
The Functional Consequences of the Novel Ribosomal Pausing Site in SARS-CoV-2 Spike Glycoprotein RNA

 , ,
, , 

Abstract
:
1. Introduction
2. Results
2.1. Computational Analysis of the CCTCGGCGGGCA (-PRRA-) Insertion
2.2. Description of Spike Protein Constructs
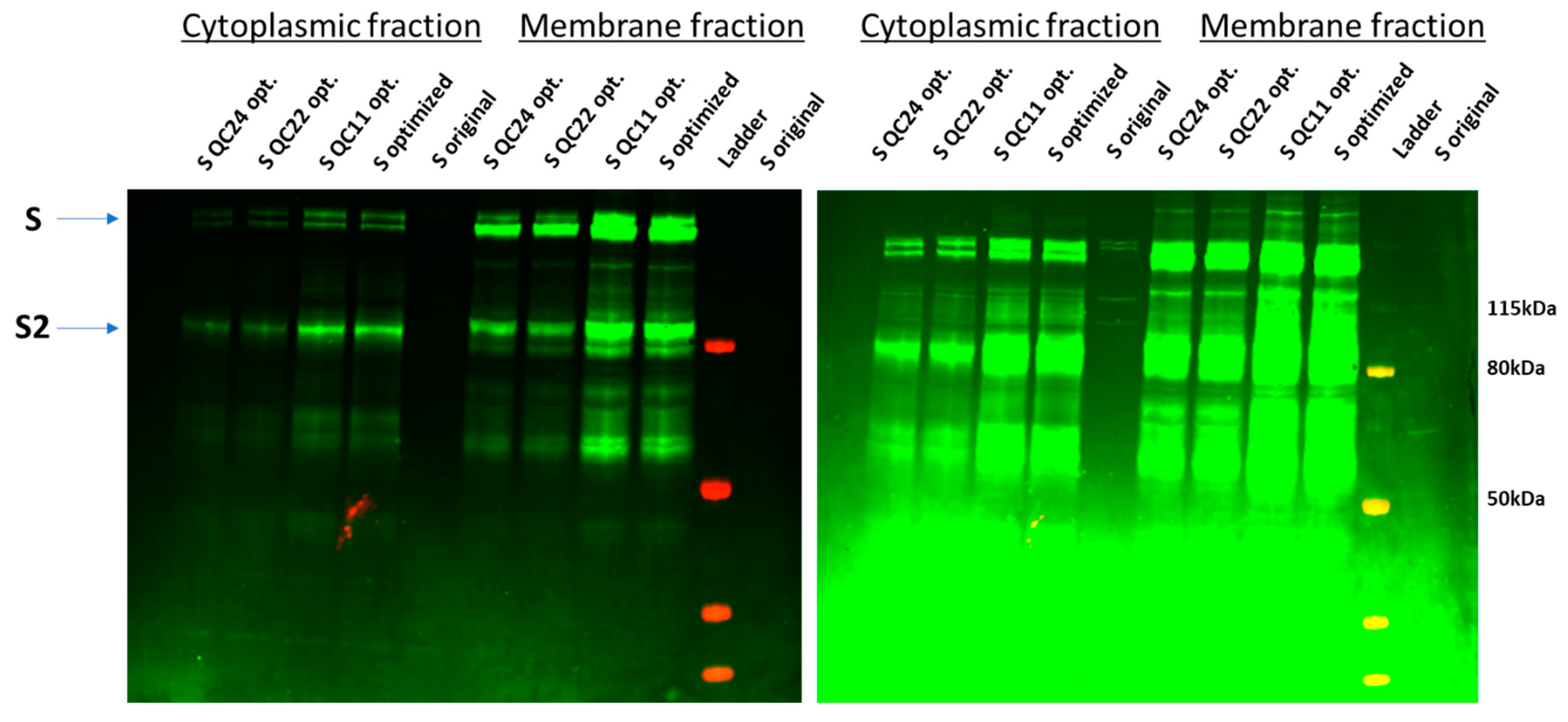
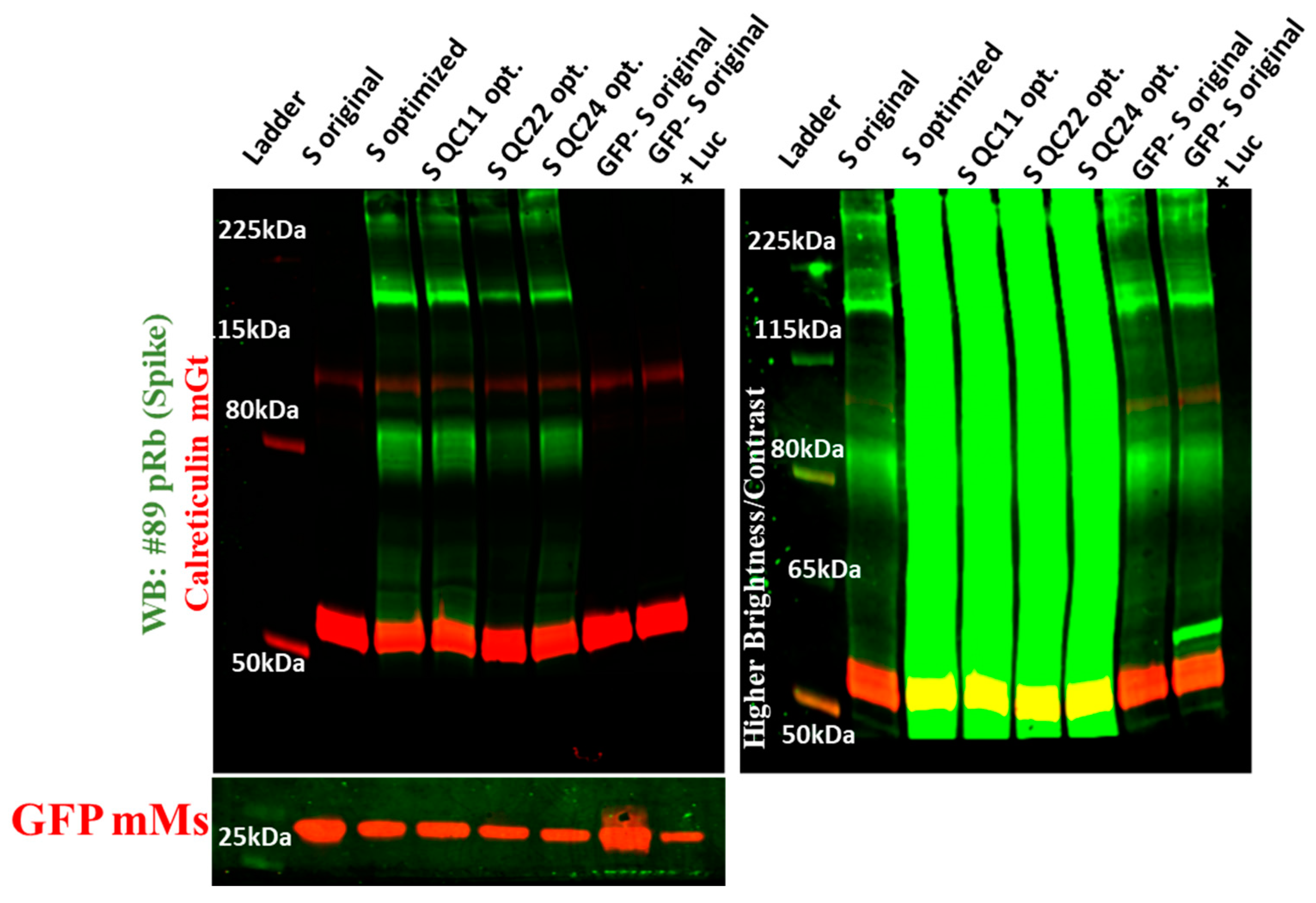
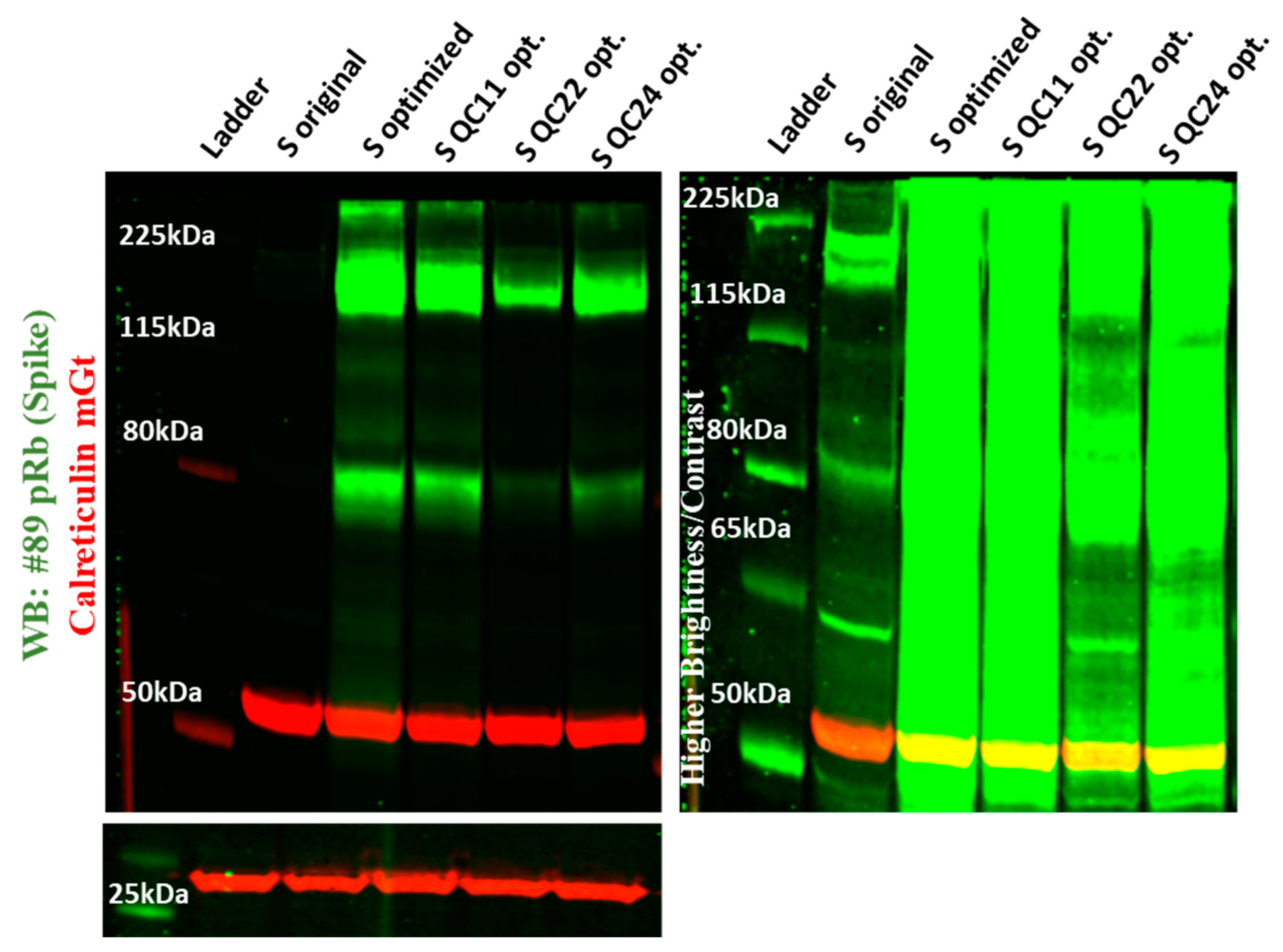
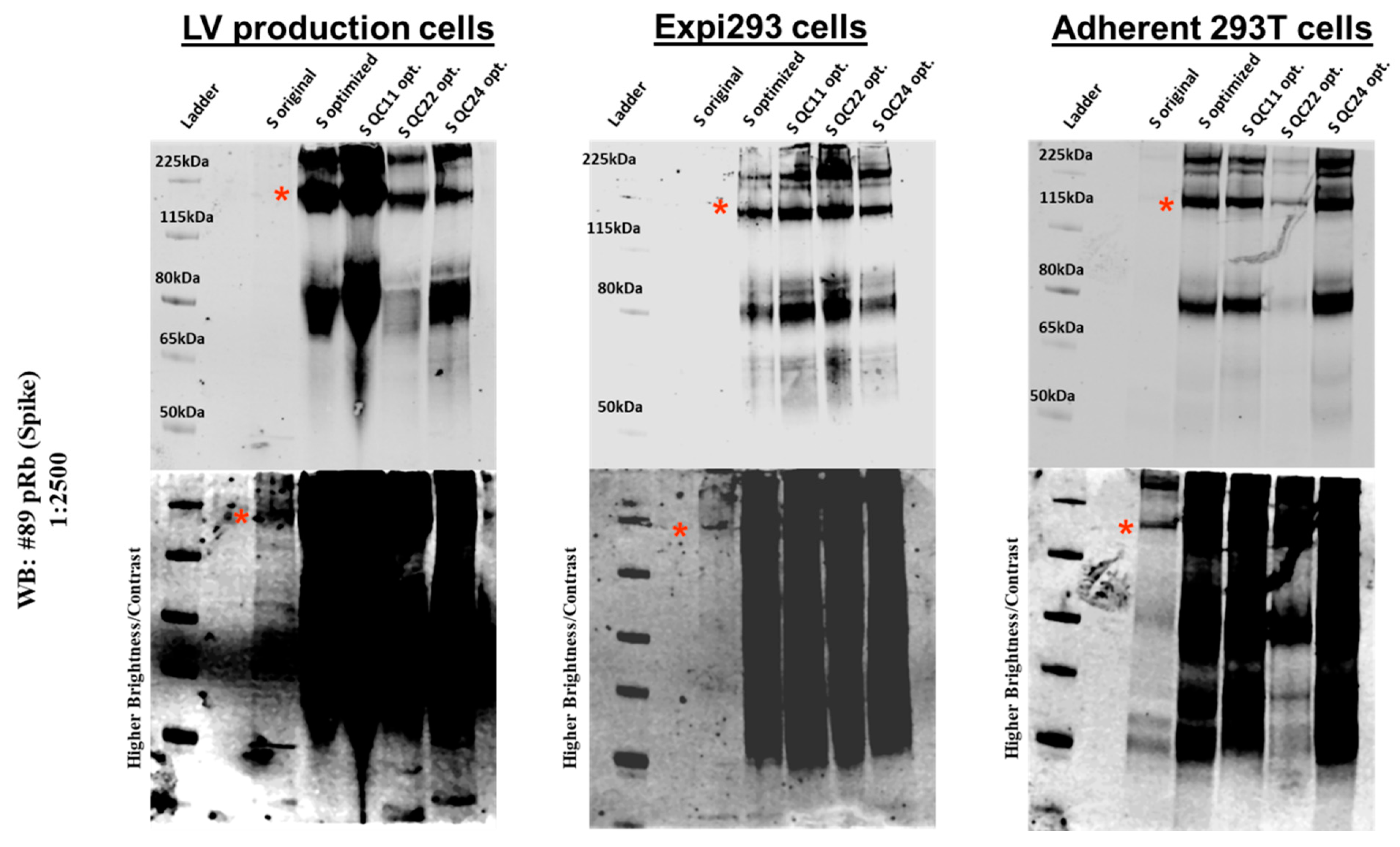
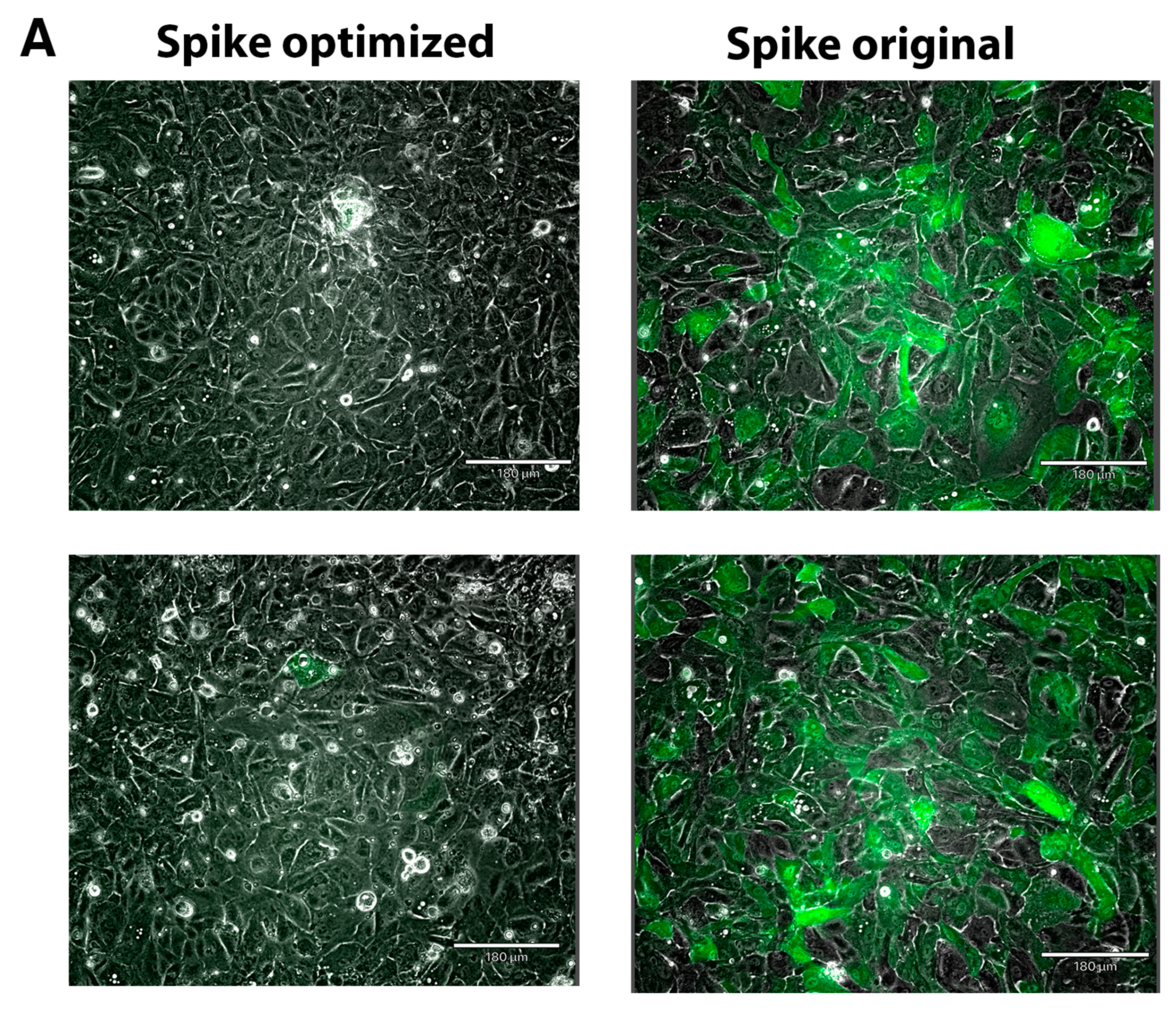
2.3. Expression of Spike Protein (Various Constructs) in Different Cell Lines
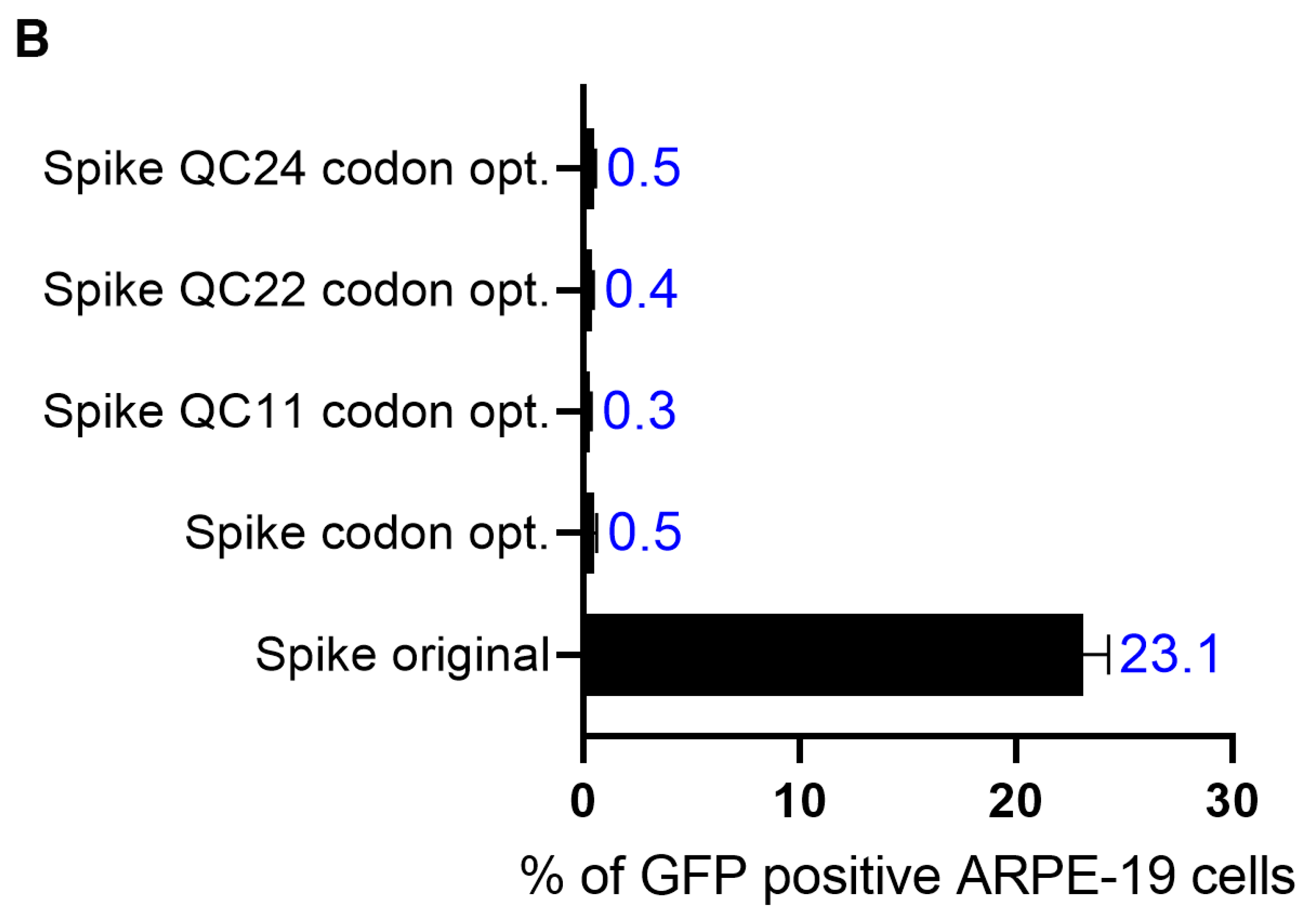
2.4. The Effect of the Novel Predicted Pausing Site on Expression of SARS-CoV-2 Spike Glycoprotein Variants in Lentiviral Pseudotypes
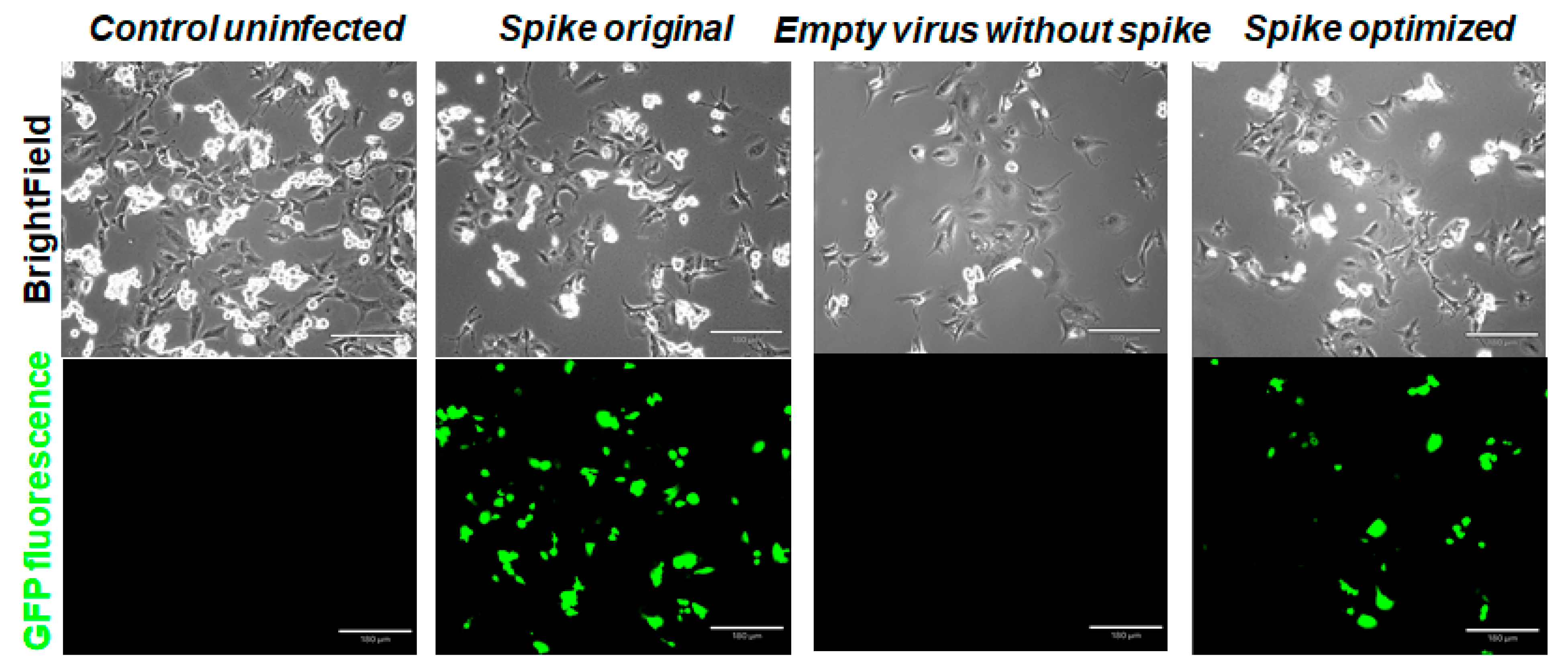
2.5. Infection of Various Cells with Spike Protein Variant Pseudotyped Particles
3. Discussion
4. Materials and Methods
4.1. Computational Analysis
4.2. Constructs and Plasmids
4.3. Cell Lines
4.4. Production of SARS-CoV-2 S Protein Pseudovirions
4.5. Measurement of Physical and Infectious Viral Titer
4.6. Immunoblot Analysis of S Proteins
4.7. Mass Spectrometry Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Cyranoski, D. Profile of a killer: The complex biology powering the coronavirus pandemic. Nature 2020, 581, 22–26. [Google Scholar] [CrossRef]
- Baranov, P.V.; Henderson, C.M.; Anderson, C.B.; Gesteland, R.F.; Atkins, J.F.; Howard, M.T. Programmed ribosomal frameshifting in decoding the SARS-CoV genome. Virology 2005, 332, 498–510. [Google Scholar] [CrossRef] [Green Version]
- Puray-Chavez, M.; Tenneti, K.; Vuong, H.R.; Lee, N.; Liu, Y.; Horani, A.; Huang, T.; Case, J.B.; Yang, W.; Diamond, M.S.; et al. The translational landscape of SARS-CoV-2 and infected cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lopinski, J.D.; Dinman, J.D.; Bruenn, J.A. Kinetics of ribosomal pausing during programmed -1 translational