.A.; Baker, A.; Theodoulou, F.L. Intrinsic acyl-CoA thioesterase activity of a peroxisomal ATP binding cassette transporter is required for transport and metabolism of fatty acids. Proc. Natl. Acad. Sci. USA 2013, 110, 1279–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, K.; Mukai, E.; Watanabe, S.; Yamashita, A.; Morita, M.; So, T.; Imanaka, T. Acyl-CoA thioesterase activity of peroxisomal ABC protein ABCD1 is required for the transport of very long-chain acyl-CoA into peroxisomes. Sci. Rep. 2021, 11, 2192. [Google Scholar] [CrossRef] [PubMed]
- van Roermund, C.W.; IJlst, L.; Baker, A.; Wanders, R.J.; Theodoulou, F.L.; Waterham, H.R. The Saccharomyces cerevisiae ABC subfamily D transporter Pxa1/Pxa2p co-imports CoASH into the peroxisome. FEBS Lett. 2021, 595, 763–772. [Google Scholar] [CrossRef]
- Baumgart, E.; Vanhooren, J.C.; Fransen, M.; Marynen, P.; Puype, M.; Vandekerckhove, J.; Leunissen, J.A.; Fahimi, H.D.; Mannaerts, G.P.; van Veldhoven, P.P. Molecular characterization of the human peroxisomal branched-chain acyl-CoA oxidase: cDNA cloning, chromosomal assignment, tissue distribution, and evidence for the absence of the protein in Zellweger syndrome. Proc. Natl. Acad. Sci. USA 1996, 93, 13748–13753. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandusse, S.; Denis, S.; van Roermund, C.W.T.; Preece, M.A.; Koster, J.; Ebberink, M.S.; Waterham, H.R.; Wanders, R.J.A. A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta 2018, 1864, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Schepers, L.; Van Veldhoven, P.P.; Casteels, M.; Eyssen, H.J.; Mannaerts, G.P. Presence of three acyl-CoA oxidases in rat liver peroxisomes. An inducible fatty acyl-CoA oxidase, a noninducible fatty acyl-CoA oxidase, and a noninducible trihydroxycoprostanoyl-CoA oxidase. J. Biol. Chem. 1990, 265, 5242–5246. [Google Scholar] [CrossRef]
- Reddy, J.K.; Hashimoto, T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: An adaptive metabolic system. Annu. Rev. Nutr. 2001, 21, 193–230. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2015, 3, 83. [Google Scholar] [CrossRef] [Green Version]
- Trompier, D.; Savary, S. X-Linked Adrenoleukodystrophy; Morgan and Claypool Life Sciences Publishers: San Rafael, CA, USA, 2013; Volume 2. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [Green Version]
- Eichler, F.; Duncan, C.; Musolino, P.L.; Orchard, P.J.; De Oliveira, S.; Thrasher, A.J.; Armant, M.; Dansereau, C.; Lund, T.C.; Miller, W.P.; et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017, 377, 1630–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, A.R.; Brosco, J.; Comeau, A.M.; Green, N.S.; Grosse, S.D.; Jones, E.; Kwon, J.M.; Lam, W.K.; Ojodu, J.; Prosser, L.A.; et al. Newborn screening for X-linked adrenoleukodystrophy: Evidence summary and advisory committee recommendation. Genet. Med. 2017, 19, 121–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, W.C.; Moser, A.B.; Liu, A.C.; Jones, R.O.; Steinberg, S.J.; Lorey, F.; Panny, S.R.; Vogt, R.F., Jr.; Macaya, D.; Turgeon, C.T.; et al. Newborn screening for X-linked adrenoleukodystrophy (X-ALD): Validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method. Mol. Genet. Metab. 2009, 97, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Jangouk, P.; Zackowski, K.M.; Naidu, S.; Raymond, G.V. Adrenoleukodystrophy in female heterozygotes: Underrecognized and undertreated. Mol. Genet. Metab. 2012, 105, 180–185. [Google Scholar] [CrossRef]
