
Differential Effects of Furin Deficiency on Insulin Receptor Processing and Glucose Control in Liver and Pancreatic β Cells of Mice

 ,
,  and
and 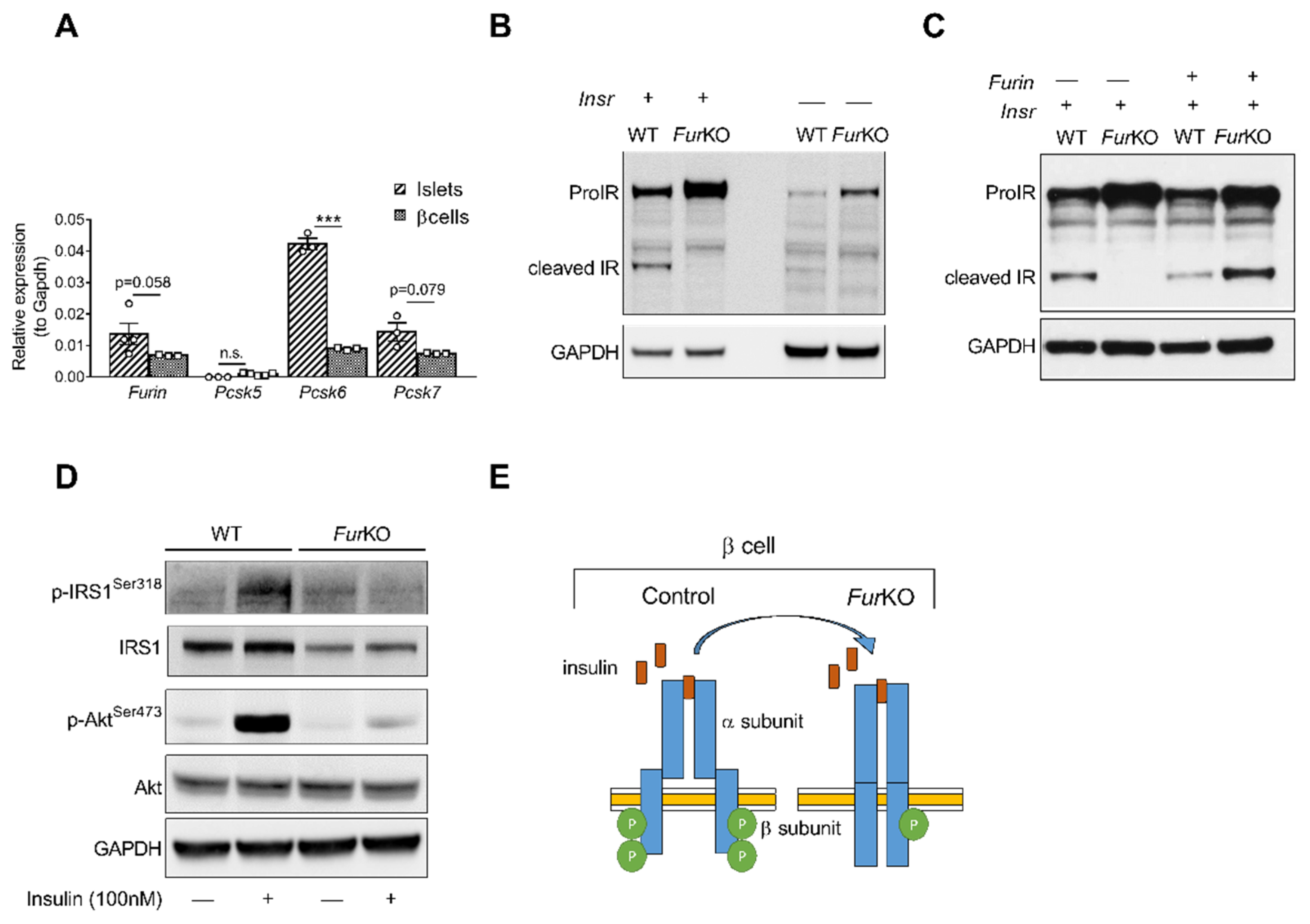{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. IR Processing and Signaling Are Severely Affected in FurKO β Cells
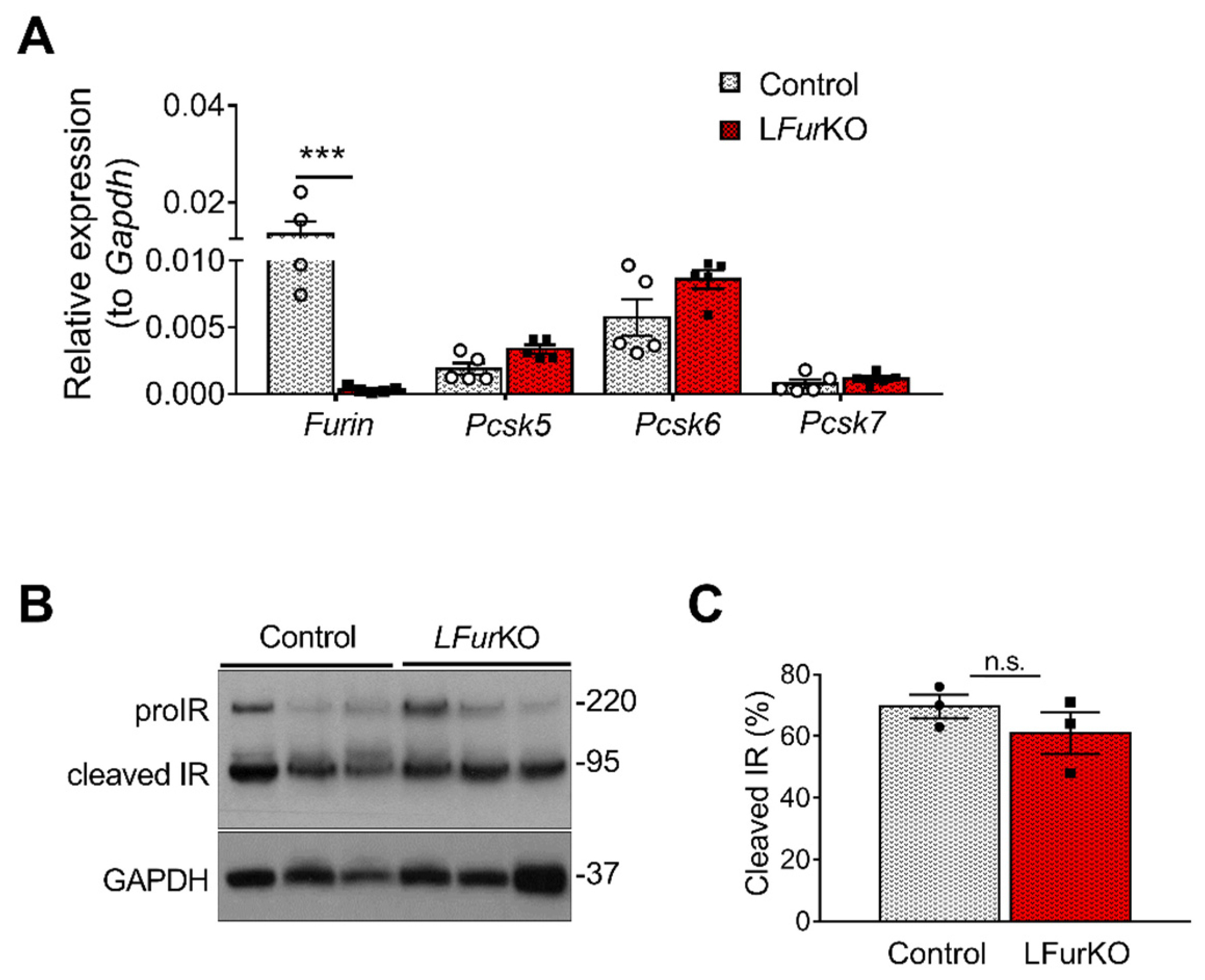
2.2. IR Proteolytic Cleavage Is Not Altered in Liver-Specific FurKO Mice
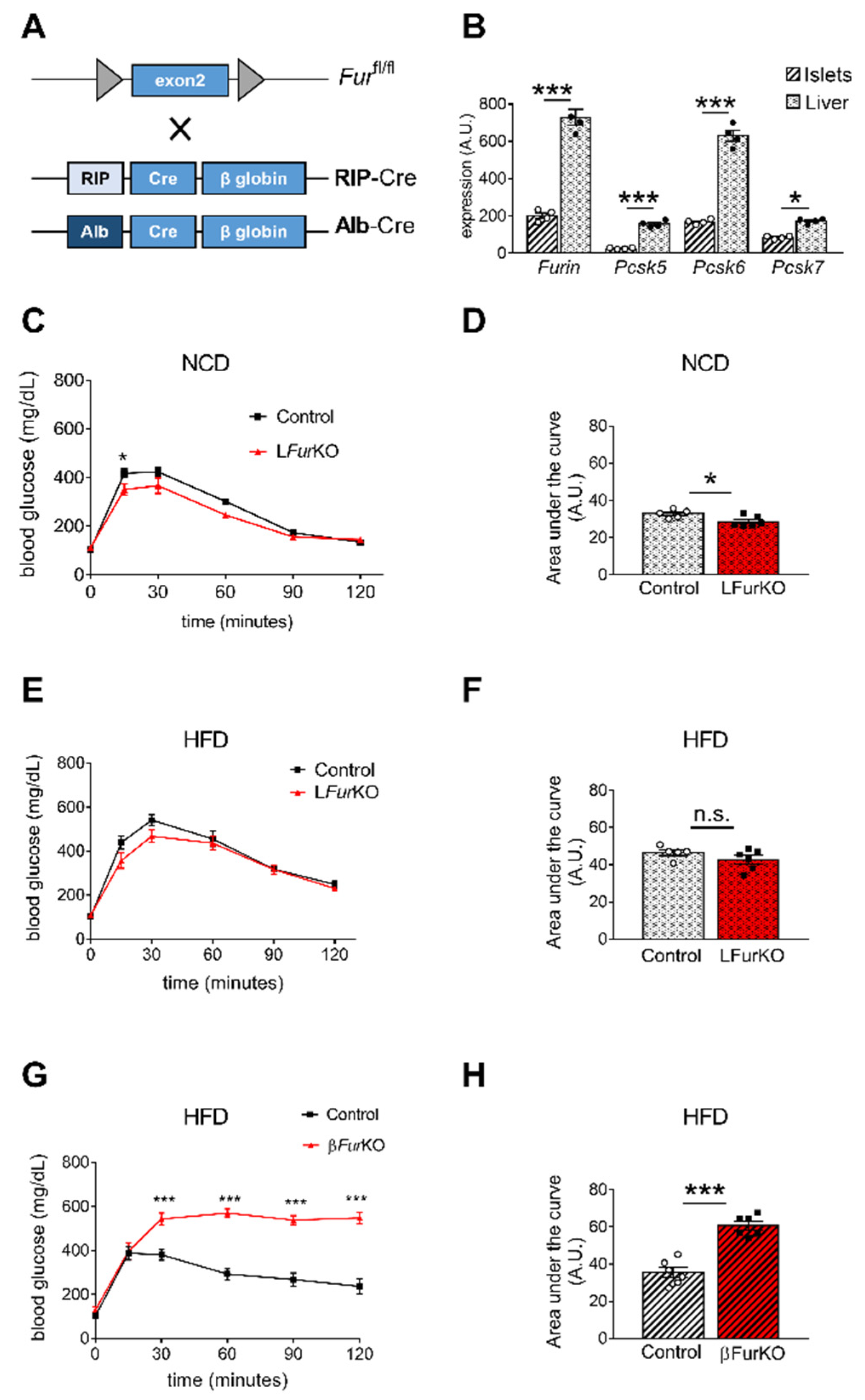
2.3. Impact of Conditional Furin Deletion in Liver and Pancreatic β Cells on Glucose Homeostasis
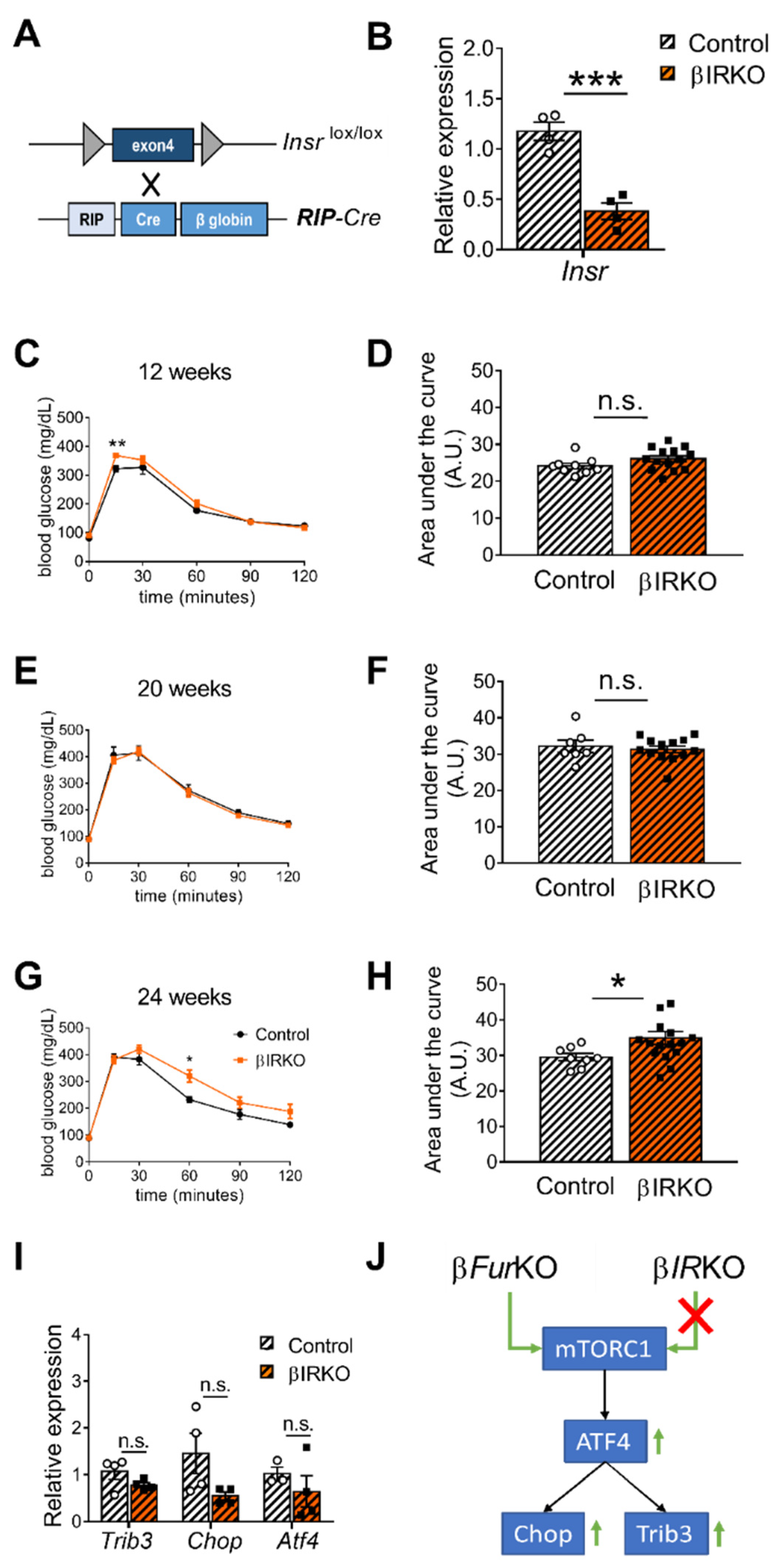
2.4. IR Deficiency in Pancreatic β Cells Does Not Induce Severe Glucose Intolerance
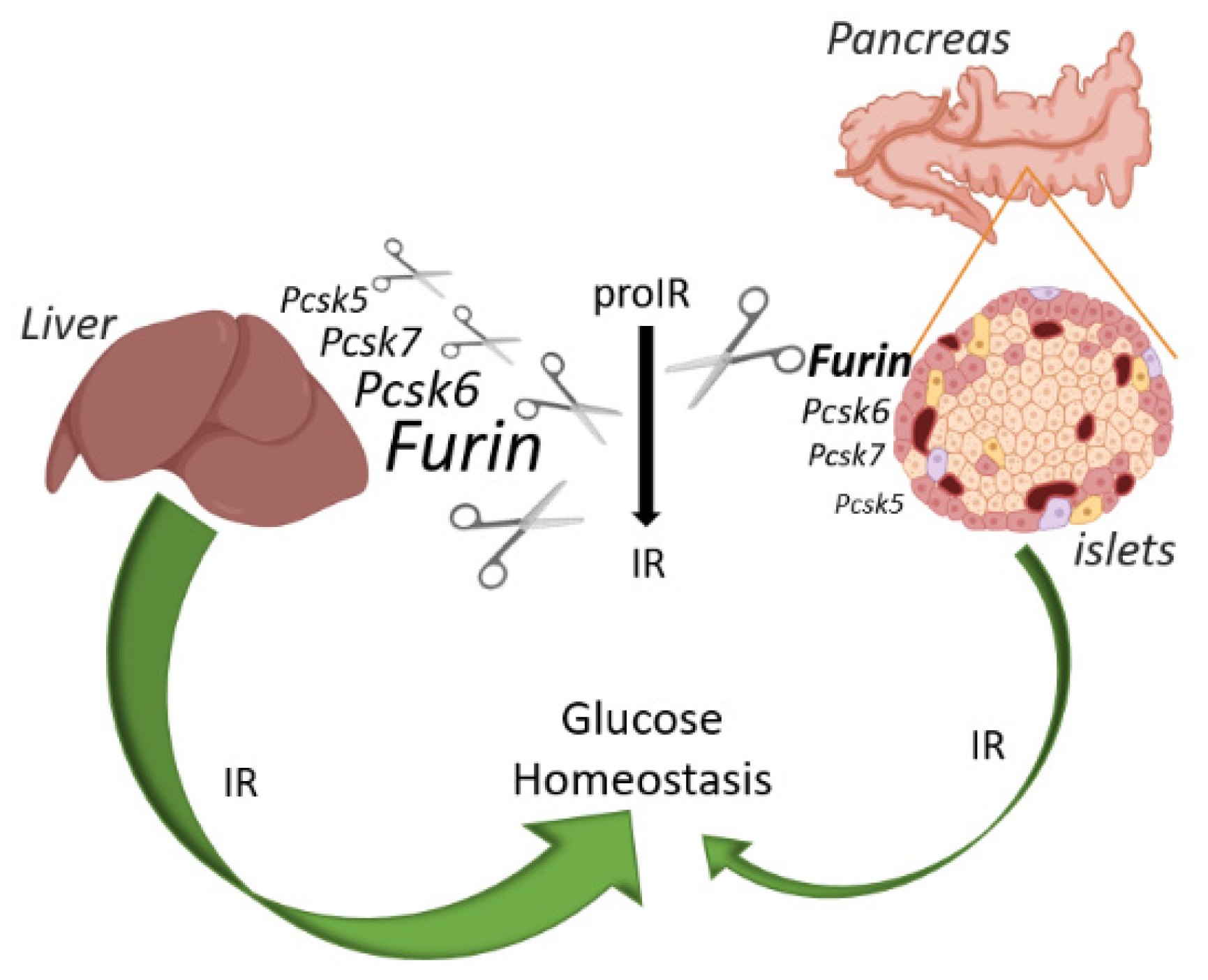
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Intraperitoneal Glucose and Insulin Tolerance Test (IPGTT and IPITT)
4.3. Islet Isolation
4.4. Cell Culture and Transfection
4.5. Generation of Furin Knockout βTC3 Cells Using the CRISPR-Cas9 Nuclease System
4.6. Microarray Analysis