frameshifting. Mol. Cell Biol. 2000, 20, 1095–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, V.; Zoller, S.; Anisimova, M. Accounting for Programmed Ribosomal Frameshifting in the Computation of Codon Usage Bias Indices. G3 (Bethesda) 2018, 8, 3173–3183. [Google Scholar] [CrossRef]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell. Biol. 2018, 19, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Chantawannakul, P.; Cutler, R.W. Convergent host-parasite codon usage between honeybee and bee associated viral genomes. J. Invertebr. Pathol. 2008, 98, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Alias, X.; Benisty, H.; Schaefer, M.H.; Serrano, L. Translational adaptation of human viruses to the tissues they infect. Cell Rep. 2021, 34, 108872. [Google Scholar] [CrossRef] [PubMed]
- Lopes, E.N.; Fonseca, V.; Frias, D.; Tosta, S.; Salgado, A.; Assuncao Vialle, R.; Paulo Eduardo, T.S.; Barreto, F.K.; Ariston de Azevedo, V.; Guarino, M.; et al. Betacoronaviruses genome analysis reveals evolution toward specific codons usage: Implications for SARS-CoV-2 mitigation strategies. J. Med. Virol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wu, P.; Deng, S.; Zhang, H.; Hou, Y.; Hu, Z.; Zhang, J.; Chen, X.; Yang, J.R. Dissimilation of synonymous codon usage bias in virus-host coevolution due to translational selection. Nat. Ecol. Evol. 2020, 4, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, Y.; Kawamata, K.; Haraguchi, T.; Chikashige, Y. Codon usage bias is correlated with gene expression levels in the fission yeast Schizosaccharomyces pombe. Genes Cells 2009, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Buragohain, L.; Borah, P. Analysis of codon usage of severe acute respiratory syndrome corona virus 2 (SARS-CoV-2) and its adaptability in dog. Virus Res. 2020, 288, 198113. [Google Scholar] [CrossRef] [PubMed]