- Ofman, R.; Dijkstra, I.M.; van Roermund, C.W.; Burger, N.; Turkenburg, M.; van Cruchten, A.; van Engen, C.E.; Wanders, R.J.; Kemp, S. The role of ELOVL1 in very long-chain fatty acid homeostasis and X-linked adrenoleukodystrophy. EMBO Mol. Med. 2010, 2, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Savary, S.; Trompier, D.; Andreoletti, P.; Le Borgne, F.; Demarquoy, J.; Lizard, G. Fatty acids—Induced lipotoxicity and inflammation. Curr. Drug Metab. 2012, 13, 1358–1370. [Google Scholar] [CrossRef]
- Singh, I.; Pujol, A. Pathomechanisms underlying X-adrenoleukodystrophy: A three-hit hypothesis. Brain Pathol. 2010, 20, 838–844. [Google Scholar] [CrossRef]
- Bergner, C.G.; van der Meer, F.; Winkler, A.; Wrzos, C.; Turkmen, M.; Valizada, E.; Fitzner, D.; Hametner, S.; Hartmann, C.; Pfeifenbring, S.; et al. Microglia damage precedes major myelin breakdown in X-linked adrenoleukodystrophy and metachromatic leukodystrophy. Glia 2019, 67, 1196–1209. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Sasidharan, N.; Laheji, F.; Frosch, M.; Musolino, P.; Tanzi, R.; Kim, D.Y.; Biffi, A.; El Khoury, J.; Eichler, F. Microglial dysfunction as a key pathological change in adrenomyeloneuropathy. Ann. Neurol. 2017, 82, 813–827. [Google Scholar] [CrossRef] [Green Version]
- Aubourg, P.; Blanche, S.; Jambaque, I.; Rocchiccioli, F.; Kalifa, G.; Naud-Saudreau, C.; Rolland, M.O.; Debre, M.; Chaussain, J.L.; Griscelli, C.; et al. Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation. N. Engl. J. Med. 1990, 322, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Casasnovas, C.; Ruiz, M.; Schlüter, A.; Naudí, A.; Fourcade, S.; Veciana, M.; Castañer, S.; Albertí, A.; Bargalló, N.; Johnson, M.; et al. Biomarker Identification, Safety, and Efficacy of High-Dose Antioxidants for Adrenomyeloneuropathy: A Phase II Pilot Study. Neurotherapeutics 2019, 16, 1167–1182. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Pascau, L.; Britti, E.; Calap-Quintana, P.; Dong, Y.N.; Vergara, C.; Delaspre, F.; Medina-Carbonero, M.; Tamarit, J.; Pallardó, F.V.; Gonzalez-Cabo, P.; et al. PPAR gamma agonist leriglitazone improves frataxin-loss impairments in cellular and animal models of Friedreich Ataxia. Neurobiol. Dis. 2021, 148, 105162. [Google Scholar] [CrossRef]
- Paton, B.C.; Heron, S.E.; Nelson, P.V.; Morris, C.P.; Poulos, A. Absence of mutations raises doubts about the role of the 70-kD peroxisomal membrane protein in Zellweger syndrome. Am. J. Hum. Genet. 1997, 60, 1535–1539. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.A. Peroxisome Metabolism in Cancer. Cells 2020, 9, 1692. [Google Scholar] [CrossRef] [PubMed]
- Hlaváč, V.; Souček, P. Role of family D ATP-binding cassette transporters (ABCD) in cancer. Biochem. Soc. Trans. 2015, 43, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Heimerl, S.; Bosserhoff, A.K.; Langmann, T.; Ecker, J.; Schmitz, G. Mapping ATP-binding cassette transporter gene expression profiles in melanocytes and melanoma cells. Melanoma Res. 2007, 17, 265–273. [Google Scholar] [CrossRef]
- Hour, T.C.; Kuo, Y.Z.; Liu, G.Y.; Kang, W.Y.; Huang, C.Y.; Tsai, Y.C.; Wu, W.J.; Huang, S.P.; Pu, Y.S. Downregulation of ABCD1 in human renal cell carcinoma. Int. J. Biol. Markers 2009, 24, 171–178. [Google Scholar] [CrossRef]
- Soucek, P.; Hlavac, V.; Elsnerova, K.; Vaclavikova, R.; Kozevnikovova, R.; Raus, K. Whole exome sequencing analysis of ABCC8 and ABCD2 genes associating with clinical course of breast carcinoma. Physiol. Res. 2015, 64, S549–S557. [Google Scholar] [CrossRef]
