
Assessment of Paroxetine Molecular Interactions with Selected Monoamine and γ-Aminobutyric Acid Transporters



Abstract
:1. Introduction
2. Results
2.1. Molecular Docking Studies
2.1.1. Structural Similarity Studies for Neurotransmitters and Their Transporters
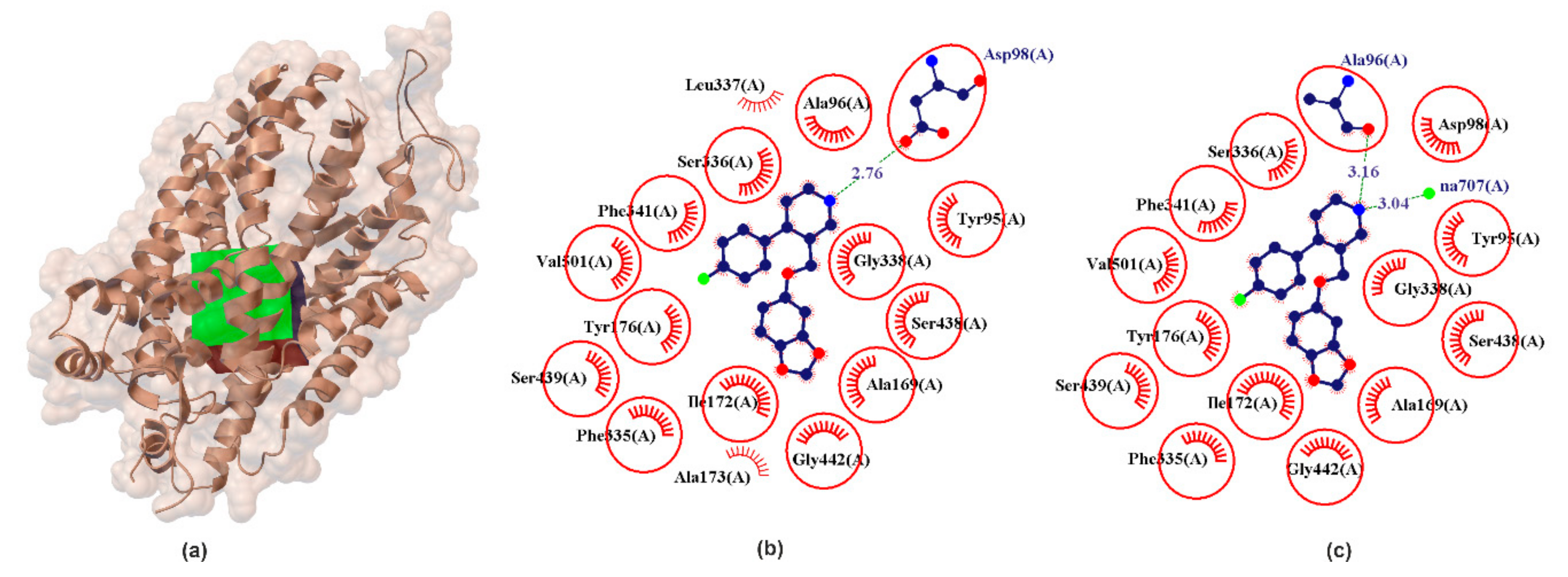
2.1.2. hSERT
2.1.3. hDAT
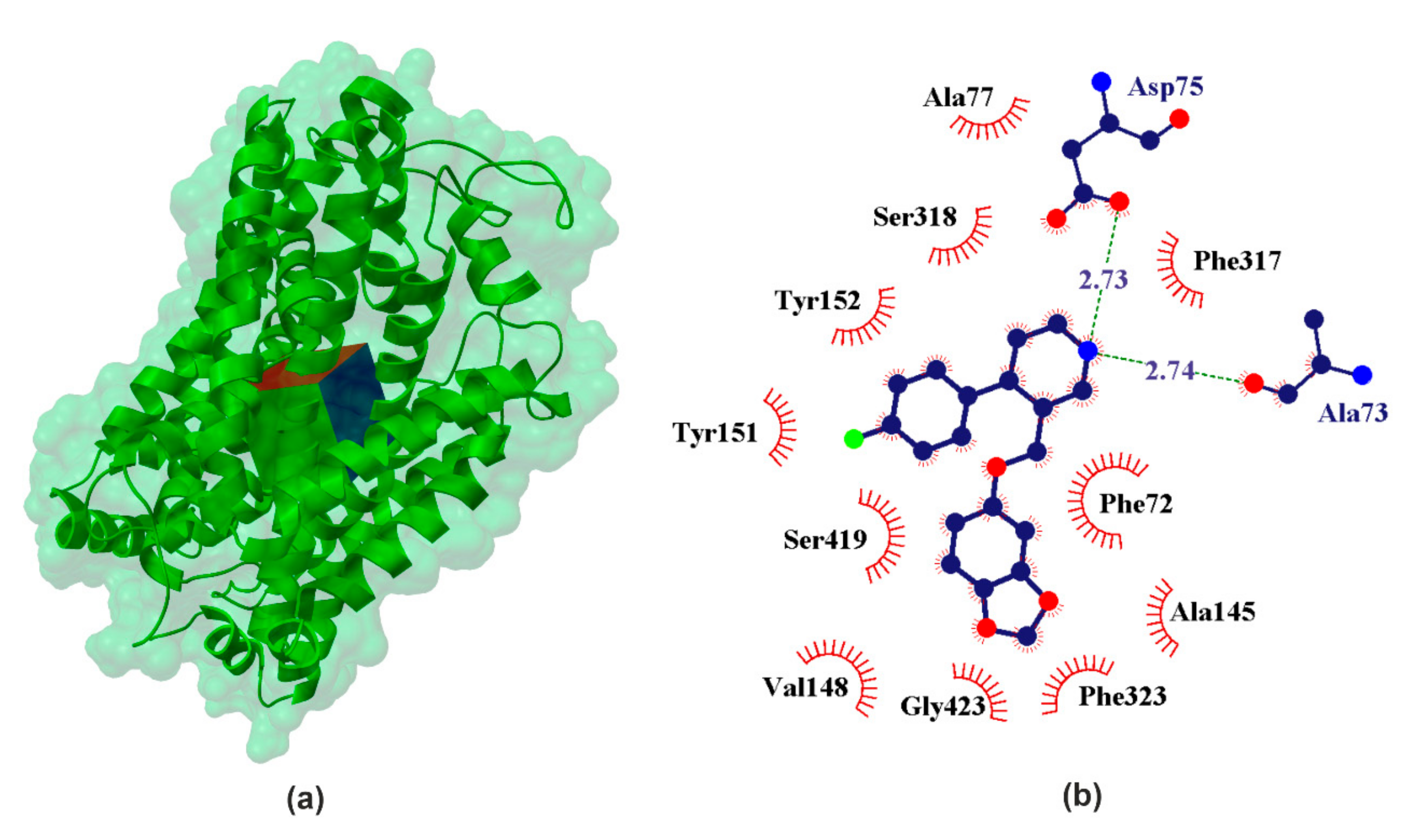
2.1.4. hNET
2.1.5. hGAT1
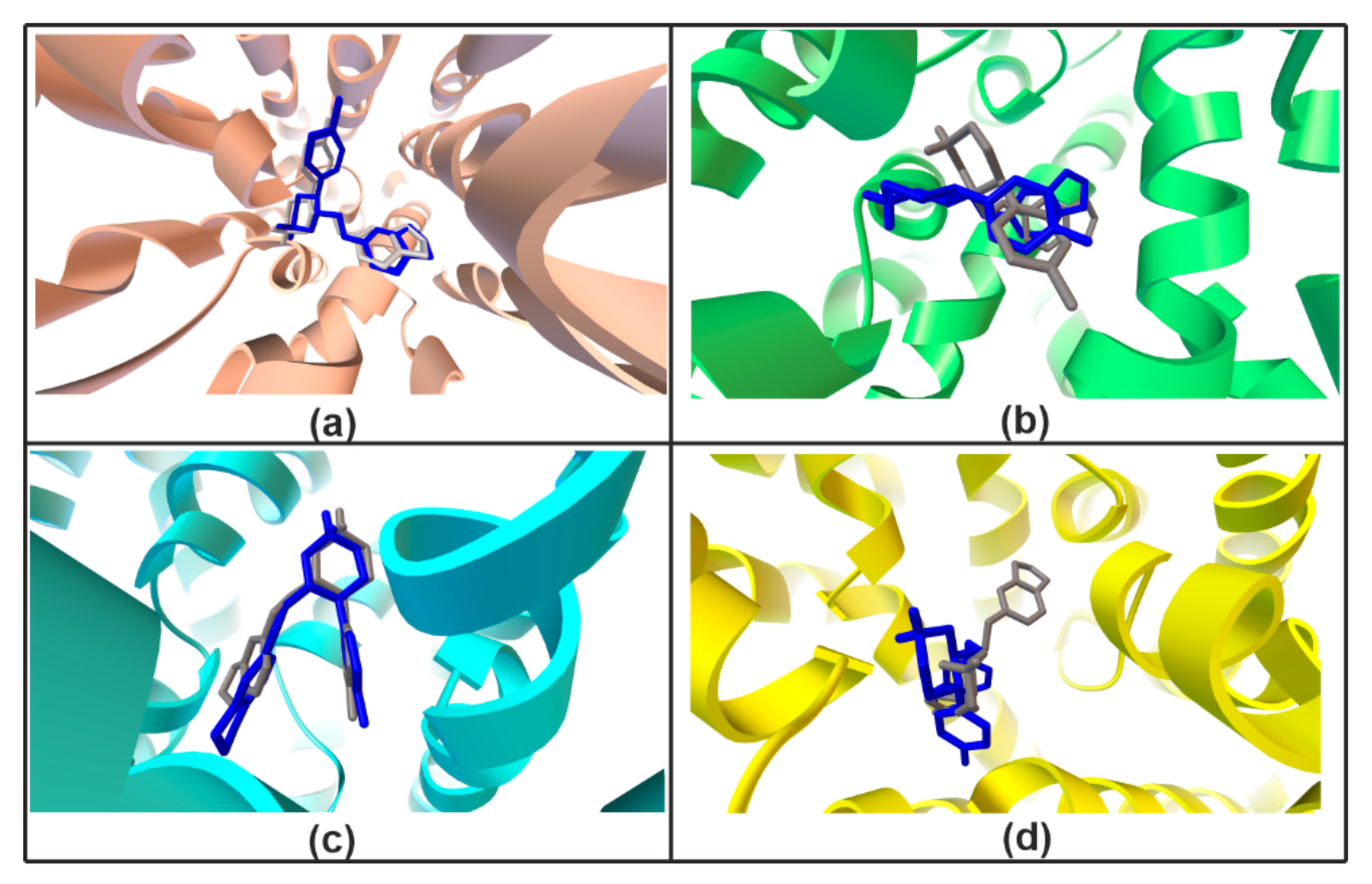
2.1.6. Validation Experiment
2.2. Inhibitory Activities of PRX, SER, NE and DA at hGATs
3. Discussion
4. Materials and Methods
4.1. Pharmacological Studies
4.2. In Silico Studies
4.2.1. Docking Study
Ligand Preparation
Monoamine and γ-Aminobutyric Acid Transporters Preparation
- hSERT
- hDAT, hNET hGAT1
Molecular Docking
4.3. Similarity and Superimposition Study
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Compliance with Ethical Standards
Abbreviations
| θ | hydrogen bond angle |
| # | hydrogen bond components: from the protein |
| % | hydrogen bond components: from the ligand |
| acc | hydrogen bond acceptor |
| ADT | AutoDockTools |
| CHO | Chinese hamster ovary |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DA | dopamine |
| DAT | proteins of dopamine transporter |
| EB | complex energy binding |
| EHB | hydrogen bond energy |
| GABA | γ-aminobutyric acid |
| GAT1 | GABA transporter isoform 1 |
| GAT2 | GABA transporter isoform 2 |
| GAT3 | GABA transporter isoform 3 |
| GDA | generalized anxiety disorder |
| hSERT | human proteins of serotonin transporter |
| hDAT | human proteins of dopamine transporter |
| hNET | human proteins of norepinephrine transporter |
| hGAT1 | human GABA transporter isoform 1 |
| hGAT2 | human GABA transporter isoform 2 |
| hGAT3 | human GABA transporter isoform 3 |
| hBGT1 | human betaine/GABA transporter |
| LHB | hydrogen bond length |
| MAT | monoamine transporters |
| MDD | major depressive disorder |
| NE | norepinephrine |
| NET | proteins of norepinephrine transporter |
| NPC | neural progenitor cells |
| NSC | neural stem cells |
| OCD | obsessive-compulsive disorder |
| PD | panic disorder |
| PDB ID | Protein Data Bank Identifier |
| pIC50xxx | inhibitory potency of xxx transporters |
| pKixxx | binding affinity of xxx transporters |
| PMDD | premenstrual dysphoric disorder |
| PRX | paroxetine |
| PTSD | post traumatic stress disorder |
| R/S-NE | R/S enantiomers norepinephrine |
| RMS | root mean square |
| SAD | social anxiety disorder |
| SER | serotonin |
| SERT | proteins of serotonin transporter |
| SNRI | serotonin norepinephrine reuptake inhibitors |
| SSRI | selective serotonin reuptake inhibitors |
| TCA | tricyclic antidepressants |
| Tsc | Tanimoto similarity coefficient |
| TM | transmembrane segments |
| TMH | transmembrane segments helix |
References
- Rantala, M.J.; Luoto, S.; Krams, I.; Karlsson, H. Depression subtyping based on evolutionary psychiatry: Proximate mechanisms and ultimate functions. Brain Behav. Immun. 2018, 69, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Butterweck, V. Mechanism of action of St John’s wort in depression. CNS Drugs 2003, 17, 539–562. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.R. A critical review of the mechanism of action for the selective serotonin reuptake inhibitors: Do these drugs possess anti-inflammatory properties and how relevant is this in the treatment of depression? Neuropharmacology 2013, 67, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.; Keshavan, M. The neurobiology of depression: An integrated view. Asian J. Psychiatry 2017, 27, 101–111. [Google Scholar] [CrossRef]