4.7. Crude Membrane Fraction from Mouse Liver
4.8. Western Blot
4.9. Quantitative RT-PCR (RT-qPCR)
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and Cellular Properties of Insulin Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Bravo, D.A.; Gleason, J.B.; Sanchez, R.I.; Roth, R.A.; Fuller, R.S. Accurate and Efficient Cleavage of the Human Insulin Proreceptor by the Human Proprotein-Processing Protease Furin. Characterization and Kinetic Parameters Using the Purified, Secreted Soluble Protease Expressed by a Recombinant Baculovirus. J. Biol. Chem. 1994, 269, 25830–25837. [Google Scholar] [CrossRef]
- Yoshimasa, Y.; Seino, S.; Whittaker, J.; Kakehi, T.; Kosaki, A.; Kuzuya, H.; Imura, H.; Bell, G.I.; Steiner, D.F. Insulin-Resistant Diabetes Due to a Point Mutation That Prevents Insulin Proreceptor Processing. Science 1988, 240, 784–787. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sasaoka, T.; Takata, Y.; Hisatomi, A.; Shigeta, Y. Insulin Resistance by Uncleaved Insulin Proreceptor. Emergence of Binding Site by Trypsin. Diabetes 1988, 37, 653–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.; Pierce, S.E.; Li, A.; Spees, K.; Anderson, G.R.; Seoane, J.A.; Lo, Y.-H.; Dubreuil, M.; Olivas, M.; Kamber, R.A.; et al. CRISPR Screens in Cancer Spheroids Identify 3D Growth-Specific Vulnerabilities. Nature 2020, 580, 136–141. [Google Scholar] [CrossRef]
- Kara, I.; Poggi, M.; Bonardo, B.; Govers, R.; Landrier, J.-F.; Tian, S.; Leibiger, I.; Day, R.; Creemers, J.W.M.; Peiretti, F. The Paired Basic Amino Acid-Cleaving Enzyme 4 (PACE4) Is Involved in the Maturation of Insulin Receptor Isoform B: An Opportunity to Reduce the Specific Insulin Receptor-Dependent Effects of Insulin-like Growth Factor 2 (IGF2). J. Biol. Chem. 2015, 290, 2812–2821. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Khatib, A.-M.; Creemers, J.W.M. Loss of Proprotein Convertase Furin in Mammary Gland Impairs ProIGF1R and ProIR Processing and Suppresses Tumorigenesis in Triple Negative Breast Cancer. Cancers 2020, 12, 2686. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Thorrez, L.; Siegfried, G.; Meulemans, S.; Evrard, S.; Tejpar, S.; Khatib, A.-M.; Creemers, J.W.M. The Proprotein Convertase Furin Is a Pro-Oncogenic Driver in KRAS and BRAF Driven Colorectal Cancer. Oncogene 2020, 39, 3571–3587. [Google Scholar] [CrossRef]
- Roebroek, A.J.M.; Taylor, N.A.; Louagie, E.; Pauli, I.; Smeijers, L.; Snellinx, A.; Lauwers, A.; Van de Ven, W.J.M.; Hartmann, D.; Creemers, J.W.M. Limited Redundancy of the Proprotein Convertase Furin in Mouse Liver. J. Biol. Chem. 2004, 279, 53442–53450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, F. Tissue-Specific Insulin Signaling in the Regulation of Metabolism and Aging. IUBMB Life 2014, 66, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najjar, S.M.; Perdomo, G. Hepatic Insulin Clearance: Mechanism and Physiology. Physiology 2019, 34, 198–215. [Google Scholar] [CrossRef]
- Michael, M.D.; Kulkarni, R.N.; Postic, C.; Previs, S.F.; Shulman, G.I.; Magnuson, M.A.; Kahn, C.R. Loss of Insulin Signaling in Hepatocytes Leads to Severe Insulin Resistance and Progressive Hepatic Dysfunction. Mol. Cell 2000, 6, 87–97. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Brüning, J.C.; Winnay, J.N.; Postic, C.; Magnuson, M.A.; Kahn, C.R. Tissue-Specific Knockout of the Insulin Receptor in Pancreatic Beta Cells Creates an Insulin Secretory Defect Similar to That in Type 2 Diabetes. Cell 1999, 96, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Liew, C.W.; Hu, J.; Hinault, C.; Michael, M.D.; Krtzfeldt, J.; Yin, C.; Holzenberger, M.; Stoffel, M.; Kulkarni, R.N. Insulin Receptors in Beta-Cells Are Critical for Islet Compensatory Growth Response to Insulin Resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 8977–8982. [Google Scholar] [CrossRef] [Green Version]
- Otani, K.; Kulkarni, R.N.; Baldwin, A.C.; Krutzfeldt, J.; Ueki, K.; Stoffel, M.; Kahn, C.R.; Polonsky, K.S. Reduced Beta-Cell Mass and Altered Glucose Sensing Impair Insulin-Secretory Function in BetaIRKO Mice. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E41–E49. [Google Scholar] [CrossRef]
- Wicksteed, B.; Brissova, M.; Yan, W.; Opland, D.M.; Plank, J.L.; Reinert, R.B.; Dickson, L.M.; Tamarina, N.A.; Philipson, L.H.; Shostak, A.; et al. Conditional Gene Targeting in Mouse Pancreatic SS-Cells: Analysis of Ectopic Cre Transgene Expression in the Brain. Diabetes 2010, 59, 3090–3098. [Google Scholar] [CrossRef] [Green Version]
- Brouwers, B.; de Faudeur, G.; Osipovich, A.B.; Goyvaerts, L.; Lemaire, K.; Boesmans, L.; Cauwelier, E.J.G.; Granvik, M.; Pruniau, V.P.E.G.; Van Lommel, L.; et al. Impaired Islet Function in Commonly Used Transgenic Mouse Lines Due to Human Growth Hormone Minigene Expression. Cell Metab. 2014, 20, 979–990. [Google Scholar] [CrossRef] [Green Version]
- Transgenic Artifacts Caused by Passenger Human Growth Hormone. Abstract—Europe PMC. Available online: https://europepmc.org/article/med/29921469 (accessed on 27 October 2020).