- Elsnerova, K.; Bartakova, A.; Tihlarik, J.; Bouda, J.; Rob, L.; Skapa, P.; Hruda, M.; Gut, I.; Mohelnikova-Duchonova, B.; Soucek, P.; et al. Gene Expression Profiling Reveals Novel Candidate Markers of Ovarian Carcinoma Intraperitoneal Metastasis. J. Cancer 2017, 8, 3598–3606. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, Y.; Wang, J.; Yang, J.; Yang, G. Abnormal expression of ABCD3 is an independent prognostic factor for colorectal cancer. Oncol. Lett. 2020, 19, 3567–3577. [Google Scholar] [CrossRef]
- Braicu, E.I.; Darb-Esfahani, S.; Schmitt, W.D.; Koistinen, K.M.; Heiskanen, L.; Poho, P.; Budczies, J.; Kuhberg, M.; Dietel, M.; Frezza, C.; et al. High-grade ovarian serous carcinoma patients exhibit profound alterations in lipid metabolism. Oncotarget 2017, 8, 102912–102922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hlaváč, V.; Brynychová, V.; Václavíková, R.; Ehrlichová, M.; Vrána, D.; Pecha, V.; Koževnikovová, R.; Trnková, M.; Gatěk, J.; Kopperová, D.; et al. The expression profile of ATP-binding cassette transporter genes in breast carcinoma. Pharmacogenomics 2013, 14, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, E.; Galzio, R.; Laurenti, G.; D’Angelo, B.; Melchiorre, E.; Cifone, M.G.; Fanelli, F.; Muzi, P.; Coletti, G.; Alecci, M.; et al. Lipid metabolism impairment in human gliomas: Expression of peroxisomal proteins in human gliomas at different grades of malignancy. Int. J. Immunopathol. Pharmacol. 2010, 23, 235–246. [Google Scholar] [CrossRef]
- Hama, K.; Fujiwara, Y.; Hayama, T.; Ozawa, T.; Nozawa, K.; Matsuda, K.; Hashiguchi, Y.; Yokoyama, K. Very long-chain fatty acids are accumulated in triacylglycerol and nonesterified forms in colorectal cancer tissues. Sci. Rep. 2021, 11, 6163. [Google Scholar] [CrossRef]
- Shani, N.; Watkins, P.A.; Valle, D. PXA1, a possible Saccharomyces cerevisiae ortholog of the human adrenoleukodystrophy gene. Proc. Natl. Acad. Sci. USA 1995, 92, 6012–6016. [Google Scholar] [CrossRef] [Green Version]
- Hettema, E.H.; vanRoermund, C.W.T.; Distel, B.; vandenBerg, M.; Vilela, C.; RodriguesPousada, C.; Wanders, R.J.A.; Tabak, H.F. The ABC transporter proteins Pat1 and Pat2 are required for import of long-chain fatty acids into peroxisomes of Saccharomyces cerevisiae. EMBO J. 1996, 15, 3813–3822. [Google Scholar] [CrossRef]
- Verleur, N.; Hettema, E.H.; van Roermund, C.W.; Tabak, H.F.; Wanders, R.J. Transport of activated fatty acids by the peroxisomal ATP-binding-cassette transporter Pxa2 in a semi-intact yeast cell system. Eur. J. Biochem. 1997, 249, 657–661. [Google Scholar] [CrossRef]
- Theodoulou, F.L.; Job, K.; Slocombe, S.P.; Footitt, S.; Holdsworth, M.; Baker, A.; Larson, T.R.; Graham, I.A. Jasmonic acid levels are reduced in COMATOSE ATP-binding cassette transporter mutants. Implications for transport of jasmonate precursors into peroxisomes. Plant. Physiol. 2005, 137, 835–840. [Google Scholar] [CrossRef] [Green Version]
- Kunz, H.H.; Scharnewski, M.; Feussner, K.; Feussner, I.; Flugge, U.I.; Fulda, M.; Gierth, M. The ABC transporter PXA1 and peroxisomal beta-oxidation are vital for metabolism in mature leaves of Arabidopsis during extended darkness. Plant Cell 2009, 21, 2733–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Footitt, S.; Dietrich, D.; Fait, A.; Fernie, A.R.; Holdsworth, M.J.; Baker, A.; Theodoulou, F.L. The COMATOSE ATP-binding cassette transporter is required for full fertility in Arabidopsis. Plant. Physiol. 2007, 144, 1467–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; De Marcos Lousa, C.; Schutte-Lensink, N.; Ofman, R.; Wanders, R.J.; Baldwin, S.A.; Baker, A.; Kemp, S.; Theodoulou, F.L. Conservation of targeting but divergence in function and quality control of peroxisomal ABC transporters: An analysis using cross-kingdom expression. Biochem. J. 2011, 436, 547–557. [Google Scholar] [CrossRef]
