
The Journey of DDR1 and DDR2 Kinase Inhibitors as Rising Stars in the Fight Against Cancer




{kind=link}
{kind=link}
{kind=link}
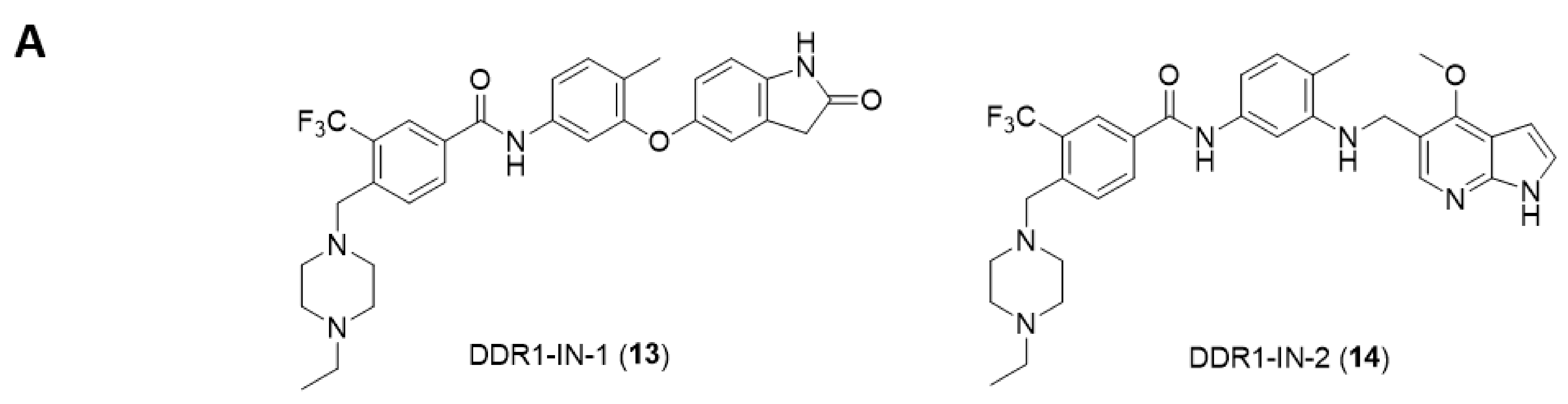{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
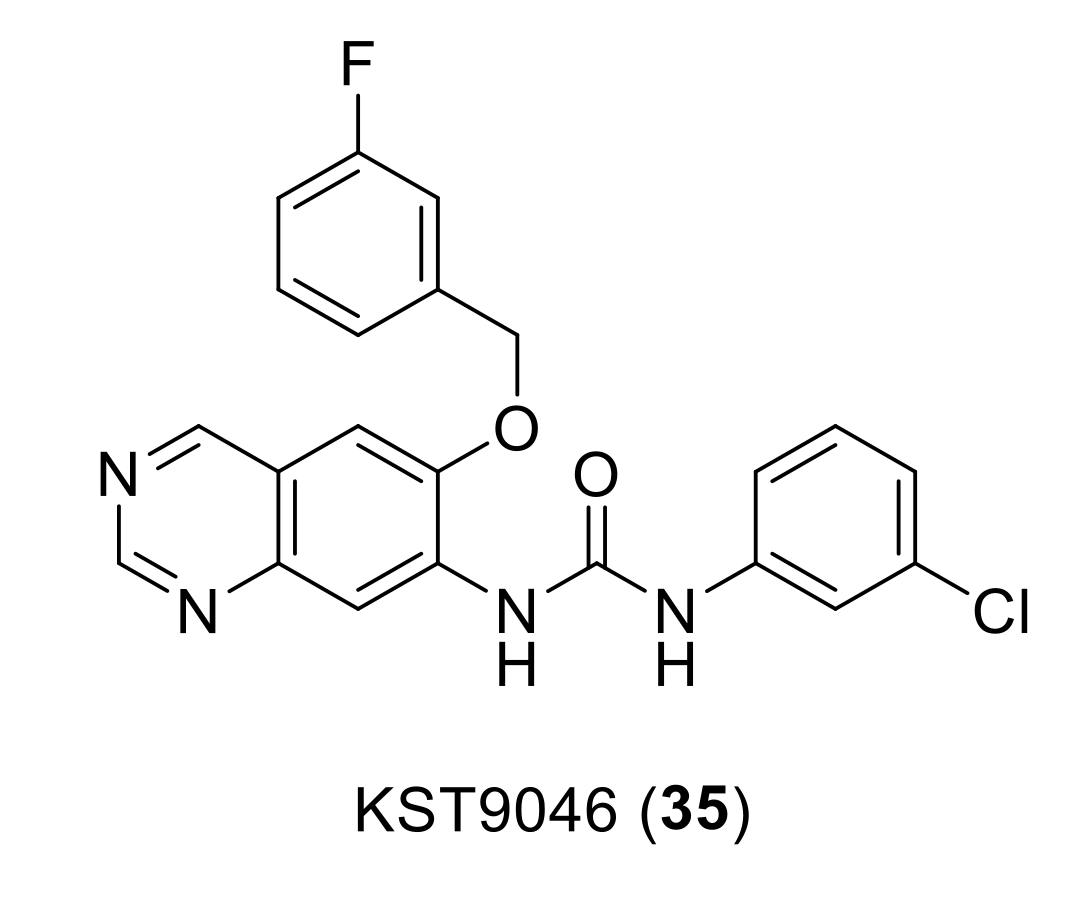{kind=link}
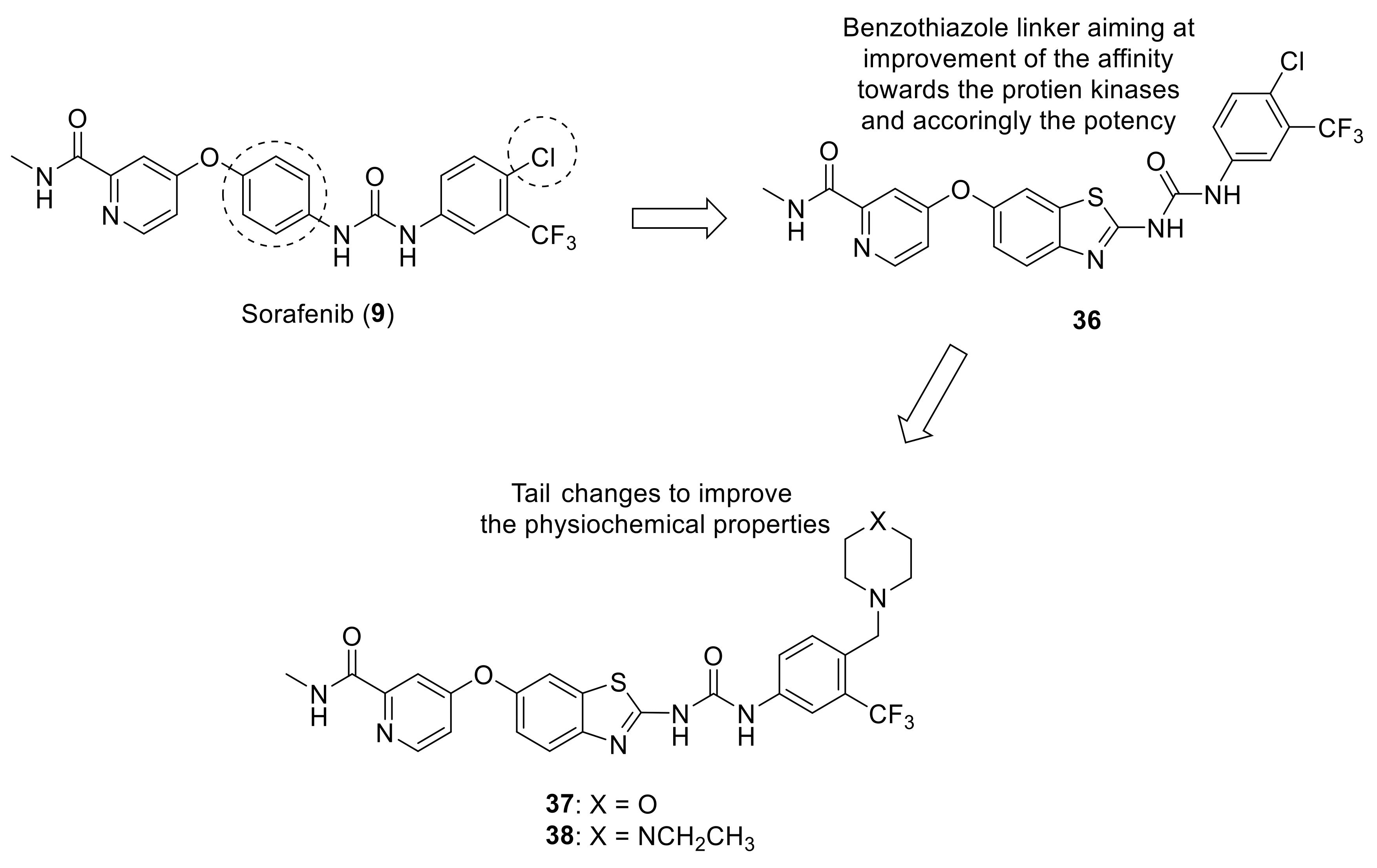{kind=link}
{kind=link}
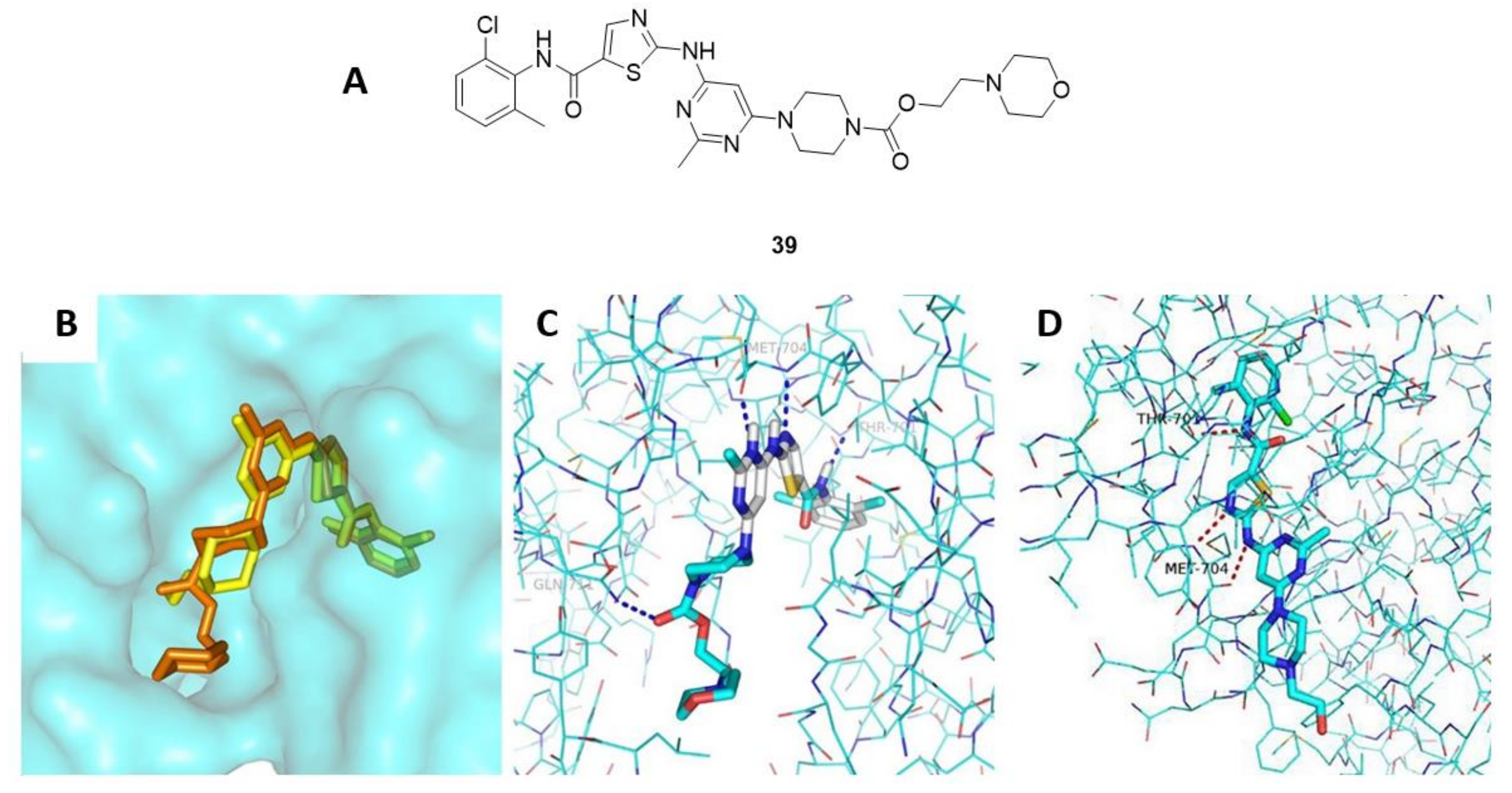{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structure and Activation of DDR Kinase
3. Biological Role of DDR
4. Role of DDR in Cancer
5. Role of DDR in Inflammation and Neurodegenerative Disorders
6. Small Molecule DDR Kinase Inhibitors
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fridman, R.; Agarwal, G. New Concepts on the Interactions of Discoidin Domain Receptors with Collagen. Biochim. Biophys. Acta BBA Mol. Cell Res. 2019, 1866, 118527. [Google Scholar] [CrossRef] [PubMed]
- Henriet, E.; Sala, M.; Abou Hammoud, A.; Tuariihionoa, A.; Di Martino, J.; Ros, M.; Saltel, F. Multitasking discoidin domain receptors are involved in several and specific hallmarks of cancer. Cell Adh. Migr. 2018, 12, 363–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, Y.-C.; Lin, H.-H.; Tang, M.-J. Dichotomy of the function of DDR1 in cells and disease progression. Biochim. Biophys. Acta BBA Mol. Cell Res 2019, 1866, 118473. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.J.; Hebron, M.; Balaraman, K.; Shi, W.; Missner, A.A.; Greenzaid, J.D.; Chiu, T.L.; Ullman, C.; Weatherdon, E.; Duka, V.; et al. Discoidin Domain Receptor 1 is a therapeutic target for neurodegenerative diseases. Hum. Mol. Genet. 2020, 29, 2882–2898. [Google Scholar] [CrossRef]
- Guo, J.; Zhang, Z.; Ding, K. A patent review of discoidin domain receptor 1 (DDR1) modulators (2014-present). Expert Opin. Ther. Pat. 2020, 30, 341–350. [Google Scholar] [CrossRef]
- Carafoli, F.; Mayer, M.C.; Shiraishi, K.; Pecheva, M.A.; Chan, L.Y.; Nan, R.; Leitinger, B.; Hohenester, E. Structure of the discoidin domain receptor 1 extracellular region bound to an inhibitory Fab fragment reveals features important for signaling. Structure 2012, 20, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Day, E.; Waters, B.; Spiegel, K.; Alnadaf, T.; Manley, P.W.; Buchdunger, E.; Walker, C.; Jarai, G. Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur. J. Pharmacol. 2008, 599, 44–53. [Google Scholar] [CrossRef]
- Canning, P.; Tan, L.; Chu, K.; Lee, S.W.; Gray, N.S.; Bullock, A.N. Structural mechanisms determining inhibition of the collagen receptor DDR1 by selective and multi-targeted type II kinase inhibitors. J. Mol. Biol. 2014, 426, 2457–2470. [Google Scholar] [CrossRef] [Green Version]
- Leitinger, B. Discoidin domain receptor functions in physiological and pathological conditions. Int. Rev. Cell Mol. Biol. 2014, 310, 39–87. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.-L.; Valiathan, R.R.; Arkwright, R.; Sohail, A.; Mihai, C.; Kumarasiri, M.; Mahasenan, K.V.; Mobashery, S.; Huang, P.; Agarwal, G.; et al. Discoidin domain receptors: Unique receptor tyrosine kinases in collagen-mediated signaling. J. Biol. Chem. 2013, 288, 7430–7437. [Google Scholar] [CrossRef] [Green Version]
- Chappell, W.H.; Candido, S.; Abrams, S.L.; Akula, S.M.; Steelman, L.S.; Martelli, A.M.; Ratti, S.; Cocco, L.; Cervello, M.; Montalto, G.; et al. Influences of TP53 and the anti-aging DDR1 receptor in controlling Raf/MEK/ERK and PI3K/Akt expression and chemotherapeutic drug sensitivity in prostate cancer cell lines. Aging 2020, 12, 10194–10210. [Google Scholar] [CrossRef]
