
Docking-Based 3D-QSAR Studies for 1,3,4-oxadiazol-2-one Derivatives as FAAH Inhibitors

 , , , and
, , , and 
Abstract
:
1. Introduction
2. Results and Discussion
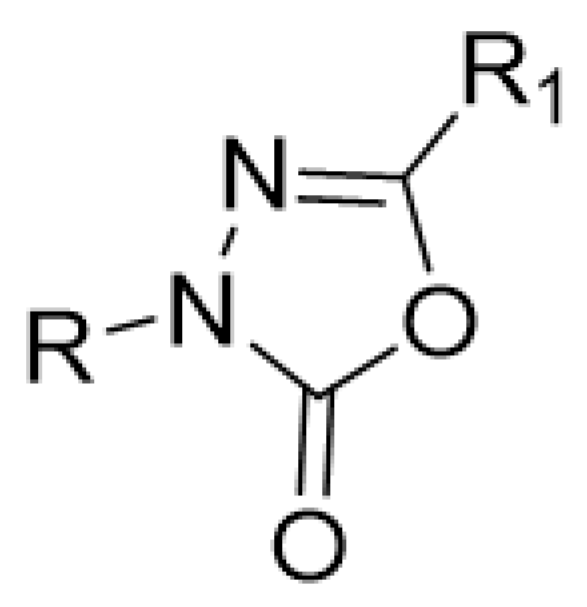
2.1. The Studied Compounds
2.2. Molecular Docking and Alignment
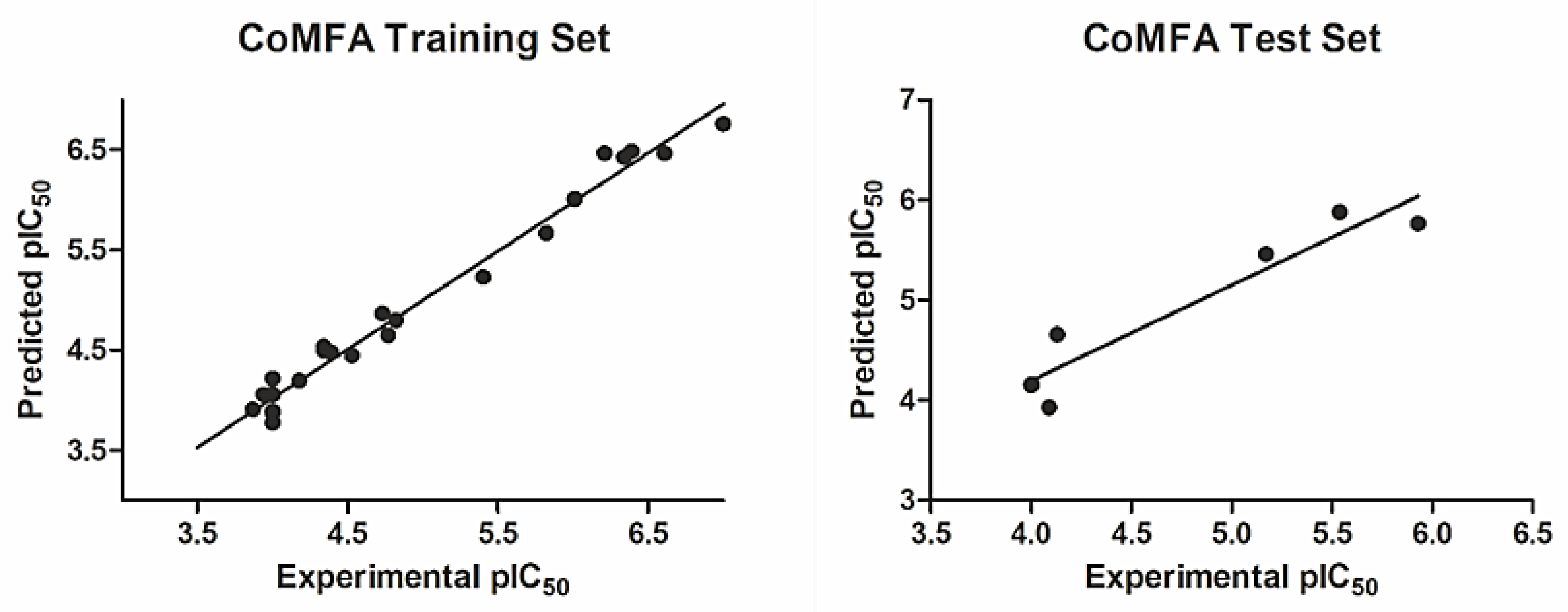
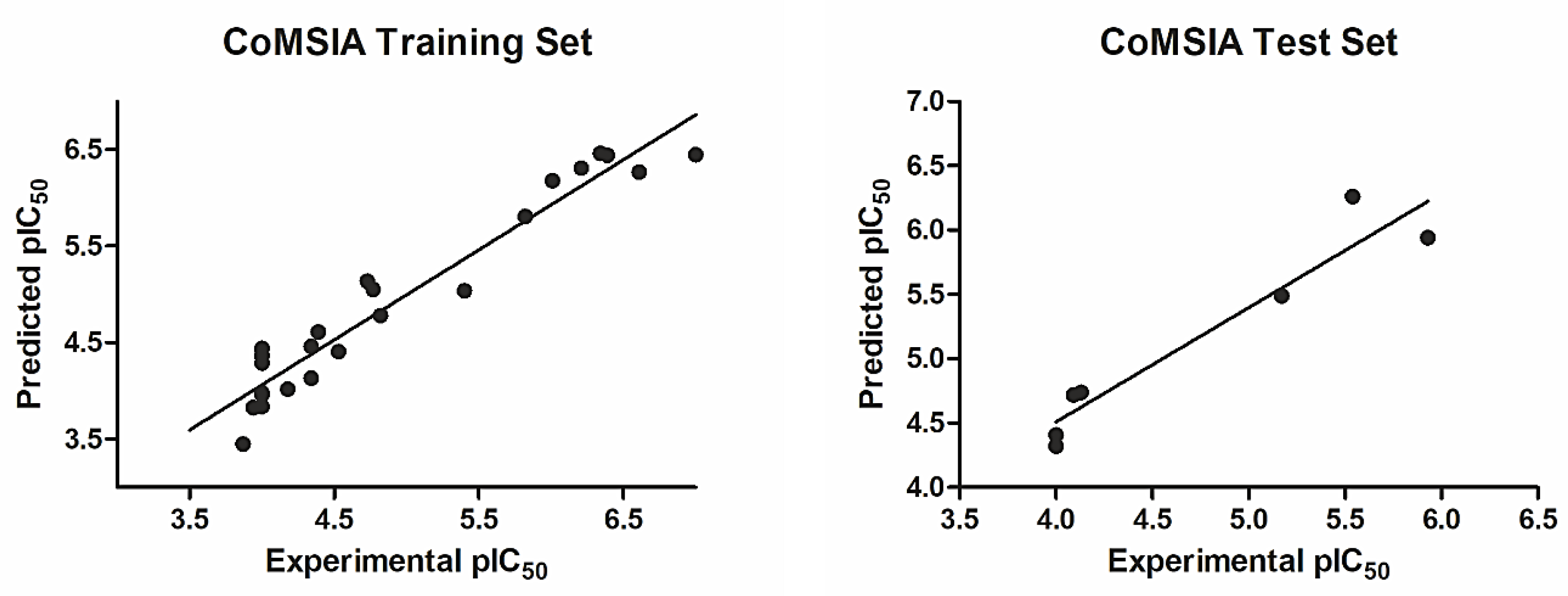
2.3. CoMFA Statistics and Validation
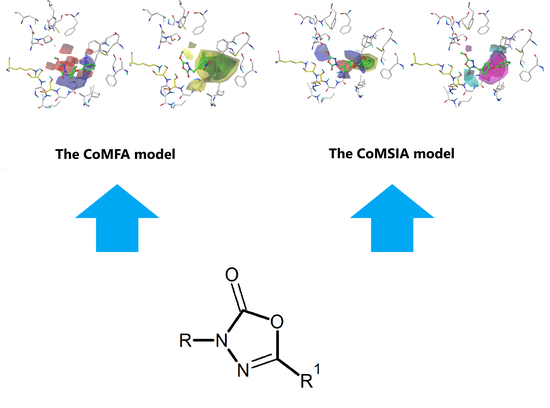
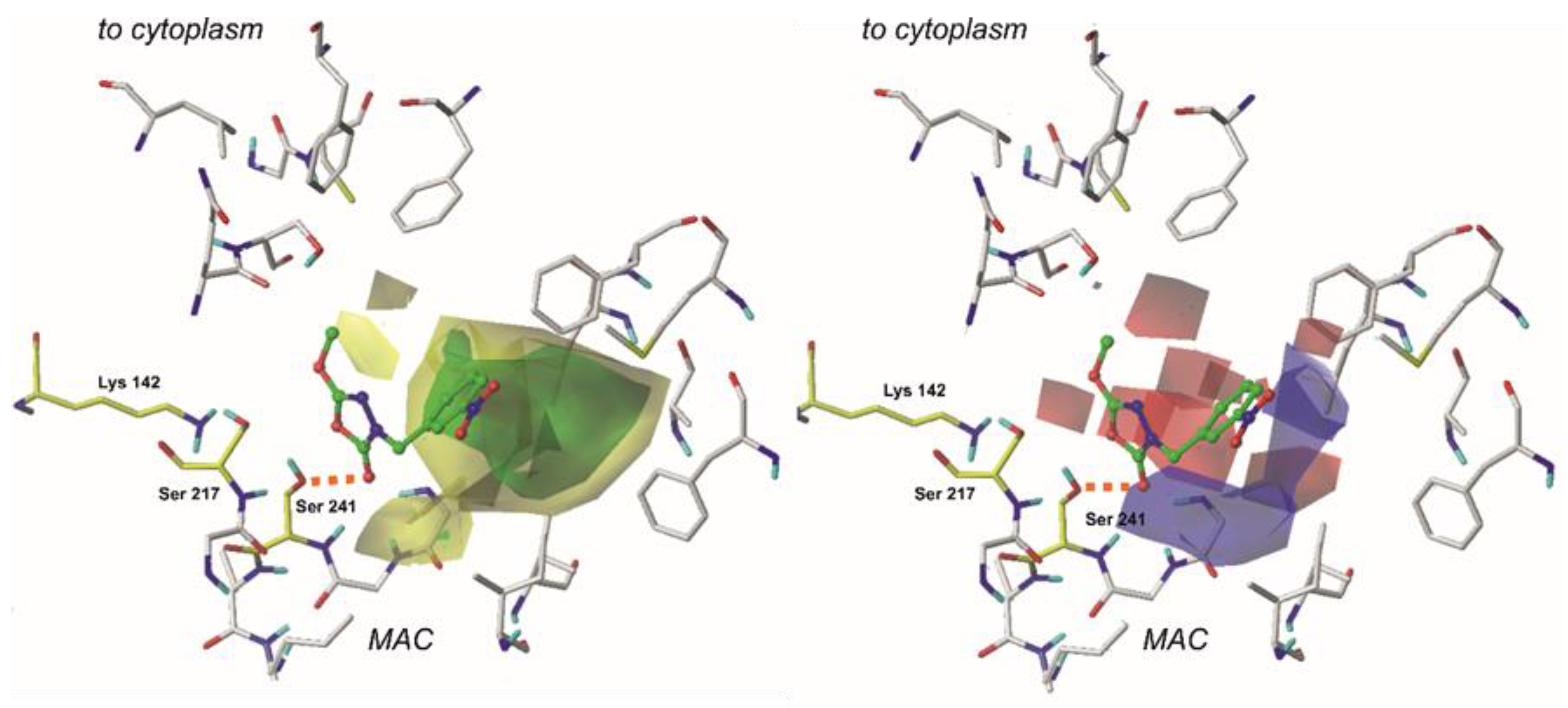
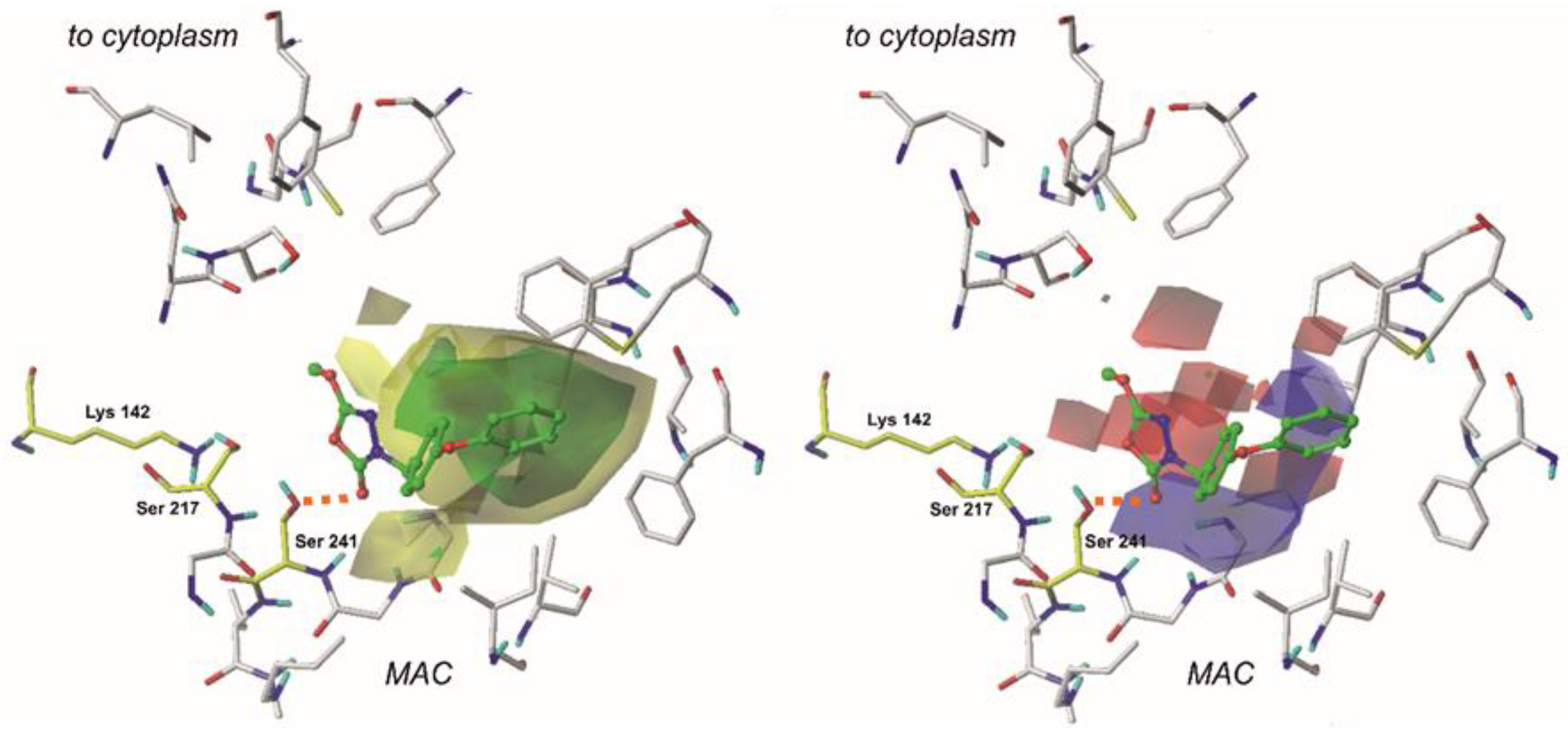
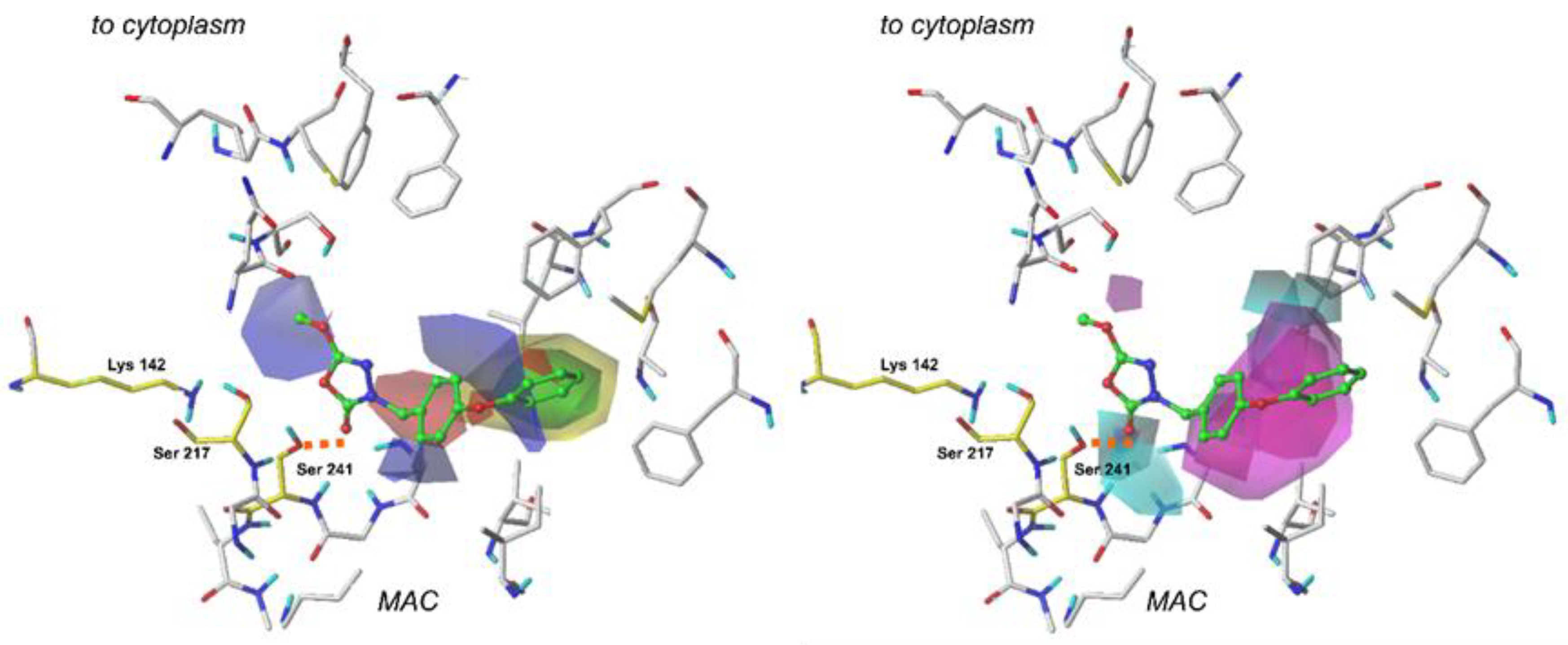
2.4. The CoMFA Contour Map and Its Mapping onto Receptor Structure
2.5. CoMSIA Model Statistics and Validation
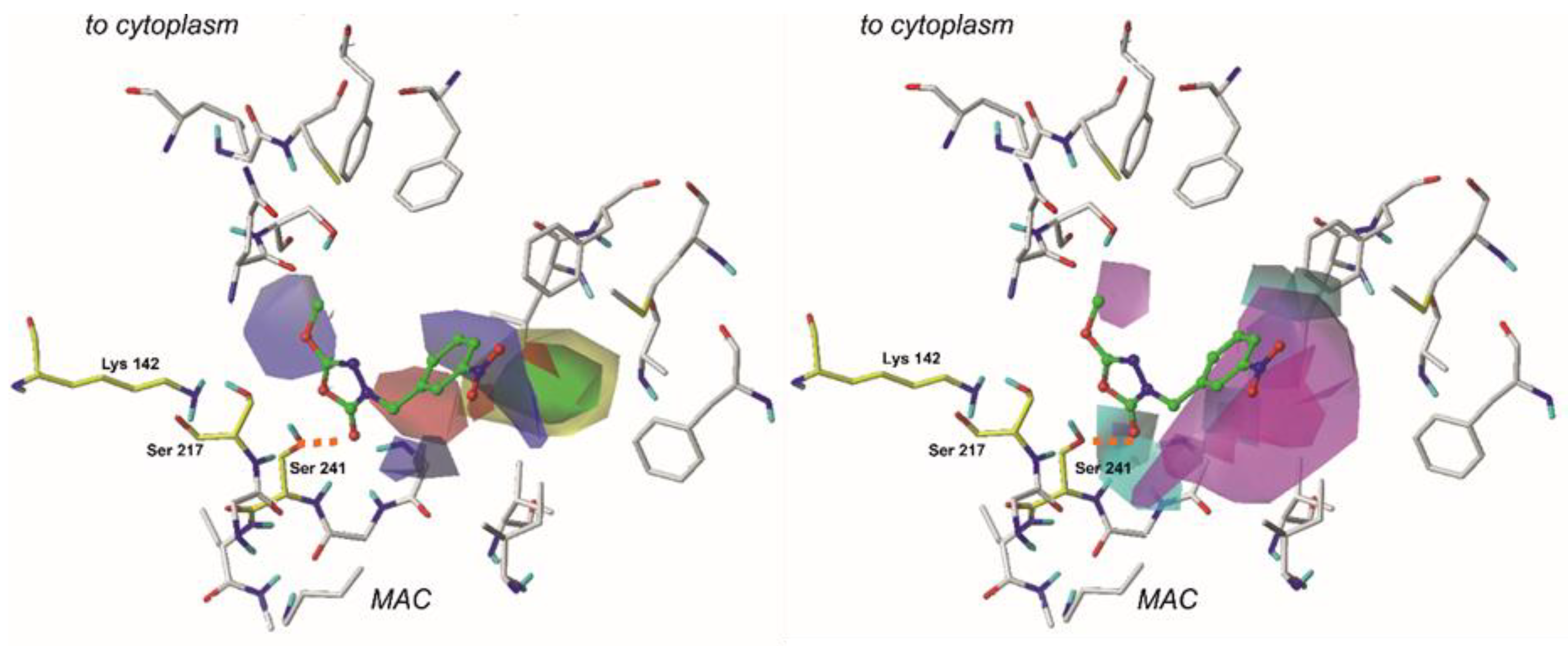
2.6. The CoMSIA Model Statistics and Validation
3. Materials and Methods
3.1. Selection and Preparation of Compounds
3.2. Molecular Docking and Alignment
3.3. CoMFA Studies
r2test-set > 0.6;
(r2 − r20)/r2 < 0.1;
0.85 ≤ k ≤ 1.15
3.4. CoMSIA Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CoMFA | Comparative Molecular Field Analysis |
| CoMSIA | Comparative Molecular Similarity Indices Analysis |
| FAAH | Fatty Acid Amide Hydrolase |
| MAC | Membrane Access Channel |
| PLS. | Partial Least Square |
| QSAR | Quantitative Structure–Activity Relationship |