- Oikonomou, G.; Shaham, S. The glia of Caenorhabditis elegans. Glia 2011, 59, 1253–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppa, A.; Guha, S.; Fourcade, S.; Parameswaran, J.; Ruiz, M.; Moser, A.B.; Schlüter, A.; Murphy, M.P.; Lizcano, J.M.; Miranda-Vizuete, A.; et al. The peroxisomal fatty acid transporter ABCD1/PMP-4 is required in the C. elegans hypodermis for axonal maintenance: A worm model for adrenoleukodystrophy. Free Radic. Biol. Med. 2020, 152, 797–809. [Google Scholar] [CrossRef]
- Gordon, H.B.; Valdez, L.; Letsou, A. Etiology and treatment of adrenoleukodystrophy: New insights from Drosophila. Dis. Model Mech. 2018, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Sivachenko, A.; Gordon, H.B.; Kimball, S.S.; Gavin, E.J.; Bonkowsky, J.L.; Letsou, A. Neurodegeneration in a Drosophila model of adrenoleukodystrophy: The roles of the Bubblegum and Double bubble acyl-CoA synthetases. Dis. Models Mech. 2016, 9, 377–387. [Google Scholar]
- Strachan, L.R.; Stevenson, T.J.; Freshner, B.; Keefe, M.D.; Miranda Bowles, D.; Bonkowsky, J.L. A zebrafish model of X-linked adrenoleukodystrophy recapitulates key disease features and demonstrates a developmental requirement for abcd1 in oligodendrocyte patterning and myelination. Hum. Mol. Genet. 2017, 26, 3600–3614. [Google Scholar] [CrossRef] [PubMed]
- Raas, Q.; van de Beek, M.C.; Forss-Petter, S.; Dijkstra, I.M.; DeSchiffart, A.; Freshner, B.C.; Stevenson, T.J.; Jaspers, Y.R.; Nagtzaam, L.M.; Wanders, R.J.; et al. Metabolic rerouting via SCD1 induction impacts X-linked adrenoleukodystrophy. J. Clin. Invest. 2021. [Google Scholar] [CrossRef]
- Gueugnon, F.; Volodina, N.; Taouil, J.; Lopez, T.; Gondcaille, C.; Sequeira-Le Grand, A.; Mooijer, P.; Kemp, S.; Wanders, R.; Savary, S. A novel cell model to study the function of the adrenoleukodystrophy-related protein. Biochem. Biophys. Res. Commun. 2006, 341, 150–157. [Google Scholar] [CrossRef]
- Singh, J.; Khan, M.; Singh, I. Silencing of Abcd1 and Abcd2 genes sensitizes astrocytes for inflammation: Implication for X-adrenoleukodystrophy. J. Lipid Res. 2009, 50, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Kruska, N.; Schonfeld, P.; Pujol, A.; Reiser, G. Astrocytes and mitochondria from adrenoleukodystrophy protein (ABCD1)-deficient mice reveal that the adrenoleukodystrophy-associated very long-chain fatty acids target several cellular energy-dependent functions. Biochim. Biophys. Acta 2015, 1852, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Toida, A.; Horiuchi, Y.; Watanabe, S.; Sasahara, M.; Kawaguchi, K.; So, T.; Imanaka, T. Generation of an immortalized astrocytic cell line from Abcd1-deficient H-2K(b)tsA58 mice to facilitate the study of the role of astrocytes in X-linked adrenoleukodystrophy. Heliyon 2021, 7, e06228. [Google Scholar] [CrossRef]
- Raas, Q.; Gondcaille, C.; Hamon, Y.; Leoni, V.; Caccia, C.; Ménétrier, F.; Lizard, G.; Trompier, D.; Savary, S. CRISPR/Cas9-mediated knockout of Abcd1 and Abcd2 genes in BV-2 cells: Novel microglial models for X-linked Adrenoleukodystrophy. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2019, 1864, 704–714. [Google Scholar] [CrossRef]
- Schaumburg, H.H.; Powers, J.M.; Suzuki, K.; Raine, C.S. Adreno-leukodystrophy (sex-linked Schilder disease). Ultrastructural demonstration of specific cytoplasmic inclusions in the central nervous system. Arch. Neurol. 1974, 31, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Kapfhammer, J.P.; Hindelang, C.; Kemp, S.; Troffer-Charlier, N.; Broccoli, V.; Callyzot, N.; Mooyer, P.; Selhorst, J.; Vreken, P.; et al. Inactivation of the peroxisomal ABCD2 transporter in the mouse leads to late-onset ataxia involving mitochondria, Golgi and endoplasmic reticulum damage. Hum. Mol. Genet. 2005, 14, 3565–3577. [Google Scholar] [CrossRef] [Green Version]