- Vella, V.; Malaguarnera, R. The Emerging Role of Insulin Receptor Isoforms in Thyroid Cancer: Clinical Implications and New Perspectives. Intern. J. Mol. Sci. 2018, 19, 3814. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhou, J.; Li, J. Discoidin domain receptors orchestrate cancer progression: A focus on cancer therapies. Cancer Sci. 2020. [Google Scholar] [CrossRef]
- Valiathan, R.R.; Marco, M.; Leitinger, B.; Kleer, C.G.; Fridman, R. Discoidin domain receptor tyrosine kinases: New players in cancer progression. Cancer Metastasis Rev. 2012, 31, 295–321. [Google Scholar] [CrossRef] [Green Version]
- Vogel, W.F.; Abdulhussein, R.; Ford, C.E. Sensing extracellular matrix: An update on discoidin domain receptor function. Cell. Signal. 2006, 18, 1108–1116. [Google Scholar] [CrossRef]
- Iwai, L.K.; Luczynski, M.T.; Huang, P.H. Discoidin domain receptors: A proteomic portrait. Cell. Mol. Life Sci. CMLS 2014, 71, 3269–3279. [Google Scholar] [CrossRef]
- Borza, C.M.; Pozzi, A. Discoidin domain receptors in disease. Matrix Biol. J. Inter. Soc. Matrix Biol. 2014, 34, 185–192. [Google Scholar] [CrossRef]
- Kothiwale, S.; Borza, C.M.; Lowe, E.W., Jr.; Pozzi, A.; Meiler, J. Discoidin domain receptor 1 (DDR1) kinase as target for structure-based drug discovery. Drug Discov. Today 2015, 20, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.C.; Lin, H.H.; Tang, M.J. A tale of two collagen receptors, integrin β1 and discoidin domain receptor 1, in epithelial cell differentiation. Am. J. Physiol. Cell Physiol. 2012, 303, C1207–C1217. [Google Scholar] [CrossRef] [Green Version]
- Kamohara, H.; Yamashiro, S.; Galligan, C.; Yoshimura, T. Discoidin domain receptor 1 isoform-a (DDR1alpha) promotes migration of leukocytes in three-dimensional collagen lattices. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 2724–2726. [Google Scholar] [CrossRef]
- Cario, M. DDR1 and DDR2 in skin. Cell Adh. Migr. 2018, 12, 386–393. [Google Scholar] [CrossRef]
- Vogel, W.F.; Aszódi, A.; Alves, F.; Pawson, T. Discoidin domain receptor 1 tyrosine kinase has an essential role in mammary gland development. Mol. Cell. Biol. 2001, 21, 2906–2917. [Google Scholar] [CrossRef] [Green Version]
- Faraci-Orf, E.; McFadden, C.; Vogel, W.F. DDR1 signaling is essential to sustain Stat5 function during lactogenesis. J. Cell. Biochem. 2006, 97, 109–121. [Google Scholar] [CrossRef]
- Meyer zum Gottesberge, A.M.; Gross, O.; Becker-Lendzian, U.; Massing, T.; Vogel, W.F. Inner ear defects and hearing loss in mice lacking the collagen receptor DDR1. Lab. Investig. J. Tech. Met. Pathol. 2008, 88, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Dorison, A.; Dussaule, J.C.; Chatziantoniou, C. The Role of Discoidin Domain Receptor 1 in Inflammation, Fibrosis and Renal Disease. Nephron 2017, 137, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Chetoui, N.; El Azreq, M.A.; Boisvert, M.; Bergeron, M.; Aoudjit, F. Discoidin domain receptor 1 expression in activated T cells is regulated by the ERK MAP kinase signaling pathway. J. Cell. Biochem. 2011, 112, 3666–3674. [Google Scholar] [CrossRef]