| SEE | Standard error of estimate |
| SP | Standard Precision |
References
- Piomelli, D. Endocannabinoids. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 194–196. ISBN 978-0-12-378631-9. [Google Scholar]
- Tripathi, R.K.P. A Perspective Review on Fatty Acid Amide Hydrolase (FAAH) Inhibitors as Potential Therapeutic Agents. Eur. J. Med. Chem. 2020, 188, 111953. [Google Scholar] [CrossRef] [PubMed]
- Leweke, F.M.; Piomelli, D.; Pahlisch, F.; Muhl, D.; Gerth, C.W.; Hoyer, C.; Klosterkötter, J.; Hellmich, M.; Koethe, D. Cannabidiol Enhances Anandamide Signaling and Alleviates Psychotic Symptoms of Schizophrenia. Transl. Psychiatry 2012, 2, e94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaetani, S.; Dipasquale, P.; Romano, A.; Righetti, L.; Cassano, T.; Piomelli, D.; Cuomo, V. Chapter 5 The Endocannabinoid System as A Target for Novel Anxiolytic and Antidepressant Drugs. In International Review of Neurobiology; Academic Press: Amsterdam, The Netherlands, 2009; Volume 85, pp. 57–72. [Google Scholar]
- Mileni, M.; Garfunkle, J.; DeMartino, J.K.; Cravatt, B.F.; Boger, D.L.; Stevens, R.C. Binding and Inactivation Mechanism of a Humanized Fatty Acid Amide Hydrolase by α-Ketoheterocycle Inhibitors Revealed from Co-Crystal Structures. J. Am. Chem. Soc. 2009, 131, 10497. [Google Scholar] [CrossRef] [Green Version]