- Pujol, A.; Hindelang, C.; Callizot, N.; Bartsch, U.; Schachner, M.; Mandel, J.L. Late onset neurological phenotype of the X-ALD gene inactivation in mice: A mouse model for adrenomyeloneuropathy. Hum. Mol. Genet. 2002, 11, 499–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourcade, S.; Lopez-Erauskin, J.; Galino, J.; Duval, C.; Naudi, A.; Jove, M.; Kemp, S.; Villarroya, F.; Ferrer, I.; Pamplona, R.; et al. Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum. Mol. Genet. 2008, 17, 1762–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuinness, M.C.; Lu, J.F.; Zhang, H.P.; Dong, G.X.; Heinzer, A.K.; Watkins, P.A.; Powers, J.; Smith, K.D. Role of ALDP (ABCD1) and mitochondria in X-linked adrenoleukodystrophy. Mol. Cell. Biol. 2003, 23, 744–753. [Google Scholar] [CrossRef] [Green Version]
- Galino, J.; Ruiz, M.; Fourcade, S.; Schluter, A.; Lopez-Erauskin, J.; Guilera, C.; Jove, M.; Naudi, A.; Garcia-Arumi, E.; Andreu, A.L.; et al. Oxidative damage compromises energy metabolism in the axonal degeneration mouse model of X-adrenoleukodystrophy. Antioxid. Redox Signal. 2011, 15, 2095–2107. [Google Scholar] [CrossRef]
- Lopez-Erauskin, J.; Galino, J.; Ruiz, M.; Cuezva, J.M.; Fabregat, I.; Cacabelos, D.; Boada, J.; Martinez, J.; Ferrer, I.; Pamplona, R.; et al. Impaired mitochondrial oxidative phosphorylation in the peroxisomal disease X-linked adrenoleukodystrophy. Hum. Mol. Genet. 2013, 22, 3296–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Launay, N.; Aguado, C.; Fourcade, S.; Ruiz, M.; Grau, L.; Riera, J.; Guilera, C.; Giros, M.; Ferrer, I.; Knecht, E.; et al. Autophagy induction halts axonal degeneration in a mouse model of X-adrenoleukodystrophy. Acta Neuropathol. 2015, 129, 399–415. [Google Scholar] [CrossRef] [Green Version]
- Morató, L.; Galino, J.; Ruiz, M.; Calingasan, N.Y.; Starkov, A.A.; Dumont, M.; Naudí, A.; Martínez, J.J.; Aubourg, P.; Portero-Otín, M.; et al. Pioglitazone halts axonal degeneration in a mouse model of X-linked adrenoleukodystrophy. Brain 2013, 136, 2432–2443. [Google Scholar] [CrossRef] [Green Version]
- Launay, N.; Ruiz, M.; Grau, L.; Ortega, F.J.; Ilieva, E.V.; Martinez, J.J.; Galea, E.; Ferrer, I.; Knecht, E.; Pujol, A.; et al. Tauroursodeoxycholic bile acid arrests axonal degeneration by inhibiting the unfolded protein response in X-linked adrenoleukodystrophy. Acta Neuropathol. 2017, 133, 283–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourcade, S.; Goicoechea, L.; Parameswaran, J.; Schlüter, A.; Launay, N.; Ruiz, M.; Seyer, A.; Colsch, B.; Calingasan, N.Y.; Ferrer, I.; et al. High-dose biotin restores redox balance, energy and lipid homeostasis, and axonal health in a model of adrenoleukodystrophy. Brain Pathol. 2020, 30, 945–963. [Google Scholar] [CrossRef] [PubMed]
- Olah, M.; Patrick, E.; Villani, A.C.; Xu, J.; White, C.C.; Ryan, K.J.; Piehowski, P.; Kapasi, A.; Nejad, P.; Cimpean, M.; et al. A transcriptomic atlas of aged human microglia. Nat. Commun. 2018, 9, 539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.F.; Barron-Casella, E.; Deering, R.; Heinzer, A.K.; Moser, A.B.; de Mesy Bentley, K.L.; Wand, G.S.; McGuinness, M.C.; Pei, Z.; Watkins, P.A.; et al. The role of peroxisomal ABC transporters in the mouse adrenal gland: The loss of Abcd2 (ALDR), Not Abcd1 (ALD), causes oxidative damage. Lab. Investig. 2007, 87, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, H.W.; Moser, A.B.; Kawamura, N.; Murphy, J.; Suzuki, K.; Schaumburg, H.; Kishimoto, Y. Adrenoleukodystrophy: Elevated C26 fatty acid in cultured skin fibroblasts. Ann. Neurol. 1980, 7, 542–549. [Google Scholar] [CrossRef]