- Li, S.W.; Prockop, D.J.; Helminen, H.; Fässler, R.; Lapveteläinen, T.; Kiraly, K.; Peltarri, A.; Arokoski, J.; Lui, H.; Arita, M.; et al. Transgenic mice with targeted inactivation of the Col2 alpha 1 gene for collagen II develop a skeleton with membranous and periosteal bone but no endochondral bone. Genes Dev. 1995, 9, 2821–2830. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Li, S.W.; Helminen, H.J.; Khillan, J.S.; Bao, Y.; Prockop, D.J. Apoptosis of chondrocytes in transgenic mice lacking collagen II. Exp. Cell Res. 1997, 235, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Labrador, J.P.; Azcoitia, V.; Tuckermann, J.; Lin, C.; Olaso, E.; Mañes, S.; Brückner, K.; Goergen, J.L.; Lemke, G.; Yancopoulos, G.; et al. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep. 2001, 2, 446–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kano, K.; Kitamura, A.; Matsuwaki, T.; Morimatsu, M.; Naito, K. Discoidin domain receptor 2 (DDR2) is required for maintenance of spermatogenesis in male mice. Mol. Reprod. Dev. 2010, 77, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Borochowitz, Z.; Langer, L.O., Jr.; Gruber, H.E.; Lachman, R.; Katznelson, M.B.; Rimoin, D.L. Spondylo-meta-epiphyseal dysplasia (SMED), short limb-hand type: A congenital familial skeletal dysplasia with distinctive features and histopathology. Am. J. Med. Genet. 1993, 45, 320–326. [Google Scholar] [CrossRef]
- Kano, K.; Marín de Evsikova, C.; Young, J.; Wnek, C.; Maddatu, T.P.; Nishina, P.M.; Naggert, J.K. A novel dwarfism with gonadal dysfunction due to loss-of-function allele of the collagen receptor gene, Ddr2, in the mouse. Mol. Endocrin. 2008, 22, 1866–1880. [Google Scholar] [CrossRef] [Green Version]
- Bargal, R.; Cormier-Daire, V.; Ben-Neriah, Z.; Le Merrer, M.; Sosna, J.; Melki, J.; Zangen, D.H.; Smithson, S.F.; Borochowitz, Z.; Belostotsky, R.; et al. Mutations in DDR2 gene cause SMED with short limbs and abnormal calcifications. Am. J. Hum. Genet. 2009, 84, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Ali, B.R.; Xu, H.; Akawi, N.A.; John, A.; Karuvantevida, N.S.; Langer, R.; Al-Gazali, L.; Leitinger, B. Trafficking defects and loss of ligand binding are the underlying causes of all reported DDR2 missense mutations found in SMED-SL patients. Hum. Mol. Genet. 2010, 19, 2239–2250. [Google Scholar] [CrossRef]
- Al-Kindi, A.; Kizhakkedath, P.; Xu, H.; John, A.; Sayegh, A.A.; Ganesh, A.; Al-Awadi, M.; Al-Anbouri, L.; Al-Gazali, L.; Leitinger, B.; et al. A novel mutation in DDR2 causing spondylo-meta-epiphyseal dysplasia with short limbs and abnormal calcifications (SMED-SL) results in defective intra-cellular trafficking. BMC Med. Genet. 2014, 15, 42. [Google Scholar] [CrossRef] [Green Version]
- Olaso, E.; Ikeda, K.; Eng, F.J.; Xu, L.; Wang, L.H.; Lin, H.C.; Friedman, S.L. DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells. J. Clin. Investig. 2001, 108, 1369–1378. [Google Scholar] [CrossRef]
- Herrera-Herrera, M.L.; Quezada-Calvillo, R. DDR2 plays a role in fibroblast migration independent of adhesion ligand and collagen activated DDR2 tyrosine kinase. Biochem. Biophys. Res. Commun. 2012, 429, 39–44. [Google Scholar] [CrossRef]
- Kim, D.; You, E.; Min, N.Y.; Lee, K.H.; Kim, H.K.; Rhee, S. Discoidin domain receptor 2 regulates the adhesion of fibroblasts to 3D collagen matrices. Int. J. Mol. Med. 2013, 31, 1113–1118. [Google Scholar] [CrossRef] [Green Version]