- Moon, J.-M.; Thapliyal, N.; Hussain, K.K.; Goyal, R.N.; Shim, Y.-B. Conducting polymer-based electrochemical biosensors for neurotransmitters: A review. Biosens. Bioelectron. 2018, 102, 540–552. [Google Scholar] [CrossRef]
- Ravna, A.W.; Sylte, I.; Dahl, S.G. Structure and localisation of drug binding sites on neurotransmitter transporters. J. Mol. Mod. 2009, 15, 1155–1164. [Google Scholar] [CrossRef]
- Zangrossi, H., Jr.; Del Ben, C.M.; Graeff, F.G.; Guimarães, F.S. Serotonin in panic and anxiety disorders. In Handbook of Behavioral Neuroscience; Elsevier: Amsterdam, The Netherlands, 2020; Volume 31, pp. 611–633. [Google Scholar]
- Whitaker-Azmitia, P.M. The discovery of serotonin and its role in neuroscience. Neuropsychopharmacology 1999, 21, 2–8. [Google Scholar] [CrossRef]
- Fijałkowski, Ł.; Sałat, K.; Podkowa, A.; Zaręba, P.; Nowaczyk, A. Potential role of selected antiepileptics used in neuropathic pain as human GABA transporter isoform 1 (GAT1) inhibitors—Molecular docking and pharmacodynamic studies. Eur. J. Pharm. Sci. 2017, 96, 362–372. [Google Scholar] [CrossRef]
- Monteleone, P.; Maj, M.; Iovino, M.; Steardo, L. GABA, depression and the mechanism of action of antidepressant drugs: A neuroendocrine approach. J. Affect. Disord. 1990, 20, 1–5. [Google Scholar] [CrossRef]
- Schwartz, T.L.; Nihalani, N. Tiagabine in anxiety disorders. Expert Opin. Pharmacother. 2006, 7, 1977–1987. [Google Scholar] [CrossRef]
- Iversen, L. Neurotransmitter transporters: Fruitful targets for CNS drug discovery. Mol. Psychiatry 2000, 5, 357–362. [Google Scholar] [CrossRef]
- Racagni, G.; Brunello, N. Physiology to functionality: The brain and neurotransmitter activity. Int. Clin. Psychopharmacol. 1999, 14, S3–S7. [Google Scholar] [CrossRef]
- Kalueff, A.V.; Nutt, D.J. Role of GABA in anxiety and depression. Depress. Anxiety 2007, 24, 495–517. [Google Scholar] [CrossRef]
- Kristensen, A.S.; Andersen, J.; Jørgensen, T.N.; Sørensen, L.; Eriksen, J.; Loland, C.J.; Strømgaard, K.; Gether, U. SLC6 neurotransmitter transporters: Structure, function, and regulation. Pharmacol. Rev. 2011, 63, 585–640. [Google Scholar] [CrossRef]
- Iversen, L. Neurotransmitter transporters and their impact on the development of psychopharmacology. Br. J. Pharmacol. 2006, 147, S82–S88. [Google Scholar] [CrossRef] [Green Version]
- Tuma, A.H.; Maser, J.D. Anxiety and the Anxiety Disorders; Routledge: New York, NY, USA, 2019. [Google Scholar]
- Boas, G.R.V.; de Lacerda, R.B.; Paes, M.M.; Gubert, P.; da Cruz Almeida, W.L.; Rescia, V.C.; de Carvalho, P.M.G.; de Carvalho, A.A.V.; Oesterreich, S.A. Molecular aspects of depression: A review from neurobiology to treatment. Eur. J. Pharmacol. 2019, 851, 99–121. [Google Scholar] [CrossRef]
- Coleman, J.A.; Navratna, V.; Antermite, D.; Yang, D.; Bull, J.A.; Gouaux, E. Chemical and structural investigation of the paroxetine-human serotonin transporter complex. eLife 2020, 9, e56427. [Google Scholar] [CrossRef]