- Griffin, D.E.; Moser, H.W.; Mendoza, Q.; Moench, T.R.; O’Toole, S.; Moser, A.B. Identification of the inflammatory cells in the central nervous system of patients with adrenoleukodystrophy. Ann. Neurol. 1985, 18, 660–664. [Google Scholar] [CrossRef]
- Lannuzel, A.; Aubourg, P.; Tardieu, M. Excessive production of tumour necrosis factor alpha by peripheral blood mononuclear cells in X-linked adrenoleukodystrophy. Eur. J. Paediatr. Neurol. 1998, 2, 27–32. [Google Scholar] [CrossRef]
- Weinhofer, I.; Zierfuss, B.; Hametner, S.; Wagner, M.; Popitsch, N.; Machacek, C.; Bartolini, B.; Zlabinger, G.; Ohradanova-Repic, A.; Stockinger, H.; et al. Impaired plasticity of macrophages in X-linked adrenoleukodystrophy. Brain 2018, 141, 2329–2342. [Google Scholar] [CrossRef]
- Weber, F.D.; Wiesinger, C.; Forss-Petter, S.; Regelsberger, G.; Einwich, A.; Weber, W.H.; Kohler, W.; Stockinger, H.; Berger, J. X-linked adrenoleukodystrophy: Very long-chain fatty acid metabolism is severely impaired in monocytes but not in lymphocytes. Hum. Mol. Genet. 2014, 23, 2542–2550. [Google Scholar] [CrossRef]
- Wang, X.M.; Yik, W.Y.; Zhang, P.; Lu, W.; Dranchak, P.K.; Shibata, D.; Steinberg, S.J.; Hacia, J.G. The gene expression profiles of induced pluripotent stem cells from individuals with childhood cerebral adrenoleukodystrophy are consistent with proposed mechanisms of pathogenesis. Stem. Cell. Res. Ther. 2012, 3, 39. [Google Scholar] [CrossRef] [Green Version]
- Baarine, M.; Khan, M.; Singh, A.; Singh, I. Functional Characterization of IPSC-Derived Brain Cells as a Model for X-Linked Adrenoleukodystrophy. PLoS ONE 2015, 10, e0143238. [Google Scholar] [CrossRef] [PubMed]
- Son, D.; Quan, Z.; Kang, P.J.; Park, G.; Kang, H.C.; You, S. Generation of two induced pluripotent stem cell (iPSC) lines from X-linked adrenoleukodystrophy (X-ALD) patients with adrenomyeloneuropathy (AMN). Stem Cell Res. 2017, 25, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.; Kang, H.C.; Kim, H.S.; Kim, J.Y.; Huh, Y.J.; Kim, D.S.; Yoo, J.E.; Lee, J.A.; Lim, B.; Lee, J.; et al. Induced pluripotent stem cell models from X-linked adrenoleukodystrophy patients. Ann. Neurol. 2011, 70, 402–409. [Google Scholar] [CrossRef]
- Jang, J.; Park, S.; Jin Hur, H.; Cho, H.J.; Hwang, I.; Pyo Kang, Y.; Im, I.; Lee, H.; Lee, E.; Yang, W.; et al. 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat. Commun. 2016, 7, 13129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.A.A.; Seo, H.S.; Armien, A.G.; Bates, F.S.; Tolar, J.; Azarin, S.M. Modeling and rescue of defective blood-brain barrier function of induced brain microvascular endothelial cells from childhood cerebral adrenoleukodystrophy patients. Fluids Barriers CNS 2018, 15, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloeckner, C.J.; Mayerhofer, P.U.; Landgraf, P.; Muntau, A.C.; Holzinger, A.; Gerber, J.K.; Kammerer, S.; Adamski, J.; Roscher, A.A. Human adrenoleukodystrophy protein and related peroxisomal ABC transporters interact with the peroxisomal assembly protein PEX19p. Biochem. Biophys. Res. Commun. 2000, 271, 144–150. [Google Scholar] [CrossRef]