- Tu, M.M.; Lee, F.Y.F.; Jones, R.T.; Kimball, A.K.; Saravia, E.; Graziano, R.F.; Coleman, B.; Menard, K.; Yan, J.; Michaud, E.; et al. Targeting DDR2 enhances tumor response to anti-PD-1 immunotherapy. Sci. Adv. 2019, 5, eaav2437. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Baek, H.A.; Lee, H.J.; Park, H.S.; Jang, K.Y.; Kang, M.J.; Lee, D.G.; Lee, Y.C.; Moon, W.S.; Chung, M.J. Discoidin domain receptor 1 is associated with poor prognosis of non-small cell lung carcinomas. Oncol. Rep. 2010, 24, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Zhu, S.; Wang, Y.; Li, Y.; Ding, J.; Dai, J.; Cai, H.; Zhang, D.; Song, Y. Discoidin domain receptor 1 is associated with poor prognosis of non-small cell lung cancer and promotes cell invasion via epithelial-to-mesenchymal transition. Med. Oncol. 2013, 30, 626. [Google Scholar] [CrossRef]
- Valencia, K.; Ormazábal, C.; Zandueta, C.; Luis-Ravelo, D.; Antón, I.; Pajares, M.J.; Agorreta, J.; Montuenga, L.M.; Martínez-Canarias, S.; Leitinger, B.; et al. Inhibition of collagen receptor discoidin domain receptor-1 (DDR1) reduces cell survival, homing, and colonization in lung cancer bone metastasis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Davies, H.; Hunter, C.; Smith, R.; Stephens, P.; Greenman, C.; Bignell, G.; Teague, J.; Butler, A.; Edkins, S.; Stevens, C.; et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005, 65, 7591–7595. [Google Scholar] [CrossRef] [Green Version]
- Hammerman, P.S.; Sos, M.L.; Ramos, A.H.; Xu, C.; Dutt, A.; Zhou, W.; Brace, L.E.; Woods, B.A.; Lin, W.; Zhang, J.; et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 2011, 1, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Wasinski, B.; Sohail, A.; Bonfil, R.D.; Kim, S.; Saliganan, A.; Polin, L.; Bouhamdan, M.; Kim, H.C.; Prunotto, M.; Fridman, R. Discoidin Domain Receptors, DDR1b and DDR2, Promote Tumour Growth within Collagen but DDR1b Suppresses Experimental Lung Metastasis in HT1080 Xenografts. Sci. Rep. 2020, 10, 2309. [Google Scholar] [CrossRef]
- Couvelard, A.; Hu, J.; Steers, G.; O’Toole, D.; Sauvanet, A.; Belghiti, J.; Bedossa, P.; Gatter, K.; Ruszniewski, P.; Pezzella, F. Identification of potential therapeutic targets by gene-expression profiling in pancreatic endocrine tumors. Gastroenterology 2006, 131, 1597–1610. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Nakamura, M.; Ishida, E.; Higuchi, T.; Yamamoto, H.; Tsujikawa, K.; Konishi, N. Prostate cancer antigen-1 contributes to cell survival and invasion though discoidin receptor 1 in human prostate cancer. Cancer Sci. 2008, 99, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Barker, K.T.; Martindale, J.E.; Mitchell, P.J.; Kamalati, T.; Page, M.J.; Phippard, D.J.; Dale, T.C.; Gusterson, B.A.; Crompton, M.R. Expression patterns of the novel receptor-like tyrosine kinase, DDR, in human breast tumours. Oncogene 1995, 10, 569–575. [Google Scholar] [PubMed]
- Neuhaus, B.; Bühren, S.; Böck, B.; Alves, F.; Vogel, W.F.; Kiefer, F. Migration inhibition of mammary epithelial cells by Syk is blocked in the presence of DDR1 receptors. Cell. Mol. Life Sci. CMLS 2011, 68, 3757–3770. [Google Scholar] [CrossRef]
- Weiner, H.L.; Rothman, M.; Miller, D.C.; Ziff, E.B. Pediatric brain tumors express multiple receptor tyrosine kinases including novel cell adhesion kinases. Pediatric Neurosurg. 1996, 25, 64–71. [Google Scholar] [CrossRef]
- Weiner, H.L.; Huang, H.; Zagzag, D.; Boyce, H.; Lichtenbaum, R.; Ziff, E.B. Consistent and selective expression of the discoidin domain receptor-1 tyrosine kinase in human brain tumors. Neurosurgery 2000, 47, 1400–1409. [Google Scholar] [CrossRef]
- Heinzelmann-Schwarz, V.A.; Gardiner-Garden, M.; Henshall, S.M.; Scurry, J.; Scolyer, R.A.; Davies, M.J.; Heinzelmann, M.; Kalish, L.H.; Bali, A.; Kench, J.G.; et al. Overexpression of the cell adhesion molecules DDR1, Claudin 3, and Ep-CAM in metaplastic ovarian epithelium and ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 4427–4436. [Google Scholar] [CrossRef] [Green Version]