- Davis, B.A.; Nagarajan, A.; Forrest, L.R.; Singh, S.K. Mechanism of paroxetine (paxil) inhibition of the serotonin transporter. Sci. Rep. 2016, 6, 23789. [Google Scholar] [CrossRef] [Green Version]
- Bourin, M.; Chue, P.; Guillon, Y. Paroxetine: A review. CNS Drug Rev. 2001, 7, 25–47. [Google Scholar] [CrossRef]
- Kowalska, M.; Nowaczyk, J.; Fijałkowski, Ł.; Nowaczyk, A. Paroxetine—Overview of the molecular mechanisms of action. Int. J. Mol. Sci. 2021, 22, 1662. [Google Scholar] [CrossRef]
- Oh, S.-J.; Cheng, J.; Jang, J.-H.; Arace, J.; Jeong, M.; Shin, C.-H.; Park, J.; Jin, J.; Greengard, P.; Oh, Y.-S. Hippocampal mossy cell involvement in behavioral and neurogenic responses to chronic antidepressant treatment. Mol. Psychiatry 2020, 25, 1215–1228. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj Manikandan, A.; Ramanathan, T.; Selvarajan, K.; Kesavanarayanan, T.L.; Salleh, M. Identification of Benzoxazolinone Derivatives Based Inhibitors for Depression and Pain Related Disorders Using Human Serotonin and Norepinephrine Transporter as Dual Therapeutic Target: A Computational Approach. Int. J. Drug Deliv. 2014, 6, 389–395. [Google Scholar]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-ray structures of Drosophila dopamine transporter in complex with nisoxetine and reboxetine. Nat. Struct. Mol. Biol. 2015, 22, 506–508. [Google Scholar] [CrossRef] [Green Version]
- Nowaczyk, A.; Fijałkowski, Ł.; Kowalska, M.; Podkowa, A.K.S. Studies on the activity of selected highly lipophilic compounds toward hGAT1 inhibition: Part II. ACS Chem. Neurosci. 2019, 10, 337–347. [Google Scholar] [CrossRef]
- Haddad, Y.; Heger, Z.; Adam, V. Guidelines for homology modeling of dopamine, norepinephrine, and serotonin transporters. ACS Chem. Neurosci. 2016, 7, 1607–1613. [Google Scholar] [CrossRef]
- Bero, S.; Muda, A.; Choo, Y.; Muda, N.; Pratama, S. Similarity measure for molecular structure: A brief review. J. Phys. 2017, 2017, 012015. [Google Scholar] [CrossRef] [Green Version]
- Koldsø, H.; Christiansen, A.B.; Sinning, S.; Schiøtt, B. Comparative modeling of the human monoamine transporters: Similarities in substrate binding. ACS Chem. Neurosci. 2013, 4, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Giros, B.; Wang, Y.-M.; Suter, S.; McLeskey, S.B.; Pifl, C.; Caron, M.G. Delineation of discrete domains for substrate, cocaine, and tricyclic antidepressant interactions using chimeric dopamine-norepinephrine transporters. J. Biol. Chem. 1994, 269, 15985–15988. [Google Scholar] [CrossRef]
- Wang, K.H.; Penmatsa, A.; Gouaux, E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 2015, 521, 322–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortore, G.; Orlandini, E.; Betti, L.; Giannaccini, G.; Mazzoni, M.R.; Camodeca, C.; Nencetti, S. Focus on Human Monoamine Transporter Selectivity. New Human DAT and NET Models, Experimental Validation, and SERT Affinity Exploration. ACS Chem. Neurosci. 2020, 11, 3214–3232. [Google Scholar] [CrossRef] [PubMed]
- Zafar, S.; Nguyen, M.E.; Muthyala, R.; Jabeen, I.; Sham, Y.Y. Modeling and Simulation of hGAT1: A Mechanistic Investigation of the GABA Transport Process. Comput. Struct. Biotechnol. J. 2019, 17, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Madhusudhan, M.; Marti-Renom, M.A.; Sanchez, R.; Sali, A. Variable gap penalty for protein sequence–structure alignment. Protein Eng. Des. Sel. 2006, 19, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.; Currie, J. Teaching with SCIGRES Molecular Modeling in Chemistry; Fujitsu Limited: Forest Grove, OR, USA, 2012. [Google Scholar]