- Pollock, D.D.; Castoe, T.A.; Perry, B.W.; Lytras, S.; Wade, K.J.; Robertson, D.L.; Holmes, E.C.; Boni, M.F.; Kosakovsky Pond, S.L.; Parry, R.; et al. Viral CpG Deficiency Provides No Evidence That Dogs Were Intermediate Hosts for SARS-CoV-2. Mol. Biol. Evol. 2020, 37, 2706–2710. [Google Scholar] [CrossRef]
- Woo, P.C.; Wong, B.H.; Huang, Y.; Lau, S.K.; Yuen, K.Y. Cytosine deamination and selection of CpG suppressed clones are the two major independent biological forces that shape codon usage bias in coronaviruses. Virology 2007, 369, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Xia, X. Extreme Genomic CpG Deficiency in SARS-CoV-2 and Evasion of Host Antiviral Defense. Mol. Biol. Evol. 2020, 37, 2699–2705. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Rogozin, I.B. Getting positive about selection. Genome Biol. 2003, 4, 331. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Klimczak, L.J.; Randall, T.A.; Saini, N.; Li, J.L.; Gordenin, D.A. Similarity between mutation spectra in hypermutated genomes of rubella virus and in SARS-CoV-2 genomes accumulated during the COVID-19 pandemic. PLoS ONE 2020, 15, e0237689. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Collart, M.A.; Weiss, B. Ribosome pausing, a dangerous necessity for co-translational events. Nucleic Acids Res. 2020, 48, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faure, G.; Ogurtsov, A.Y.; Shabalina, S.A.; Koonin, E.V. Adaptation of mRNA structure to control protein folding. RNA Biol. 2017, 14, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A. A pause for thought along the co-translational folding pathway. Trends Biochem. Sci. 2009, 34, 16–24. [Google Scholar] [CrossRef]