- Quan, J.; Yahata, T.; Adachi, S.; Yoshihara, K.; Tanaka, K. Identification of receptor tyrosine kinase, discoidin domain receptor 1 (DDR1), as a potential biomarker for serous ovarian cancer. Int. J. Mol. Sci. 2011, 12, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Cicinnati, V.R.; Zhang, X.; Iacob, S.; Weber, F.; Sotiropoulos, G.C.; Radtke, A.; Lu, M.; Paul, A.; Gerken, G.; et al. Role of microRNA-199a-5p and discoidin domain receptor 1 in human hepatocellular carcinoma invasion. Mol. Cancer 2010, 9, 227. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo-Carcedo, C.; Hooper, S.; Chaudhry, S.I.; Williamson, P.; Harrington, K.; Leitinger, B.; Sahai, E. Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat. Cell Biol. 2011, 13, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Matada, G.S.P.; Das, A.; Dhiwar, P.S.; Ghara, A. DDR1 and DDR2: A review on signaling pathway and small molecule inhibitors as an anticancer agent. Med. Chem. Res. 2021, 30, 535–551. [Google Scholar] [CrossRef]
- Gadiya, M.; Chakraborty, G. Signaling by discoidin domain receptor 1 in cancer metastasis. Cell Adh. Migr. 2018, 12, 315–323. [Google Scholar] [CrossRef]
- Chen, Y.L.; Tsai, W.H.; Ko, Y.C.; Lai, T.Y.; Cheng, A.J.; Shiah, S.G.; Hsiao, J.R.; Chang, J.Y.; Lin, S.F. Discoidin Domain Receptor-1 (DDR1) is Involved in Angiolymphatic Invasion in Oral Cancer. Cancers 2020, 12, 841. [Google Scholar] [CrossRef] [Green Version]
- Baltes, F.; Caspers, J.; Henze, S.; Schlesinger, M.; Bendas, G. Targeting Discoidin Domain Receptor 1 (DDR1) Signaling and Its Crosstalk with β(1)-integrin Emerges as a Key Factor for Breast Cancer Chemosensitization upon Collagen Type 1 Binding. Int. J. Mol. Sci. 2020, 21, 4956. [Google Scholar] [CrossRef] [PubMed]
- Reger de Moura, C.; Prunotto, M.; Sohail, A.; Battistella, M.; Jouenne, F.; Marbach, D.; Lebbé, C.; Fridman, R.; Mourah, S. Discoidin Domain Receptors in Melanoma: Potential Therapeutic Targets to Overcome MAPK Inhibitor Resistance. Front. Oncol. 2020, 10, 1748. [Google Scholar] [CrossRef]
- Lafitte, M.; Sirvent, A.; Roche, S. Collagen Kinase Receptors as Potential Therapeutic Targets in Metastatic Colon Cancer. Front. Oncol. 2020, 10, 125. [Google Scholar] [CrossRef]
- Ford, C.E.; Lau, S.K.; Zhu, C.Q.; Andersson, T.; Tsao, M.S.; Vogel, W.F. Expression and mutation analysis of the discoidin domain receptors 1 and 2 in non-small cell lung carcinoma. Br. J. Cancer 2007, 96, 808–814. [Google Scholar] [CrossRef]
- Reger de Moura, C.; Battistella, M.; Sohail, A.; Caudron, A.; Feugeas, J.P.; Podgorniak, M.P.; Pages, C.; Mazouz Dorval, S.; Marco, O.; Menashi, S.; et al. Discoidin domain receptors: A promising target in melanoma. Pigment Cell Melanoma Res. 2019, 32, 697–707. [Google Scholar] [CrossRef]
- Ma, Y.-S.; Wu, Z.-J.; Bai, R.-Z.; Dong, H.; Xie, B.-X.; Wu, X.-H.; Hang, X.-S.; Liu, A.-N.; Jiang, X.-H.; Wang, G.-R.; et al. DRR1 promotes glioblastoma cell invasion and epithelial-mesenchymal transition via regulating AKT activation. Cancer Lett. 2018, 423, 86–94. [Google Scholar] [CrossRef]