- Van Esbroeck, A.C.M.; Janssen, A.P.A.; Cognetta, A.B.; Ogasawara, D.; Shpak, G.; van der Kroeg, M.; Kantae, V.; Baggelaar, M.P.; de Vrij, F.M.S.; Deng, H.; et al. Activity-Based Protein Profiling Reveals off-Target Proteins of the FAAH Inhibitor BIA 10-2474. Science 2017, 356, 1084–1087. [Google Scholar] [CrossRef] [Green Version]
- Roy, K. Advances in QSAR Modeling: Applications in Pharmaceutical, Chemical, Food, Agricultural and Environmental Sciences; Springer: New York, NY, USA, 2017; ISBN 978-3-31-986018-3. [Google Scholar]
- Roy, K.; Kar, S.; Das, R.N. Chapter 8—Introduction to 3D-QSAR. In Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Roy, K., Kar, S., Das, R.N., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 291–317. ISBN 978-0-12-801505-6. [Google Scholar]
- Beliaev, A.; Ferreira, H.S.; Learmonth, D.A.; Bonifácio, M.J.; Torrão, L.; Pires, N.M.; Soares-da-Silva, P.; Kiss, L.E. Synthesis and Structure–Activity Relationships of Ionizable 1,3,4-Oxadiazol-2(3H)-Ones as Peripherally Selective FAAH Inhibitors with Improved Aqueous Solubility. Pure Appl. Chem. 2016, 88, 341–347. [Google Scholar] [CrossRef]