- Waudby, C.A.; Dobson, C.M.; Christodoulou, J. Nature and Regulation of Protein Folding on the Ribosome. Trends Biochem. Sci. 2019, 44, 914–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeuchi, K.; Izawa, T.; Inada, T. Recent Progress on the Molecular Mechanism of Quality Controls Induced by Ribosome Stalling. Front. Genet. 2018, 9, 743. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Peterson, A.; Zinshteyn, B.; Regot, S.; Green, R. Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell 2020, 182, 404–416.e14. [Google Scholar] [CrossRef]
- Arpat, A.B.; Liechti, A.; De Matos, M.; Dreos, R.; Janich, P.; Gatfield, D. Transcriptome-wide sites of collided ribosomes reveal principles of translational pausing. Genome Res. 2020, 30, 985–999. [Google Scholar] [CrossRef]
- Zhao, J.; Qin, B.; Nikolay, R.; Spahn, C.M.T.; Zhang, G. Translatomics: The Global View of Translation. Int. J. Mol. Sci. 2019, 20, 212. [Google Scholar] [CrossRef] [Green Version]
- Rodnina, M.V. The ribosome in action: Tuning of translational efficiency and protein folding. Protein Sci. 2016, 25, 1390–1406. [Google Scholar] [CrossRef] [Green Version]
- Hussmann, J.A.; Patchett, S.; Johnson, A.; Sawyer, S.; Press, W.H. Understanding Biases in Ribosome Profiling Experiments Reveals Signatures of Translation Dynamics in Yeast. PLoS Genet. 2015, 11, e1005732. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, D.E.; Shah, P.; Eichhorn, S.W.; Hussmann, J.A.; Plotkin, J.B.; Bartel, D.P. Improved Ribosome-Footprint and mRNA Measurements Provide Insights into Dynamics and Regulation of Yeast Translation. Cell Rep. 2016, 14, 1787–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albers, S.; Czech, A. Exploiting tRNAs to Boost Virulence. Life 2016, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Weringh, A.; Ragonnet-Cronin, M.; Pranckeviciene, E.; Pavon-Eternod, M.; Kleiman, L.; Xia, X. HIV-1 modulates the tRNA pool to improve translation efficiency. Mol. Biol. Evol. 2011, 28, 1827–1834. [Google Scholar] [CrossRef] [Green Version]