- Chothia, C.; Lesk, A.M. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986, 5, 823–826. [Google Scholar] [CrossRef]
- Fujitsu Limited. CAChe for Windows User Guide; Fujitsu Limited: Tokyo, Japan; Oxford Molecular Ltd.: Oxford, UK, 2003. [Google Scholar]
- Navratna, V.; Tosh, D.K.; Jacobson, K.A.; Gouaux, E. Thermostabilization and purification of the human dopamine transporter (hDAT) in an inhibitor and allosteric ligand bound conformation. PLoS ONE 2018, 13, e0200085. [Google Scholar] [CrossRef] [Green Version]
- Mattson, R.J.; Catt, J.D.; Denhart, D.J.; Deskus, J.A.; Ditta, J.L.; Higgins, M.A.; Marcin, L.R.; Sloan, C.P.; Beno, B.R.; Gao, Q. Conformationally restricted homotryptamines. 2. Indole cyclopropylmethylamines as selective serotonin reuptake inhibitors. J. Med. Chem. 2005, 48, 6023–6034. [Google Scholar] [CrossRef]
- Owens, M.J.; Krulewicz, S.; Simon, J.S.; Sheehan, D.V.; Thase, M.E.; Carpenter, D.J.; Plott, S.J.; Nemeroff, C.B. Estimates of serotonin and norepinephrine transporter inhibition in depressed patients treated with paroxetine or venlafaxine. Neuropsychopharmacology 2008, 33, 3201–3212. [Google Scholar] [CrossRef]
- Tatsumi, M.; Groshan, K.; Blakely, R.D.; Richelson, E. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur. J. Pharmacol. 1997, 340, 249–258. [Google Scholar] [CrossRef]
- Andersen, J.; Ringsted, K.B.; Bang-Andersen, B.; Strømgaard, K.; Kristensen, A.S. Binding site residues control inhibitor selectivity in the human norepinephrine transporter but not in the human dopamine transporter. Sci. Rep. 2015, 5, 15650. [Google Scholar] [CrossRef] [Green Version]
- Skovstrup, S.; Taboureau, O.; Bräuner-Osborne, H.; Jørgensen, F.S. Homology modelling of the GABA transporter and analysis of tiagabine binding. ChemMedChem 2010, 5, 986–1000. [Google Scholar] [CrossRef]
- Abramyan, A.M.; Slack, R.D.; Meena, S.; Davis, B.A.; Newman, A.H.; Singh, S.K.; Shi, L. Computation-guided analysis of paroxetine binding to hSERT reveals functionally important structural elements and dynamics. Neuropharmacology 2019, 161, 107411. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Grundt, P.; Kopajtic, T.; Katz, J.L.; Newman, A.H. Structure—activity relationships at monoamine transporters for a series of N-substituted 3α-(bis [4-fluorophenyl] methoxy) tropanes: Comparative molecular field analysis, synthesis, and pharmacological evaluation. J. Med. Chem. 2004, 47, 3388–3398. [Google Scholar] [CrossRef]
- Beuming, T.; Kniazeff, J.; Bergmann, M.L.; Shi, L.; Gracia, L.; Raniszewska, K.; Newman, A.H.; Javitch, J.A.; Weinstein, H.; Gether, U. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat. Neurosci. 2008, 11, 780–789. [Google Scholar] [CrossRef] [Green Version]
- Rounsaville, B.J. Treatment of cocaine dependence and depression. Biol. Psychiatry 2004, 56, 803–809. [Google Scholar] [CrossRef]
- Xu, L.; Chen, L.Y. Identification of a new allosteric binding site for cocaine in dopamine transporter. J. Chem. Inf. Model. 2020, 60, 3958–3968. [Google Scholar] [CrossRef]
- Wilson, M.; Tripp, J. Clomipramine. Statpearls. 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK541006/ (accessed on 1 June 2021).