- Oropeza, D.; Jouvet, N.; Budry, L.; Campbell, J.E.; Bouyakdan, K.; Lacombe, J.; Perron, G.; Bergeron, V.; Neuman, J.C.; Brar, H.K.; et al. Phenotypic Characterization of MIP-CreERT1Lphi Mice with Transgene-Driven Islet Expression of Human Growth Hormone. Diabetes 2015, 64, 3798–3807. [Google Scholar] [CrossRef] [Green Version]
- Oakie, A.; Zhou, L.; Rivers, S.; Cheung, C.; Li, J.; Wang, R. Postnatal Knockout of Beta Cell Insulin Receptor Impaired Insulin Secretion in Male Mice Exposed to High-Fat Diet Stress. Mol. Cell Endocrinol. 2020, 499, 110588. [Google Scholar] [CrossRef] [PubMed]
- Skovsø, S.; Panzhinskiy, E.; Kolic, J.; Dionne, D.A.; Dai, X.-Q.; Sharma, R.B.; Elghazi, L.; Cen, H.H.; Ellis, C.E.; Faulkner, K.; et al. Beta-Cell specific insulin resistance promotes glucose-stimulated insulin hypersecretion. bioRxiv 2020. [Google Scholar] [CrossRef]
- Brouwers, B.; Coppola, I.; Vints, K.; Dislich, B.; Jouvet, N.; Lommel, L.V.; Segers, C.; Gounko, N.V.; Thorrez, L.; Schuit, F.; et al. Loss of furin in β Cells induces an MTORC1-ATF4 anabolic pathway that leads to β cell dysfunction. Diabetes 2020. [Google Scholar] [CrossRef]
- Herrera, P.L. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 2000, 127, 2317–2322. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using cre recombinase. J. Biol. Chem. 1999, 274, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Liew, C.W.; Bochenski, J.; Kawamori, D.; Hu, J.; Leech, C.A.; Wanic, K.; Malecki, M.; Warram, J.H.; Qi, L.; Krolewski, A.S.; et al. The pseudokinase tribbles homolog 3 interacts with atf4 to negatively regulate insulin exocytosis in human and mouse β cells. J. Clin. Investig. 2010, 120, 2876–2888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creemers, J.W.M.; Khatib, A.-M. Knock-out mouse models of proprotein convertases: Unique functions or redundancy? Front. Biosci. 2008, 13, 4960–4971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuliawat, P.; Arvan, P. Protein targeting via the “constitutive-like” secretory pathway in isolated pancreatic islets: Passive sorting in the immature granule compartment. J. Cell Biol. 1992, 118, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Mbikay, M.; Arimura, A. Pituitary Adenylate Cyclase-Activating Polypeptide Precursor Is Processed Solely by Prohormone Convertase 4 in the Gonads. Endocrinology 2000, 141, 3723–3730. [Google Scholar] [CrossRef]
- Li, M.; Mbikay, M.; Nakayama, K.; Miyata, A.; Arimura, A. Prohormone convertase PC4 processes the precursor of PACAP in the testis. Ann. N. Y. Acad. Sci. 2000, 921, 333–339. [Google Scholar] [CrossRef]
- Li, M.; Shuto, Y.; Somogyvári-Vigh, A.; Arimura, A. Prohormone convertases 1 and 2 process ProPACAP and generate matured, bioactive PACAP38 and PACAP27 in transfected rat pituitary GH4C1 cells. Neuroendocrinology 1999, 69, 217–226. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Engel, J.L.; Wiley, S.E.; Xiao, J.; Gonzalez, D.J.; Appaiah, H.N.; Koller, A.; Nizet, V.; White, K.E.; Dixon, J.E. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 5520–5525. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Ramos-Molina, B.; Lick, A.N.; Prideaux, M.; Albornoz, V.; Bonewald, L.; Lindberg, I. Posttranslational processing of FGF23 in osteocytes during the osteoblast to osteocyte transition. Bone 2016, 84, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.F.; McClain, D.A.; Dull, T.J.; Ullrich, A.; Olefsky, J.M. Characterization of an insulin receptor mutant lacking the subunit processing site. J. Biol. Chem. 1990, 265, 8463–8469. [Google Scholar] [CrossRef]