- Davidson, A.D.; Williamson, M.K.; Lewis, S.; Shoemark, D.; Carroll, M.W.; Heesom, K.J.; Zambon, M.; Ellis, J.; Lewis, P.A.; Hiscox, J.A.; et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; et al. The coding capacity of SARS-CoV-2. Nature 2021, 589, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, L.; Luo, M.; Liang, B. Genomic characterization and evolution of SARS-CoV-2 of a Canadian population. PLoS ONE 2021, 16, e0247799. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortola, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef]
- Plante, J.A.; Mitchell, B.M.; Plante, K.S.; Debbink, K.; Weaver, S.C.; Menachery, V.D. The variant gambit: COVID-19’s next move. Cell Host Microbe 2021, 29, 508–515. [Google Scholar] [CrossRef]
- Davidson, S.K.; Hunt, L.A. Unusual neutral oligosaccharides in mature Sindbis virus glycoproteins are synthesized from truncated precursor oligosaccharides in Chinese hamster ovary cells. J. Gen. Virol. 1983, 64 Pt 3, 613–625. [Google Scholar] [CrossRef]
- Davidson, S.K.; Hunt, L.A. Sindbis virus glycoproteins are abnormally glycosylated in Chinese hamster ovary cells deprived of glucose. J. Gen. Virol. 1985, 66, 1457–1468. [Google Scholar] [CrossRef]
- Yang, Q.; Hughes, T.A.; Kelkar, A.; Yu, X.; Cheng, K.; Park, S.; Huang, W.C.; Lovell, J.F.; Neelamegham, S. Inhibition of SARS-CoV-2 viral entry upon blocking N- and O-glycan elaboration. Elife 2020, 9. [Google Scholar] [CrossRef]
- Zhang, L.; Mann, M.; Syed, Z.; Reynolds, H.M.; Tian, E.; Samara, N.L.; Zeldin, D.C.; Tabak, L.A.; Ten Hagen, K.G. Furin cleavage of the SARS-CoV-2 spike is modulated by O-glycosylation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Alexaki, A.; Kames, J.; Holcomb, D.D.; Athey, J.; Santana-Quintero, L.V.; Lam, P.V.N.; Hamasaki-Katagiri, N.; Osipova, E.; Simonyan, V.; Bar, H.; et al. Codon and Codon-Pair Usage Tables (CoCoPUTs): Facilitating Genetic Variation Analyses and Recombinant Gene Design. J. Mol. Biol. 2019, 431, 2434–2441. [Google Scholar] [CrossRef]