- Shibata, H.; Kashiwayama, Y.; Imanaka, T.; Kato, H. Domain architecture and activity of human Pex19p, a chaperone-like protein for intracellular trafficking of peroxisomal membrane proteins. J. Biol. Chem. 2004, 279, 38486–38494. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Liu, J.; Lester, J.D.; Pijut, S.S.; Graf, G.A. ABCD2 identifies a subclass of peroxisomes in mouse adipose tissue. Biochem. Biophys. Res. Commun. 2015, 456, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.K.; Hettema, E.H. Multiple pathways for protein transport to peroxisomes. J. Mol. Biol. 2015, 427, 1176–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillebrand, M.; Gersting, S.W.; Lotz-Havla, A.S.; Schafer, A.; Rosewich, H.; Valerius, O.; Muntau, A.C.; Gartner, J. Identification of a new fatty acid synthesis-transport machinery at the peroxisomal membrane. J. Biol. Chem. 2012, 287, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Makkar, R.S.; Contreras, M.A.; Paintlia, A.S.; Smith, B.T.; Haq, E.; Singh, I. Molecular organization of peroxisomal enzymes: Protein-protein interactions in the membrane and in the matrix. Arch. Biochem. Biophys. 2006, 451, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Fecik, R.A.; Patterson, J.E.; Miyauchi, H.; Patricelli, M.P.; Cravatt, B.F. Fatty acid amide hydrolase substrate specificity. Bioorg. Med. Chem. Lett. 2000, 10, 2613–2616. [Google Scholar] [CrossRef]
- Wei, B.Q.; Mikkelsen, T.S.; McKinney, M.K.; Lander, E.S.; Cravatt, B.F. A second fatty acid amide hydrolase with variable distribution among placental mammals. J. Biol. Chem. 2006, 281, 36569–36578. [Google Scholar] [CrossRef] [Green Version]
- Brown, I.; Cascio, M.G.; Wahle, K.W.; Smoum, R.; Mechoulam, R.; Ross, R.A.; Pertwee, R.G.; Heys, S.D. Cannabinoid receptor-dependent and -independent anti-proliferative effects of omega-3 ethanolamides in androgen receptor-positive and -negative prostate cancer cell lines. Carcinogenesis 2010, 31, 1584–1591. [Google Scholar] [CrossRef] [Green Version]
- Havugimana, P.C.; Hart, G.T.; Nepusz, T.; Yang, H.; Turinsky, A.L.; Li, Z.; Wang, P.I.; Boutz, D.R.; Fong, V.; Phanse, S.; et al. A census of human soluble protein complexes. Cell 2012, 150, 1068–1081. [Google Scholar] [CrossRef] [Green Version]
- Young, P.A.; Senkal, C.E.; Suchanek, A.L.; Grevengoed, T.J.; Lin, D.D.; Zhao, L.; Crunk, A.E.; Klett, E.L.; Füllekrug, J.; Obeid, L.M.; et al. Long-chain acyl-CoA synthetase 1 interacts with key proteins that activate and direct fatty acids into niche hepatic pathways. J. Biol. Chem. 2018, 293, 16724–16740. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandusse, S.; Falkenberg, K.D.; Koster, J.; Mooyer, P.A.; Jones, R.; van Roermund, C.W.T.; Pizzino, A.; Schrader, M.; Wanders, R.J.A.; Vanderver, A.; et al. ACBD5 deficiency causes a defect in peroxisomal very long-chain fatty acid metabolism. J. Med. Genet. 2017, 54, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Ewing, R.M.; Chu, P.; Elisma, F.; Li, H.; Taylor, P.; Climie, S.; McBroom-Cerajewski, L.; Robinson, M.D.; O’Connor, L.; Li, M.; et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol. Syst. Biol. 2007, 3, 89. [Google Scholar] [CrossRef]
- Chang, C.L.; Weigel, A.V.; Ioannou, M.S.; Pasolli, H.A.; Xu, C.S.; Peale, D.R.; Shtengel, G.; Freeman, M.; Hess, H.F.; Blackstone, C.; et al. Spastin tethers lipid droplets to peroxisomes and directs fatty acid trafficking through ESCRT-III. J. Cell Biol. 2019, 218, 2583–2599. [Google Scholar] [CrossRef]
- Cuevas-Fernández, B.; Fuentes-Almagro, C.; Peragón, J. Proteomics Analysis Reveals the Implications of Cytoskeleton and Mitochondria in the Response of the Rat Brain to Starvation. Nutrients 2019, 11, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, M.; Grille, S.; Fahimi, H.D.; Islinger, M. Peroxisome interactions and cross-talk with other subcellular compartments in animal cells. Subcell. Biochem. 2013, 69, 1–22. [Google Scholar]