- O’Donnell, J.; Zeppenfeld, D.; McConnell, E.; Pena, S.; Nedergaard, M. Norepinephrine: A neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem. Res. 2012, 37, 2496–2512. [Google Scholar] [CrossRef] [Green Version]
- Eliwa, H.; Belzung, C.; Surget, A. Adult hippocampal neurogenesis: Is it the alpha and omega of antidepressant action? Biochem. Pharmacol. 2017, 141, 86–99. [Google Scholar] [CrossRef]
- Pidathala, S.; Mallela, A.K.; Joseph, D.; Penmatsa, A. Structural basis of norepinephrine recognition and transport inhibition in neurotransmitter transporters. bioRxiv 2020. [Google Scholar] [CrossRef]
- Altova, E.P.; Rykov, A.N.; Abaev, M.A.; Marochkin, I.I.; Vilkova, A.L.; Popik, M.V.; Shishkov, I.F. Molecular structure of noradrenaline according to gas-phase electron diffraction data and quantum chemical calculations. Mendeleev Commun. 2019, 29, 411–413. [Google Scholar] [CrossRef]
- Andersen, A. Structural studies of metabolic products of dopamine. IV. Crystal and molecular structure of (—)-noradrenaline. Acta Chem. Scand. 1975, 29, 871–876. [Google Scholar] [CrossRef]
- Pandit-Taskar, N.; Modak, S. Norepinephrine transporter as a target for imaging and therapy. J. Nucl. Med. 2017, 58, 39S–53S. [Google Scholar] [CrossRef] [Green Version]
- Moret, C.; Briley, M. The importance of norepinephrine in depression. Neuropsychiatry Dis. Treat. 2011, 7, 9. [Google Scholar]
- Brambilla, P.; Perez, J.; Barale, F.; Schettini, G.; Soares, J.C. GABAergic dysfunction in mood disorders. Mol. Psychiatry 2003, 8, 721–737. [Google Scholar] [CrossRef] [Green Version]
- Kickinger, S.; Hellsberg, E.; Schousboe, A.; Ecker, G.F.; Wellendorph, P. Structural and molecular aspects of betaine-GABA transporter 1 (BGT1) and its relation to brain function. Neuropharmacology 2019, 161, 107644. [Google Scholar] [CrossRef]
- Kvist, T.; Christiansen, B.; Jensen, A.A.; Brauner-Osborne, H. The four human γ-aminobutyric acid (GABA) transporters: Pharmacological characterization and validation of a highly efficient screening assay. Comb. Chem. High Throughput Screen. 2009, 12, 241–249. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [Green Version]
- Shah, K.; Mujwar, S.; Gupta, J.K.; Shrivastava, S.K.; Mishra, P. Molecular Docking and In Silico Cogitation Validate Mefenamic Acid Prodrugs as Human Cyclooxygenase-2 Inhibitor. Assay Drug Dev. Technol. 2019, 17, 285–291. [Google Scholar] [CrossRef]
- Stahl, S.M. Serotonin: It's possible to have too much of a good thing. J. Clin. Psychiatry 1997, 58, 520–521. [Google Scholar] [CrossRef] [Green Version]
- Möhler, H. The GABA system in anxiety and depression and its therapeutic potential. Neuropharmacology 2012, 62, 42–53. [Google Scholar] [CrossRef]
- Wang, S.; Du, L.; Peng, G.; Li, W. GABA inhibits proliferation and self-renewal of mouse retinal progenitor cell. Cell Death Discov. 2019, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-C. Neurogenesis and antidepressant action. Cell Tissue Res. 2019, 377, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Earnheart, J.C.; Schweizer, C.; Crestani, F.; Iwasato, T.; Itohara, S.; Mohler, H.; Lüscher, B. GABAergic control of adult hippocampal neurogenesis in relation to behavior indicative of trait anxiety and depression states. J. Neurosci. 2007, 27, 3845–3854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, C.L.; Bolzan, J.A.; Surget, A.; Belzung, C. Do antidepressants promote neurogenesis in adult hippocampus? A systematic review and meta-analysis on naive rodents. Pharmacol. Ther. 2020, 210, 107515. [Google Scholar] [CrossRef]
- Huffman, J.; Taylor, G.T. Stress, Neurogenesis, and Mood: An Introduction to the Neurogenic Hypothesis of Depression; Wiley: New York, NY, USA, 2020; pp. 37–42. [Google Scholar]
- Kempermann, G.; Song, H.; Gage, F.H. Neurogenesis in the adult hippocampus. Cold Spring Harb. Perspect. Biol. 2015, 7, a018812. [Google Scholar] [CrossRef] [Green Version]