- Hur, H.; Ham, I.-H.; Lee, D.; Jin, H.; Aguilera, K.Y.; Oh, H.J.; Han, S.-U.; Kwon, J.E.; Kim, Y.-B.; Ding, K.; et al. Discoidin domain receptor 1 activity drives an aggressive phenotype in gastric carcinoma. BMC Cancer 2017, 17, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrogio, C.; Nadal, E.; Villanueva, A.; Gómez-López, G.; Cash, T.P.; Barbacid, M.; Santamaría, D. KRAS-driven lung adenocarcinoma: Combined DDR1/Notch inhibition as an effective therapy. ESMO Open 2016, 1, e000076. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Zhang, W.; Sun, T. DDR1 promotes breast tumor growth by suppressing antitumor immunity. Oncol. Rep. 2019, 42, 2844–2854. [Google Scholar] [CrossRef]
- Rodrigues, R.; Roque, L.; Espadinha, C.; Pinto, A.; Domingues, R.; Dinis, J.; Catarino, A.; Pereira, T.; Leite, V. Comparative genomic hybridization, BRAF, RAS, RET, and oligo-array analysis in aneuploid papillary thyroid carcinomas. Oncol. Rep. 2007, 18, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Squire, J.A.; Bayani, J.; Luk, C.; Unwin, L.; Tokunaga, J.; MacMillan, C.; Irish, J.; Brown, D.; Gullane, P.; Kamel-Reid, S. Molecular cytogenetic analysis of head and neck squamous cell carcinoma: By comparative genomic hybridization, spectral karyotyping, and expression array analysis. Head Neck 2002, 24, 874–887. [Google Scholar] [CrossRef]
- Renné, C.; Willenbrock, K.; Küppers, R.; Hansmann, M.L.; Bräuninger, A. Autocrine- and paracrine-activated receptor tyrosine kinases in classic Hodgkin lymphoma. Blood 2005, 105, 4051–4059. [Google Scholar] [CrossRef] [Green Version]
- Willenbrock, K.; Küppers, R.; Renné, C.; Brune, V.; Eckerle, S.; Weidmann, E.; Bräuninger, A.; Hansmann, M.L. Common features and differences in the transcriptome of large cell anaplastic lymphoma and classical Hodgkin’s lymphoma. Haematologica 2006, 91, 596–604. [Google Scholar]
- Yan, Z.; Jin, S.; Wei, Z.; Huilian, H.; Zhanhai, Y.; Yue, T.; Juan, L.; Jing, L.; Libo, Y.; Xu, L. Discoidin domain receptor 2 facilitates prostate cancer bone metastasis via regulating parathyroid hormone-related protein. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2014, 1842, 1350–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Lu, W.; Zhang, S.; Zhu, C.; Ren, T.; Zhu, T.; Zhao, H.; Liu, Y.; Su, J. Overexpression of DDR2 contributes to cell invasion and migration in head and neck squamous cell carcinoma. Cancer Biol. Ther. 2014, 15, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Corsa, C.A.; Ponik, S.M.; Prior, J.L.; Piwnica-Worms, D.; Eliceiri, K.W.; Keely, P.J.; Longmore, G.D. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat. Cell Biol. 2013, 15, 677–687. [Google Scholar] [CrossRef]
- Ren, T.; Zhang, J.; Zhang, J.; Liu, X.; Yao, L. Increased expression of discoidin domain receptor 2 (DDR2): A novel independent prognostic marker of worse outcome in breast cancer patients. Med. Oncol. 2013, 30, 397. [Google Scholar] [CrossRef]
- Ren, T.; Zhang, W.; Liu, X.; Zhao, H.; Zhang, J.; Zhang, J.; Li, X.; Zhang, Y.; Bu, X.; Shi, M.; et al. Discoidin domain receptor 2 (DDR2) promotes breast cancer cell metastasis and the mechanism implicates epithelial-mesenchymal transition programme under hypoxia. J. Pathol. 2014, 234, 526–537. [Google Scholar] [CrossRef]
- Loriaux, M.M.; Levine, R.L.; Tyner, J.W.; Fröhling, S.; Scholl, C.; Stoffregen, E.P.; Wernig, G.; Erickson, H.; Eide, C.A.; Berger, R.; et al. High-throughput sequence analysis of the tyrosine kinome in acute myeloid leukemia. Blood 2008, 111, 4788–4796. [Google Scholar] [CrossRef] [Green Version]
- Tomasson, M.H.; Xiang, Z.; Walgren, R.; Zhao, Y.; Kasai, Y.; Miner, T.; Ries, R.E.; Lubman, O.; Fremont, D.H.; McLellan, M.D.; et al. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood 2008, 111, 4797–4808. [Google Scholar] [CrossRef] [Green Version]