- Kimchi-Sarfaty, C.; Oh, J.M.; Kim, I.W.; Sauna, Z.E.; Calcagno, A.M.; Ambudkar, S.V.; Gottesman, M.M. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 2007, 315, 525–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, C.; Carrea, A.; Diambra, L. Bicodon bias can determine the role of synonymous SNPs in human diseases. BMC Genom. 2017, 18, 227. [Google Scholar] [CrossRef] [Green Version]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Torre-Ubieta, L.; Won, H.; Stein, J.L.; Geschwind, D.H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 2016, 22, 345–361. [Google Scholar] [CrossRef]
- Poliakov, E.; Koonin, E.V.; Rogozin, I.B. Impairment of translation in neurons as a putative causative factor for autism. Biol. Direct 2014, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogozin, I.B.; Gertz, E.M.; Baranov, P.V.; Poliakov, E.; Schaffer, A.A. Genome-Wide Changes in Protein Translation Efficiency Are Associated with Autism. Genome Biol. Evol. 2018, 10, 1902–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech, A.; Fedyunin, I.; Zhang, G.; Ignatova, Z. Silent mutations in sight: Co-variations in tRNA abundance as a key to unravel consequences of silent mutations. Mol. Biosyst. 2010, 6, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Kames, J.; Holcomb, D.D.; Kimchi, O.; DiCuccio, M.; Hamasaki-Katagiri, N.; Wang, T.; Komar, A.A.; Alexaki, A.; Kimchi-Sarfaty, C. Sequence analysis of SARS-CoV-2 genome reveals features important for vaccine design. Sci. Rep. 2020, 10, 15643. [Google Scholar] [CrossRef]
- Keller, T.L.; Zocco, D.; Sundrud, M.S.; Hendrick, M.; Edenius, M.; Yum, J.; Kim, Y.J.; Lee, H.K.; Cortese, J.F.; Wirth, D.F.; et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 2012, 8, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, D.R.; Clausen, T.M.; Nora, C.; Cribbs, A.P.; Denardo, A.; Clark, A.E.; Garretson, A.F.; Coker, J.K.C.; Narayanan, A.; Majowicz, S.A.; et al. The Prolyl-tRNA Synthetase Inhibitor Halofuginone Inhibits SARS-CoV-2 Infection. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gould, S.J. Tempo and mode in the macroevolutionary reconstruction of Darwinism. Proc. Natl. Acad. Sci. USA 1994, 91, 6764–6771. [Google Scholar] [CrossRef] [Green Version]
- Heasley, L.R.; Sampaio, N.M.V.; Argueso, J.L. Systemic and rapid restructuring of the genome: A new perspective on punctuated equilibrium. Curr. Genet. 2020. [Google Scholar] [CrossRef]
- Gussow, A.B.; Auslander, N.; Faure, G.; Wolf, Y.I.; Zhang, F.; Koonin, E.V. Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. Proc. Natl. Acad. Sci. USA 2020, 117, 15193–15199. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic. Acids. Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Geraerts, M.; Willems, S.; Baekelandt, V.; Debyser, Z.; Gijsbers, R. Comparison of lentiviral vector titration methods. BMC Biotechnol. 2006, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Yu, J.; Liu, T.; Defnet, A.E.; Zalesak, S.; Farese, A.M.; MacVittie, T.J.; Kane, M.A. Proteomics of Non-human Primate Plasma after Partial-body Radiation with Minimal Bone Marrow Sparing. Health Phys. 2020, 119, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.K.; Fischer, B.; Grossmann, J.; Maccoss, M.J. A fast SEQUEST cross correlation algorithm. J. Proteome Res. 2008, 7, 4598–4602. [Google Scholar] [CrossRef] [PubMed]
- Dorfer, V.; Pichler, P.; Stranzl, T.; Stadlmann, J.; Taus, T.; Winkler, S.; Mechtler, K. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J. Proteome Res 2014, 13, 3679–3684. [Google Scholar] [CrossRef] [PubMed]
- Kall, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4, 923–925. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 48 Hours | LV Cells | Expi 293 Cells | HEK293T Cells |
|---|---|---|---|
| qPCR (WPRE) | 0.99961 | 0.99951 | 0.87737 |