- Catavero, C.; Bao, H.; Song, J. Neural mechanisms underlying GABAergic regulation of adult hippocampal neurogenesis. Cell Tissue Res. 2018, 371, 33–46. [Google Scholar] [CrossRef]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and functional implications of adult neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef] [Green Version]
- Pabba, M.; Sibille, E. GABA, depression and suicide. In Biological Aspects of Suicidal Behavior; Karger Publishers: Basel, Switzerland, 2016; Volume 30, pp. 37–50. [Google Scholar]
- Pytka, K.; Dziubina, A.; Młyniec, K.; Dziedziczak, A.; Żmudzka, E.; Furgała, A.; Olczyk, A.; Sapa, J.; Filipek, B. The role of glutamatergic, GABA-ergic, and cholinergic receptors in depression and antidepressant-like effect. Pharmacol. Rep. 2016, 68, 443–450. [Google Scholar] [CrossRef]
- Frazer, A.; Benmansour, S. Delayed pharmacological effects of antidepressants. Mol. Psychiatry 2002, 7, S23–S28. [Google Scholar] [CrossRef]
- Snyder, J.S. Recalibrating the relevance of adult neurogenesis. Trends Neurosci. 2019, 42, 164–178. [Google Scholar] [CrossRef]
- Jahromi, M.; Razavi, S.; Amirpour, N.; Khosravizadeh, Z. Paroxetine can enhance neurogenesis during neurogenic differentiation of human adipose-derived stem cells. Avicenna J. Med. Biotechnol. 2016, 8, 152. [Google Scholar]
- Schousboe, A.; Madsen, K.K. Delineation of the Role of Astroglial GABA Transporters in Seizure Control. Neurochem. Res. 2017, 42, 2019–2023. [Google Scholar] [CrossRef]
- Schousboe, A.; Wellendorph, P.; Frølund, B.; Clausen, R.P.; Krogsgaard-Larsen, P. Astrocytic GABA Transporters: Pharmacological Properties and Targets for Antiepileptic Drugs. In Glial Amino Acid Transporters; Springer: Cham, Switzerland, 2017; pp. 283–296. [Google Scholar]
- Annalisa, S. Structure, function, and plasticity of GABA transporters. Front. Cell. Neurosci. 2014, 8, 1–14. [Google Scholar]
- Lie, M.E.K.; Al-Khawaja, A.; Damgaard, M.; Haugaard, A.S.; Schousboe, A.; Clarkson, A.N.; Wellendorph, P. Glial GABA Transporters as Modulators of Inhibitory Signalling in Epilepsy and Stroke. Adv. Neurobiol. 2017, 16, 137–167. [Google Scholar]
- Forrest, L.R.; Tavoulari, S.; Zhang, Y.-W.; Rudnick, G.; Honig, B. Identification of a chloride ion binding site in Na+/Cl−-dependent transporters. Proc. Natl. Acad. Sci. USA 2007, 104, 12761–12766. [Google Scholar] [CrossRef] [Green Version]
- Kempson, S.A.; Zhou, Y.; Danbolt, N.C. The betaine/GABA transporter and betaine: Roles in brain, kidney, and liver. Front. Physiol. 2014, 5, 159. [Google Scholar] [CrossRef] [Green Version]
- Rusakov, D.A.; Saitow, F.; Lehre, K.P.; Konishi, S. Modulation of presynaptic Ca2+ entry by AMPA receptors at individual GABAergic synapses in the cerebellum. J. Neurosci. 2005, 25, 4930–4940. [Google Scholar] [CrossRef] [Green Version]
- Herculano-Houzel, S. Numbers of neurons as biological correlates of cognitive capability. Curr. Opin. Behav. Sci. 2017, 16, 1–7. [Google Scholar] [CrossRef]
- Drachman, D.A. Do We Have Brain to Spare? AAN Enterprises: Worcester, MA, USA, 2005. [Google Scholar]
- Al-Khawaja, A.; Petersen, J.G.; Damgaard, M.; Jensen, M.H.; Vogensen, S.B.; Lie, M.E.K.; Kragholm, B.; Bräuner-Osborne, H.; Clausen, R.P.; Frølund, B.; et al. Pharmacological identification of a guanidine-containing β-alanine analogue with low micromolar potency and selectivity for the betaine/GABA transporter 1 (BGT1). Neurochem. Res. 2014, 39, 1988–1996. [Google Scholar] [CrossRef]
- Furgała-Wojas, A.; Kowalska, M.; Nowaczyk, A.; Fijałkowski, Ł.; Sałat, K. Comparison of Bromhexine and its Active Metabolite-Ambroxol as Potential Analgesics Reducing Oxaliplatin-induced Neuropathic Pain-Pharmacodynamic and Molecular Docking Studies. Curr. Drug Metab. 2020, 21, 548–561. [Google Scholar] [CrossRef]
- Kowalska, M.; Nowaczyk, J.; Nowaczyk, A. KV11.1, NaV1.5, and CaV1.2 Transporter Proteins as Antitarget for Drug Cardiotoxicity. Int. J. Mol. Sci. 2020, 21, 8099. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.; Stratmann, R.E.; Burant, S.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.-C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; ‘Donovan, C.; Phan, I. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 1, 365–370. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | EB | pKi | Amino Acid Residues | HB | Angle | LHB | EHB | ||