- Pitini, V.; Arrigo, C.; Di Mirto, C.; Mondello, P.; Altavilla, G. Response to dasatinib in a patient with SQCC of the lung harboring a discoid-receptor-2 and synchronous chronic myelogenous leukemia. Lung Cancer 2013, 82, 171–172. [Google Scholar] [CrossRef] [PubMed]
- Vogel, W. Discoidin domain receptors: Structural relations and functional implications. FASEB J. 1999, 13, S77–S82. [Google Scholar] [CrossRef] [PubMed]
- Castro-Sanchez, L.; Soto-Guzman, A.; Guaderrama-Diaz, M.; Cortes-Reynosa, P.; Salazar, E.P. Role of DDR1 in the gelatinases secretion induced by native type IV collagen in MDA-MB-231 breast cancer cells. Clin. Exp. Metastasis 2011, 28, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; Del Río Hernández, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [Green Version]
- Sadoughi, F.; Mirsafaei, L.; Dana, P.M.; Hallajzadeh, J.; Asemi, Z.; Mansournia, M.A.; Montazer, M.; Hosseinpour, M.; Yousefi, B. The role of DNA damage response in chemo- and radio-resistance of cancer cells: Can DDR inhibitors sole the problem? DNA Rep. 2021, 101, 103074. [Google Scholar] [CrossRef]
- Katoueezadeh, M.; Pilehvari, N.; Fatemi, A.; Hassanshahi, G.; Torabizadeh, S.A. Inhibition of DNA damage response pathway using combination of DDR pathway inhibitors and radiation in treatment of acute lymphoblastic leukemia cells. Future Oncol. 2021. [Google Scholar] [CrossRef]
- Nokin, M.J.; Darbo, E.; Travert, C.; Drogat, B.; Lacouture, A.; San José, S.; Cabrera, N.; Turcq, B.; Prouzet-Mauleon, V.; Falcone, M.; et al. Inhibition of DDR1 enhances in vivo chemosensitivity in KRAS-mutant lung adenocarcinoma. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Majo, S.; Auguste, P. The Yin and Yang of Discoidin Domain Receptors (DDRs): Implications in Tumor Growth and Metastasis Development. Cancers 2021, 13, 1725. [Google Scholar] [CrossRef]
- Mehta, V.; Chander, H.; Munshi, A. Complex roles of discoidin domain receptor tyrosine kinases in cancer. Clin. Trans. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2021. [Google Scholar] [CrossRef]
- Li, Y.; Lu, X.; Ren, X.; Ding, K. Small molecule discoidin domain receptor kinase inhibitors and potential medical applications. J. Med. Chem. 2015, 58, 3287–3301. [Google Scholar] [CrossRef]
- Ferri, N.; Carragher, N.O.; Raines, E.W. Role of discoidin domain receptors 1 and 2 in human smooth muscle cell-mediated collagen remodeling: Potential implications in atherosclerosis and lymphangioleiomyomatosis. Am. J. Pathol 2004, 164, 1575–1585. [Google Scholar] [CrossRef]
- Sannomiya, Y.; Kaseda, S.; Kamura, M.; Yamamoto, H.; Yamada, H.; Inamoto, M.; Kuwazuru, J.; Niino, S.; Shuto, T.; Suico, M.A.; et al. The role of discoidin domain receptor 2 in the renal dysfunction of alport syndrome mouse model. Ren. Fail. 2021, 43, 510–519. [Google Scholar] [CrossRef]
- Matsuyama, W.; Wang, L.; Farrar, W.L.; Faure, M.; Yoshimura, T. Activation of discoidin domain receptor 1 isoform b with collagen up-regulates chemokine production in human macrophages: Role of p38 mitogen-activated protein kinase and NF-kappa B. J. Immunol. 2004, 172, 2332–2340. [Google Scholar] [CrossRef] [Green Version]
- Flamant, M.; Placier, S.; Rodenas, A.; Curat, C.A.; Vogel, W.F.; Chatziantoniou, C.; Dussaule, J.C. Discoidin domain receptor 1 null mice are protected against hypertension-induced renal disease. J. Am. Soc. Nephrol. JASN 2006, 17, 3374–3381. [Google Scholar] [CrossRef]