- Käsnänen, H.; Myllymäki, M.J.; Minkkilä, A.; Kataja, A.O.; Saario, S.M.; Nevalainen, T.; Koskinen, A.M.P.; Poso, A. 3-Heterocycle-Phenyl N-Alkylcarbamates as FAAH Inhibitors: Design, Synthesis and 3D-QSAR Studies. ChemMedChem 2010, 5, 213–231. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, M.; Huang, B.; Ji, H.; Yuan, M. Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) Studies on A1A-Adrenergic Receptor Antagonists Based on Pharmacophore Molecular Alignment. Int. J. Mol. Sci. 2011, 12, 7022–7037. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Wang, B.; Jin, H.; Wang, H.; Chen, M. Design, Synthesis and CoMFA Studies of OEA Derivatives as FAAH Inhibitors. Med. Chem. Res. 2017, 26, 2951–2966. [Google Scholar] [CrossRef]
- Lorca, M.; Valdes, Y.; Chung, H.; Romero-Parra, J.; Pessoa-Mahana, C.D.; Mella, J. Three-Dimensional Quantitative Structure-Activity Relationships (3D-QSAR) on a Series of Piperazine-Carboxamides Fatty Acid Amide Hydrolase (FAAH) Inhibitors as a Useful Tool for the Design of New Cannabinoid Ligands. Int. J. Mol. Sci. 2019, 20, 2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.Z.; Parkkari, T.; Laitinen, T.; Kaczor, A.A.; Saario, S.M.; Savinainen, J.R.; Navia-Paldanius, D.; Cipriano, M.; Leppänen, J.; Koshevoy, I.O.; et al. Chiral 1,3,4-Oxadiazol-2-Ones as Highly Selective FAAH Inhibitors. J. Med. Chem. 2013, 56, 8484–8496. [Google Scholar] [CrossRef]