|---|---|---|---|---|---|---|---|---|---|
| Protein | Ligand | kcal/mol | Donor | Acc | θ | Å | kcal/mol | ||
| hSERT | PRX | −13.92 | 10.20 | ASP98 | %NH2+ | # COO | 139.95 | 2.76 | −1.68 |
| SER | −9.7 | 7.11 | ALA96 | %NH1+ | # CONH | 129.52 | 1.91 | −0.27 | |
| ASP98 | %NH2+ | # COO | 132.83 | 1.78 | −0.15 | ||||
| SER336 | %NH3+ | # OH | 164.35 | 1.86 | −2.54 | ||||
| SER438 | %IndOH | # CONH | 138.77 | 1.92 | −0.27 | ||||
| R−NE | −5.45 | 3.99 | ASP98 | %OH | # COO | 134.19 | 1.81 | −0.12 | |
| PHE335 | %NH3+ | # CONH | 174.95 | 2.09 | −3.46 | ||||
| GLU493 | %m-Ph-OH | # COO | 134.94 | 1.85 | −0.05 | ||||
| S−NE | −5.31 | 3.89 | TYR95 | # Ph-OH | %m-PH-OH | 156.36 | 1.77 | −5.21 | |
| ASP98 | %NH3+ | # COO | 147.64 | 1.81 | −1.97 | ||||
| ASP98 | %OH | # COO | 141.19 | 1.64 | −6.26 | ||||
| DA | −5.45 | 3.99 | PHE335 | %NH3+ | # CONH | 159.93 | 2.11 | −0.87 | |
| GLU493 | %p-Ph-OH | # COO | 154.89 | 1.96 | −2.34 | ||||
| GLU493 | %m-PH-OH | # COO | 155.26 | 2.01 | −5.80 | ||||
| GABA | −6.77 | 3.86 | LYS490 | # NH1+ | %COO1 | 150.89 | 1.68 | −0.62 | |
| LYS490 | # NH2+ | %COO2 | 126.72 | 2.24 | −0.05 | ||||
| hDAT/cocaine | PRX | −8.35 | 6.12 | ASP476 | %NH2+ | # COO | 144.54 | 2.64 | −0.02 |
| SER | −7.77 | 5.70 | ASP79 | %NH1+ | # COO | 142.74 | 1.99 | −0.12 | |
| ASN157 | %IndOH | # CONH2 | 120.70 | 2.17 | −0.23 | ||||
| SER422 | %NH2+ | # CONH | 135.29 | 2.03 | −2.57 | ||||
| R−NE | −6.01 | 4.40 | PHE76 | %OH | # CONH | 154.98 | 2.12 | −1.02 | |
| ALA77 | %NH3+ | # CONH | 139.52 | 1.85 | −1.81 | ||||
| ASP79 | %NH3+ | # COO | 149.09 | 1.81 | −0.38 | ||||
| ASP79 | %m-PH-OH | # COO | 145.33 | 2.15 | −1.00 | ||||
| S−NE | −6.01 | 4.41 | ASP79 | %p-PH-OH | # COO | 168.08 | 1.89 | −3.85 | |
| ASP79 | %m-PH-OH | # COO | 153.47 | 1.73 | −1.02 | ||||
| ASP476 | %OH | # COO | 122.98 | 1.95 | −0.26 | ||||
| ASP476 | %NH3+ | # COO | 127.31 | 2.12 | −0.01 | ||||
| ASP476 | %NH3+ | # COO | 138.16 | 1.78 | −0.12 | ||||
| DA | −7.22 | 5.29 | ASP79 | %NH1 | # COO | 131.46 | 2.12 | −2.93 | |
| SER149 | %p-Ph-OH | # CONH | 158.76 | 2.19 | −3.40 | ||||
| TYR156 | %NH2 | # Ph-OH | 159.81 | 2.12 | 0.032 | ||||
| GABA | −4.62 | 3.38 | ASP79 | %NH1+ | # COO | 144.11 | 1.74 | −1.10 | |
| hDAT/clomipramine | PRX | −8.02 | 5.88 | ASP476 | %NH2+ | # COO | 155.89 | 2.75 | −0.04 |
| SER | −7.05 | 5.17 | ASP476 | %NH1+ | # COO | 124.04 | 2.19 | −0.08 | |
| ASP476 | %NH2+ | # COO | 141.21 | 1.87 | −0.51 | ||||
| ASP476 | %IndOH | # CONH | 165.74 | 1.83 | −4.42 | ||||
| R−NE | −6.04 | 4.43 | ASP385 | %p-Ph-OH | # COO | 159.51 | 1.77 | −0.46 | |
| ASP385 | %m-PH-OH | # COO | 171.74 | 1.96 | −2.65 | ||||
| ASP476 | %OH | # COO | 156.16 | 1.96 | −1.07 | ||||
| S−NE | −6.07 | 4.45 | ASP385 | %p-Ph-OH | # COO | 160.65 | 1.83 | −1.50 | |
| ASP385 | %m-PH-OH | # COO | 158.36 | 2.11 | −1.50 | ||||
| ASP476 | %OH | # COO | 168.50 | 1.84 | −1.29 | ||||
| DA | −6.72 | 4.92 | ASP385 | %p-Ph-OH | # COO | 156.93 | 1.87 | −1.38 | |
| ASP385 | %m-PH-OH | # COO | 162.14 | 2.00 | −4.85 | ||||
| ASP476 | %NH3+ | # COO | 127.69 | 1.91 | −0.10 | ||||
| GABA | −4.34 | 3.18 | ASN93 | %NH1+ | # CONH2 | 126.49 | 1.99 | −0.85 | |
| SER309 | %NH2+ | # OH | 137.67 | 2.15 | −2.84 | ||||
| hNET | PRX | −10.38 | 7.61 | ALA73 | %NH1+ | # CONH | 125.52 | 2.73 | −1.06 |
| ASP75 | %NH2+ | # COO | 162.17 | 2.74 | −0.12 | ||||
| SER | −5.91 | 4.33 | PHE72 | %NH1+ | # CONH | 152.37 | 2.18 | −4.06 | |
| ASP75 | %NH2+ | # COO | 142.29 | 2.02 | −0.04 | ||||
| SER419 | %NH3+ | # OH | 135.90 | 1.96 | −2.85 | ||||
| SER420 | %IndOH | # OH | 168.89 | 2.20 | −4.18 | ||||
| R−NE | −5.62 | 4.12 | ASP75 | %p-Ph-OH | # COO | 170.95 | 1.72 | −3.11 | |
| ASP75 | %m-PH-OH | # COO | 153.61 | 1.74 | −1.64 | ||||
| ASP473 | %OH | # COO | 143.25 | 2.01 | −0.01 | ||||