| qPCR (LTR) | 0.99866 | 0.99578 | 0.55399 |
| p24 | 0.99995 | 0.98458 | 0.64395 |
| Cell Line | Total DNA µg | Media mL | Transfection Reagent |
|---|---|---|---|
| HEK293T | 30 | 30 | FuGENE 6® |
| LV-MAX | 70 | 30 | LV-MAX |
| Expi293F | 30 | 30 | 293fectin™ |
| NAME | SEQUENCE 5′-3′ |
|---|---|
| LTR-fw | TGTGTGCCCGTCTGTTGTGT |
| LTR-rev | GAGTCCTGCGTCGAGAGAGC |
| LTR-probe | 5′-FAM-CAGTGGCGCCCGAACAGGGA-TAMRA-3 |
| WPRE-fw | CCGTTGTCAGGCAACGTG |
| WPRE-rev | AGCTGACAGGTGGTGGCAAT |
| WPRE-probe | 5′-FAM- TGCTGACGCAACCCCCACTGGT-TAMRA-3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Postnikova, O.A.; Uppal, S.; Huang, W.; Kane, M.A.; Villasmil, R.; Rogozin, I.B.; Poliakov, E.; Redmond, T.M. The Functional Consequences of the Novel Ribosomal Pausing Site in SARS-CoV-2 Spike Glycoprotein RNA. Int. J. Mol. Sci. 2021, 22, 6490. https://doi.org/10.3390/ijms22126490
Postnikova OA, Uppal S, Huang W, Kane MA, Villasmil R, Rogozin IB, Poliakov E, Redmond TM. The Functional Consequences of the Novel Ribosomal Pausing Site in SARS-CoV-2 Spike Glycoprotein RNA. International Journal of Molecular Sciences. 2021; 22(12):6490. https://doi.org/10.3390/ijms22126490
Chicago/Turabian StylePostnikova, Olga A., Sheetal Uppal, Weiliang Huang, Maureen A. Kane, Rafael Villasmil, Igor B. Rogozin, Eugenia Poliakov, and T. Michael Redmond. 2021. "The Functional Consequences of the Novel Ribosomal Pausing Site in SARS-CoV-2 Spike Glycoprotein RNA" International Journal of Molecular Sciences 22, no. 12: 6490. https://doi.org/10.3390/ijms22126490
APA StylePostnikova, O. A., Uppal, S., Huang, W., Kane, M. A., Villasmil, R., Rogozin, I. B., Poliakov, E., & Redmond, T. M. (2021). The Functional Consequences of the Novel Ribosomal Pausing Site in SARS-CoV-2 Spike Glycoprotein RNA. International Journal of Molecular Sciences, 22(12), 6490. https://doi.org/10.3390/ijms22126490