- Patel, J.Z.; van Bruchem, J.; Laitinen, T.; Kaczor, A.A.; Navia-Paldanius, D.; Parkkari, T.; Savinainen, J.R.; Laitinen, J.T.; Nevalainen, T.J. Revisiting 1,3,4-Oxadiazol-2-Ones: Utilization in the Development of ABHD6 Inhibitors. Bioorg. Med. Chem. 2015, 23, 6335–6345. [Google Scholar] [CrossRef] [PubMed]
- Use of an Inhibitor to Identify Members of the Hormone-Sensitive Lipase Family. Biochemistry. Available online: https://pubs.acs.org/doi/abs/10.1021/bi0613978 (accessed on 9 March 2021).
- Muccioli, G.G.; Labar, G.; Lambert, D.M. CAY10499, a Novel Monoglyceride Lipase Inhibitor Evidenced by an Expeditious MGL Assay. ChemBioChem 2008, 9, 2704–2710. [Google Scholar] [CrossRef]
- Point, V.; Pavan Kumar, K.V.P.; Marc, S.; Delorme, V.; Parsiegla, G.; Amara, S.; Carrière, F.; Buono, G.; Fotiadu, F.; Canaan, S.; et al. Analysis of the Discriminative Inhibition of Mammalian Digestive Lipases by 3-Phenyl Substituted 1,3,4-Oxadiazol-2(3H)-Ones. Eur. J. Med. Chem. 2012, 58, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Delorme, V.; Diomandé, S.V.; Dedieu, L.; Cavalier, J.