| S−NE | −7.75 | 5.59 | PHE72 | %NH1+ | # CONH | 121.16 | 2.06 | −0.56 | |
| ASP75 | %NH2+ | # COO | 153.53 | 2.19 | −0.53 | ||||
| SER318 | %OH | # CONH | 135.06 | 1.94 | −1.34 | ||||
| LEU319 | %m-PH-OH | # CONH | 131.15 | 2.14 | −0.61 | ||||
| DA | −7.25 | 5.31 | ASP75 | %NH1+ | # COO | 121.36 | 1.99 | −0.13 | |
| ALA145 | %p-Ph-OH | # CONH | 134.7 | 2.11 | −0.93 | ||||
| TYR152 | %NH2+ | # Ph-OH | 169.39 | 2.18 | −3.64 | ||||
| SER419 | %NH3+ | # OH | 139.35 | 2.25 | −2.08 | ||||
| GABA | −4.51 | 3.31 | ASP75 | %NH1+ | # COO | 151.09 | 1.81 | −1.60 | |
| hGAT1 | PRX | −5.93 | 4.35 | ASP451 | %NH2+ | # CONH | 151.56 | 2.91 | −3.70 |
| ASP451 | %NH2+ | # COO | 147.39 | 2.93 | −3.13 | ||||
| SER | −4.86 | 3.56 | TYR139 | %NH1 | # Ph-OH | 172.42 | 1.99 | −5.78 | |
| ASP451 | %NH2 | # COO | 143.11 | 1.86 | −2.83 | ||||
| ASP451 | %NH3 | # CONH | 122.09 | 1.87 | 0.023 | ||||
| SER456 | %m-PH-OH | # CONH | 139.31 | 2.06 | −2.94 | ||||
| MET458 | # CONH | %m-PH-OH | 150.79 | 1.90 | −4.45 | ||||
| SER459 | # CONH | %m-PH-OH | 164.48 | 2.08 | −5.04 | ||||
| R−NE | −5.28 | 3.87 | TYR139 | %NH1 | # Ph-OH | 130.79 | 2.21 | −1.60 | |
| ASP451 | %NH2 | # CONH | 148.66 | 1.85 | −1.96 | ||||
| MET458 | # CONH | %p-PH-OH | 163.77 | 1.99 | −5.67 | ||||
| SER459 | # CONH | %p-PH-OH | 151.54 | 1.93 | −4.51 | ||||
| LEU460 | # CONH | %m-PH-OH | 151.03 | 2.24 | −2.73 | ||||
| S−NE | −5.28 | 3.86 | ASP451 | %NH1 | # COO | 135.12 | 1.76 | −0.47 | |
| ASP451 | %OH | # CONH | 171.27 | 1.65 | −2.93 | ||||
| SER456 | %m-PH-OH | # CONH | 152.49 | 2.06 | −3.09 | ||||
| MET458 | # CONH | %m-PH-OH | 153.65 | 1.78 | −4.80 | ||||
| SER459 | # CONH | %m-PH-OH | 162.42 | 2.005 | −5.5 | ||||
| DA | −4.85 | 3.55 | ASP451 | %NH1 | # COO | 123.39 | 1.941 | −0.503 | |
| ASP451 | %NH2 | # CONH | 127.83 | 2.017 | −1.36 | ||||
| SER456 | %m-PH-OH | # CONH | 172.05 | 1.937 | −4.819 | ||||
| MET458 | # CONH | %m-PH-OH | 151.59 | 2.026 | −4.052 | ||||
| SER459 | # CONH | %m-PH-OH | 162.66 | 2.124 | −4.493 | ||||
| LEU460 | # CONH | %p-PH-OH | 155.46 | 1.797 | −5.329 | ||||
| GABA | −6.24 | 4.49 | ASP451 | %NH1+ | # COO | 142.52 | 2.503 | −0.026 | |
| ASP451 | %NH2+ | # CONH | 156.84 | 2.451 | −3.32 | ||||
| Protein | Residues |
|---|---|
| hSERT | Tyr95, Ala96, Asp98, Gly100, Ala169, Ile172, Ala173, Tyr176, Phe335, Ser336, Gly338, Phe341, Val343, Ser438, Thr439, Ala441, Gly442, Val489, Lys490, Glu493, Glu494, Thr497, Gly498, Pro499, Leu502 [6] |
| hNET | Ala145, Tyr151, Ile315, Phe316, Ser420, Ala426 [44]. |
| hDAT | Ala81, Tyr88, Asp385, Asp385, Asp476, Ala480, Phe472, Thr473, Asp476, His477, Ala480, Gly481, Thr482, Leu485 [6] |
| hGAT1 | Tyr60, Ala61, Gly63, Gly65, Leu136, Tyr140, Phe294, Ser295, Tyr296, Gly297, Leu300, Thr400 [45] |
| IC50 (pIC50 ± S.E.M.) (µM) | ||||
|---|---|---|---|---|
| hGAT1 | hGAT2 | hGAT3 | hBGT1 | |
| PRX | 256.1 (3.6 ± 0.03) | 122.4 (3.9 ± 0.08) | 110.6 (4.0 ± 0.04) | 85.6 (4.1 ± 0.05) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalska, M.; Fijałkowski, Ł.; Nowaczyk, A. Assessment of Paroxetine Molecular Interactions with Selected Monoamine and γ-Aminobutyric Acid Transporters. Int. J. Mol. Sci. 2021, 22, 6293. https://doi.org/10.3390/ijms22126293
Kowalska M, Fijałkowski Ł, Nowaczyk A. Assessment of Paroxetine Molecular Interactions with Selected Monoamine and γ-Aminobutyric Acid Transporters. International Journal of Molecular Sciences. 2021; 22(12):6293. https://doi.org/10.3390/ijms22126293
Chicago/Turabian StyleKowalska, Magdalena, Łukasz Fijałkowski, and Alicja Nowaczyk. 2021. "Assessment of Paroxetine Molecular Interactions with Selected Monoamine and γ-Aminobutyric Acid Transporters" International Journal of Molecular Sciences 22, no. 12: 6293. https://doi.org/10.3390/ijms22126293
APA StyleKowalska, M., Fijałkowski, Ł., & Nowaczyk, A. (2021). Assessment of Paroxetine Molecular Interactions with Selected Monoamine and γ-Aminobutyric Acid Transporters. International Journal of Molecular Sciences, 22(12), 6293. https://doi.org/10.3390/ijms22126293