- Sugibayashi, M.; Shigeta, Y.; Teraoka, H.; Kobayashi, M. Characterization of Unprocessed Insulin Proreceptors in COS 7 Cells Transfected with CDNA with Arg735 → Ser735 Point Mutation at the Cleavage Site. Metabolism 1992, 41, 820–826. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Ristow, M.; Lin, X.; White, M.F.; Magnuson, M.A.; Hennighausen, L. RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J. Biol. Chem. 2006, 281, 2649–2653. [Google Scholar] [CrossRef] [Green Version]
- Pomplun, D.; Florian, S.; Schulz, T.; Pfeiffer, A.F.H.; Ristow, M. Alterations of pancreatic beta-cell mass and islet number due to Ins2-Controlled expression of cre recombinase: RIP-Cre revisited; Part 2. Horm. Metab. Res. 2007, 39, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The MTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef]
- Yoon, M.-S. The Role of Mammalian Target of Rapamycin (MTOR) in Insulin Signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef]
- Dakin, R.S.; Walker, B.R.; Seckl, J.R.; Hadoke, P.W.F.; Drake, A.J. Estrogens Protect Male Mice from Obesity Complications and Influence Glucocorticoid Metabolism. Int. J. Obes. 2015, 39, 1539–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riant, E.; Waget, A.; Cogo, H.; Arnal, J.-F.; Burcelin, R.; Gourdy, P. Estrogens Protect against High-Fat Diet-Induced Insulin Resistance and Glucose Intolerance in Mice. Endocrinology 2009, 150, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coppola, I.; Brouwers, B.; Meulemans, S.; Ramos-Molina, B.; Creemers, J.W.M. Differential Effects of Furin Deficiency on Insulin Receptor Processing and Glucose Control in Liver and Pancreatic β Cells of Mice. Int. J. Mol. Sci. 2021, 22, 6344. https://doi.org/10.3390/ijms22126344
Coppola I, Brouwers B, Meulemans S, Ramos-Molina B, Creemers JWM. Differential Effects of Furin Deficiency on Insulin Receptor Processing and Glucose Control in Liver and Pancreatic β Cells of Mice. International Journal of Molecular Sciences. 2021; 22(12):6344. https://doi.org/10.3390/ijms22126344
Chicago/Turabian StyleCoppola, Ilaria, Bas Brouwers, Sandra Meulemans, Bruno Ramos-Molina, and John W. M. Creemers. 2021. "Differential Effects of Furin Deficiency on Insulin Receptor Processing and Glucose Control in Liver and Pancreatic β Cells of Mice" International Journal of Molecular Sciences 22, no. 12: 6344. https://doi.org/10.3390/ijms22126344
APA StyleCoppola, I., Brouwers, B., Meulemans, S., Ramos-Molina, B., & Creemers, J. W. M. (2021). Differential Effects of Furin Deficiency on Insulin Receptor Processing and Glucose Control in Liver and Pancreatic β Cells of Mice. International Journal of Molecular Sciences, 22(12), 6344. https://doi.org/10.3390/ijms22126344