-F.; Carrière, F.; Kremer, L.; Leclaire, J.; Fotiadu, F.; Canaan, S. MmPPOX Inhibits Mycobacterium Tuberculosis Lipolytic Enzymes Belonging to the Hormone-Sensitive Lipase Family and Alters Mycobacterial Growth. PLoS ONE 2012, 7, e46493. [Google Scholar] [CrossRef] [PubMed]
- Käsnänen, H.; Minkkilä, A.; Taupila, S.; Patel, J.Z.; Parkkari, T.; Lahtela-Kakkonen, M.; Saario, S.M.; Nevalainen, T.; Poso, A. 1,3,4-Oxadiazol-2-Ones as Fatty-Acid Amide Hydrolase and Monoacylglycerol Lipase Inhibitors: Synthesis, in Vitro Evaluation and Insight into Potency and Selectivity Determinants by Molecular Modelling. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2013, 49, 423–433. [Google Scholar] [CrossRef]
- Sarma, B.K.; Liu, X.; Kodadek, T. Identification of Selective Covalent Inhibitors of Platelet Activating Factor Acetylhydrolase 1B2 from the Screening of an Oxadiazolone-Capped Peptoid–Azapeptoid Hybrid Library. Bioorg. Med. Chem. 2016, 24, 3953–3963. [Google Scholar] [CrossRef] [Green Version]
- Screening of Various Hormone-Sensitive Lipase Inhibitors as Endocannabinoid-Hydrolyzing Enzyme Inhibitors—Minkkilä—2009—ChemMedChem—Wiley Online Library. Available online: https://chemistry-europe.onlinelibrary.wiley.com/doi/full/10.1002/cmdc.200900137 (accessed on 9 March 2021).