- Chen, C.; Li, J.; Qin, X.; Wang, W. Peroxisomal Membrane Contact Sites in Mammalian Cells. Front. Cell Dev. Biol. 2020, 8, 512. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [Green Version]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deakin, S.; Leviev, I.; Gomaraschi, M.; Calabresi, L.; Franceschini, G.; James, R.W. Enzymatically active paraoxonase-1 is located at the external membrane of producing cells and released by a high affinity, saturable, desorption mechanism. J. Biol. Chem. 2002, 277, 4301–4308. [Google Scholar] [CrossRef] [Green Version]
- Gonzalvo, M.C.; Gil, F.; Hernandez, A.F.; Rodrigo, L.; Villanueva, E.; Pla, A. Human liver paraoxonase (PON1): Subcellular distribution and characterization. J. Biochem. Mol. Toxicol. 1998, 12, 61–69. [Google Scholar] [CrossRef]
- Reichert, C.O.; Levy, D.; Bydlowski, S.P. Paraoxonase Role in Human Neurodegenerative Diseases. Antioxidants 2020, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.S.C.; DiGiovanni, L.; Kumar, R.; Carmichael, R.E.; Kim, P.K.; Schrader, M. Maintaining social contacts: The physiological relevance of organelle interactions. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118800. [Google Scholar] [CrossRef] [PubMed]
- Islinger, M.; Voelkl, A.; Fahimi, H.D.; Schrader, M. The peroxisome: An update on mysteries 2.0. Histochem. Cell Biol. 2018, 150, 443–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, A.; Dean, J.M.; Lodhi, I.J. Peroxisomes as Cellular Adaptors to Metabolic and Environmental Stress. Trends Cell Biol. 2021. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Protein Accession | Protein Name | Protein Probability | Fold Change a | |
|---|---|---|---|---|
| Q9QY44 | ABCD2 | ATP-binding cassette sub-family D member 2 | 1 | 12.42 |
| P97612 | FAAH1 | Fatty-acid amide hydrolase 1 | 1 | 5.02 |
| P11507 | AT2A2 | Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 | 1 | 4.71 |
| P07340 | AT1B1 | Sodium/potassium-transporting ATPase subunit beta | 1 | 2.48 |
| P55159 | PON1 | Serum paraoxonase/arylesterase 1 | 1 | 2.32 |
| D3ZHR2 | ABCD1 | ATP-binding cassette sub-family D member 1 | 1 | <2 |
| P16970 | ABCD3 | ATP-binding cassette sub-family D member 3 | 1 | <2 |
| Q7TS56 | CBR4 | Carbonyl reductase family member 4 | 1 | <2 |
| P11505 | AT2B1 | Plasma membrane calcium-transporting ATPase 1 | 1 | <2 |
| P16086 | SPTN1 | Spectrin alpha chain, non-erythrocytic 1 | 1 | <2 |
| Q63151 | ACSL3 | Long-chain acyl-CoA synthetase 3 | 0.9997 | <2 |
| O88813 | ACSL5 | Long-chain acyl-CoA synthetase 5 | 0.9994 | <2 |
| P14408 | FUMH | Fumarate hydratase, mitochondrial | 0.8013 | <2 |
| P25235 | RPN2 | Dolichyl-diphosphooligosaccharide—protein glycosyltransferase subunit 2 | 0.7224 | <2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tawbeh, A.; Gondcaille, C.; Trompier, D.; Savary, S. Peroxisomal ABC Transporters: An Update. Int. J. Mol. Sci. 2021, 22, 6093. https://doi.org/10.3390/ijms22116093
Tawbeh A, Gondcaille C, Trompier D, Savary S. Peroxisomal ABC Transporters: An Update. International Journal of Molecular Sciences. 2021; 22(11):6093. https://doi.org/10.3390/ijms22116093
Chicago/Turabian StyleTawbeh, Ali, Catherine Gondcaille, Doriane Trompier, and Stéphane Savary. 2021. "Peroxisomal ABC Transporters: An Update" International Journal of Molecular Sciences 22, no. 11: 6093. https://doi.org/10.3390/ijms22116093
APA StyleTawbeh, A., Gondcaille, C., Trompier, D., & Savary, S. (2021). Peroxisomal ABC Transporters: An Update. International Journal of Molecular Sciences, 22(11), 6093. https://doi.org/10.3390/ijms22116093