- Bank, R.P.D. RCSB PDB—3QK5: Crystal Structure of Fatty Acid Amide Hydrolase with Small Molecule Inhibitor. Available online: https://www.rcsb.org/structure/3QK5 (accessed on 14 January 2021).
- Urniaż, R.D.; Jóźwiak, K. X-Ray Crystallographic Structures as a Source of Ligand Alignment in 3D-QSAR. J. Chem. Inf. Model. 2013, 53, 1406–1414. [Google Scholar] [CrossRef]
- Hevener, K.E.; Ball, D.M.; Buolamwini, J.K.; Lee, R.E. Quantitative Structure-Activity Relationship Studies on Nitrofuranyl Antitubercular Agents. Bioorg. Med. Chem. 2008, 16, 8042–8053. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.D.; Fox, P.C. Statistical Variation in Progressive Scrambling. J. Comput. Aided Mol. Des. 2004, 18, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Luco, J.M.; Ferretti, F.H. QSAR Based on Multiple Linear Regression and PLS Methods for the Anti-HIV Activity of a Large Group of HEPT Derivatives. J. Chem. Inf. Comput. Sci. 1997, 37, 392–401. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of Q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.-D.; Lee, K.-H.; Tropsha, A. Rational Selection of Training and Test Sets for the Development of Validated QSAR Models. J. Comput. Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Murumkar, P.R.; Gupta, S.D.; Zambre, V.P.; Giridhar, R.; Yadav, M.R. Development of Predictive 3D-QSAR CoMFA and CoMSIA Models for β-Aminohydroxamic Acid-Derived Tumor Necrosis Factor-α Converting Enzyme Inhibitors. Chem. Biol. Drug Des. 2009, 73, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, P.; Inupakutika, M.; Devireddy, A.R.; Gunda, S.K.; Willmon, T.M.; Ranjana, K.; Shrestha, U.; Ranaweera, I.; Hernandez, A.J.; Barr, S.; et al. 3D-QSAR and contour map analysis of tariquidar analogues as multidrug resistance protein-1 (MRP1) inhibitors. Int. J. Pharm. Sci. Res. 2016, 7, 554–572. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Suzuki, A.; Kohno, Y. High-Throughput Screening for the Assessment of Time-Dependent Inhibitions of New Drug Candidates on Recombinant CYP2D6 and CYP3A4 Using a Single Concentration Method. Xenobiotica 2004, 34, 87–101. [Google Scholar] [CrossRef]
- Kaczor, A.; Targowska-Duda, K.; Patel, J.; Laitinen, T.; Parkkari, T.; Adams, Y.; Nevalainen, T.; Poso, A. Comparative Molecular Field Analysis and Molecular Dynamics Studies of α/β Hydrolase Domain Containing 6 (ABHD6) Inhibitors. J. Mol. Model. 2015, 21, 2789. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Zhuang, J.; Hu, S.; Li, H.; Xu, J.; Hu, Y.; Xiong, X.; Chen, Y.; Lu, T. Molecular Modeling of Exquisitely Selective C-Met Inhibitors through 3D-QSAR and Molecular Dynamics Simulations. J. Chem. Inf. Model. 2014, 54, 2544–2554. [Google Scholar] [CrossRef]
- QSAR and CoMFA Manual, Basics Manual Sybyl ®-X 1.1; Tripos Inc.: St. Louis, MO, USA, 2010.
- Flower, D.R. Immunoinformatics: Predicting Immunogenicity In Silico; Springer Science & Business Media: Totowa, NJ, USA, 2007; ISBN 978-1-58829-699-3. [Google Scholar]
- Tropsha, A.; Cho, S.J. Cross-Validated R2 Guided Region Selection for CoMFA Studies. In 3D QSAR in Drug Design: Recent Advances; Kubinyi, H., Folkers, G., Martin, Y.C., Eds.; Three-Dimensional Quantitative Structure Activity Relationships; Springer: Dordrecht, The Netherlands, 1998; pp. 57–69. ISBN 978-0-306-46858-2. [Google Scholar]
- Zheng, J.; Xiao, G.; Guo, J.; Zheng, Y.; Gao, H.; Zhao, S.; Zhang, K.; Sun, P. Exploring QSARs for 5-Lipoxygenase (5-LO) Inhibitory Activity of 2-Substituted 5-Hydroxyindole-3-Carboxylates by CoMFA and CoMSIA. Chem. Biol. Drug Des. 2011, 78, 314–321. [Google Scholar] [CrossRef]
- Balasubramanian, P.K.; Balupuri, A.; Gadhe, C.G.; Cho, S.J. 3D QSAR Modeling Study on 7-Aminofuro [2,3-c] Pyridine Derivatives as TAK1 Inhibitors Using CoMFA and COMSIA. Med. Chem. Res. 2015, 24, 2347–2365. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Chemical Structure | Experiment pIC50 | CoMFA | CoMSIA | Residual CoMFA | Residual CoMSIA |
|---|---|---|---|---|---|---|
| Training Set | ||||||
| 1 |  | 7 | 6.76 | 6.45 | 0.24 | 0.55 |
| 2 |  | 6.61 | 6.47 | 6.27 | 0.14 | 0.34 |
| 3 |  | 6.39 | 6.49 | 6.44 | −0.10 | −0.05 |
| 4 |  | 6.34 | 6.43 | 6.46 | −0.09 | −0.12 |
| 5 |  | 6.21 | 6.47 | 6.31 | −0.26 | −0.10 |
| 6 |  | 6.01 | 6.01 | 6.18 | 0.00 | −0.17 |
| 8 |  | 5.82 | 5.67 | 5.81 | 0.15 | 0.01 |
| 10 |  | 5.40 | 5.23 | 5.04 | 0.17 | 0.36 |
| 12 |  | 4.82 | 4.80 | 4.78 | 0.02 | 0.04 |
| 13 |  | 4.77 | 4.65 | 5.05 | 0.12 | −0.28 |
| 14 |  | 4.73 | 4.87 | 5.14 | −0.14 | −0.41 |
| 15 |  | 4.53 | 4.45 | 4.41 | 0.08 | 0.12 |
| 16 |  | 4.39 | 4.48 | 4.61 | −0.09 | −0.22 |
| 17 |  | 4.34 | 4.50 | 4.13 | −0.16 | 0.21 |
| 18 |  | 4.34 | 4.54 | 4.46 | −0.20 | −0.12 |
| 19 |  | 4.18 | 4.20 | 4.02 | −0.02 | 0.16 |
| 22 |  | 4.00 | 3.88 | 3.96 | 0.12 | 0.04 |
| 23 |  | 4.00 | 3.89 | 4.37 | 0.11 | −0.37 |
| 25 |  | 4.00 | 4.06 | 3.98 | −0.06 | 0.02 |
| 26 |  | 4.00 | 4.22 | 4.29 | −0.22 | −0.29 |
| 28 |  | 4.00 | 3.89 | 3.84 | 0.11 | 0.16 |
| 29 |  | 4.00 | 3.78 | 4.44 | 0.22 | −0.44 |
| 30 |  | 3.94 | 4.06 | 3.83 | −0.12 | 0.11 |
| 31 |  | 3.87 | 3.91 | 3.45 | −0.04 | 0.42 |
| Test Set | ||||||
| 7 |  | 5.93 | 5.77 | 5.94 | 0.16 | −0.01 |
| 9 |  | 5.54 | 5.88 | 6.26 | −0.34 | −0.72 |
| 11 |  | 5.17 | 5.46 | 5.49 | −0.29 | −0.32 |
| 20 |  | 4.13 | 4.66 | 4.74 | −0.53 | −0.61 |
| 21 |  | 4.09 | 3.93 | 4.72 | 0.16 | −0.63 |
| 24 |  | 4.00 | 4.15 | 4.32 | −0.15 | −0.32 |
| 27 |  | 4.00 | 4.16 | 4.41 | −0.16 | −0.41 |
| Parameter | R2 | Q2 | SEE | F-Value | Components | r2test-set | r20 | k | (r2 − r20)/r2 |
|---|---|---|---|---|---|---|---|---|---|
| CoMFA | 0.98 | 0.61 | 0.16 | 234.68 | 4 | 0.91 | 0.98 | 0.95 | 0.08 |
| CoMSIA | 0.93 | 0.64 | 0.28 | 92.82 | 3 | 0.91 | 0.98 | 0.89 | 0.08 |
| Number of Components | 5 | 4 | 3 | 2 | 1 |
|---|---|---|---|---|---|
| Q2 | 0.51 | 0.51 | 0.47 | 0.45 | 0.34 |
| cSDEP | 0.80 | 0.78 | 0.70 | 0.79 | 0.84 |
| DQ2/dR2yy | 1.12 | 0.99 | 1.04 | 0.77 | 0.78 |
| Number of Components | 5 | 4 | 3 | 2 | 1 |
|---|---|---|---|---|---|
| Q2 | 0.47 | 0.50 | 0.51 | 0.46 | 0.32 |
| cSDEP | 0.83 | 0.79 | 0.76 | 0.79 | 0.86 |
| DQ2/dR2yy | 1.54 | 1.28 | 1.18 | 1.01 | 0.78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zięba, A.; Laitinen, T.; Patel, J.Z.; Poso, A.; Kaczor, A.A. Docking-Based 3D-QSAR Studies for 1,3,4-oxadiazol-2-one Derivatives as FAAH Inhibitors. Int. J. Mol. Sci. 2021, 22, 6108. https://doi.org/10.3390/ijms22116108
Zięba A, Laitinen T, Patel JZ, Poso A, Kaczor AA. Docking-Based 3D-QSAR Studies for 1,3,4-oxadiazol-2-one Derivatives as FAAH Inhibitors. International Journal of Molecular Sciences. 2021; 22(11):6108. https://doi.org/10.3390/ijms22116108
Chicago/Turabian StyleZięba, Agata, Tuomo Laitinen, Jayendra Z. Patel, Antti Poso, and Agnieszka A. Kaczor. 2021. "Docking-Based 3D-QSAR Studies for 1,3,4-oxadiazol-2-one Derivatives as FAAH Inhibitors" International Journal of Molecular Sciences 22, no. 11: 6108. https://doi.org/10.3390/ijms22116108
APA StyleZięba, A., Laitinen, T., Patel, J. Z., Poso, A., & Kaczor, A. A. (2021). Docking-Based 3D-QSAR Studies for 1,3,4-oxadiazol-2-one Derivatives as FAAH Inhibitors. International Journal of Molecular Sciences, 22(11), 6108. https://doi.org/10.3390/ijms22116108