- Gross, O.; Girgert, R.; Beirowski, B.; Kretzler, M.; Kang, H.G.; Kruegel, J.; Miosge, N.; Busse, A.C.; Segerer, S.; Vogel, W.F.; et al. Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease. Matrix Biol. J. Int. Soc. Matrix Biol. 2010, 29, 346–356. [Google Scholar] [CrossRef]
- Avivi-Green, C.; Singal, M.; Vogel, W.F. Discoidin domain receptor 1-deficient mice are resistant to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 420–427. [Google Scholar] [CrossRef]
- Xu, L.; Servais, J.; Polur, I.; Kim, D.; Lee, P.L.; Chung, K.; Li, Y. Attenuation of osteoarthritis progression by reduction of discoidin domain receptor 2 in mice. Arthritis Rheum. 2010, 62, 2736–2744. [Google Scholar] [CrossRef] [Green Version]
- Hebron, M.; Peyton, M.; Liu, X.; Gao, X.; Wang, R.; Lonskaya, I.; Moussa, C.E.-H. Discoidin domain receptor inhibition reduces neuropathology and attenuates inflammation in neurodegeneration models. J. Neuroimmunol. 2017, 311, 1–9. [Google Scholar] [CrossRef]
- Zhu, M.; Xing, D.; Lu, Z.; Fan, Y.; Hou, W.; Dong, H.; Xiong, L.; Dong, H. DDR1 may play a key role in destruction of the blood-brain barrier after cerebral ischemia-reperfusion. Neurosci. Res. 2015, 96, 14–19. [Google Scholar] [CrossRef]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Anjum, M.; Arellano, J. Nilotinib effects on safety, tolerability, and potential biomarkers in Parkinson disease: A phase 2 randomized clinical trial. JAMA Neurol. 2020, 77, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Arellano, J.; Howard, H.H.; et al. Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson’s disease. Pharmaco. Res. Perspect. 2019, 7, e00470. [Google Scholar] [CrossRef] [PubMed]
- Hebron, M.L.; Javidnia, M.; Moussa, C.E. Tau clearance improves astrocytic function and brain glutamate-glutamine cycle. J. Neurol. Sci. 2018, 391, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies. J. Parkinson Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Lonskaya, I.; Hebron, M.L.; Selby, S.T.; Turner, R.S.; Moussa, C.E. Nilotinib and bosutinib modulate pre-plaque alterations of blood immune markers and neuro-inflammation in Alzheimer’s disease models. Neuroscience 2015, 304, 316–327. [Google Scholar] [CrossRef]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Schachter, J.B.; Moussa, C.E. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J. Mol. Med. 2014, 92, 373–386. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson’s disease models. Hum. Mol. Geneti. 2013, 22, 3315–3328. [Google Scholar] [CrossRef] [Green Version]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Bilal, M.; Raza, A.; Khan, M.I.; Mehmood, S.; Hayat, U.; Hassan, S.T.S.; Iqbal, H.M.N. Tyrosine kinase inhibitors and their unique therapeutic potentialities to combat cancer. Int. J. Biol. Macromol. 2021, 168, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ko, Y.T. Small molecule tyrosine kinase inhibitors in glioblastoma. Arch. Pharmacol. Res. 2020, 43, 385–394. [Google Scholar] [CrossRef]
- Liu, T.; Song, S.; Wang, X.; Hao, J. Small-molecule inhibitors of breast cancer-related targets: Potential therapeutic agents for breast cancer. Eur. J. Med. Chem. 2021, 210, 112954. [Google Scholar] [CrossRef]
- Sun, X.; Xu, S.; Yang, Z.; Zheng, P.; Zhu, W. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors for the treatment of non-small cell lung cancer: A patent review (2014-present). Expert Opin. Ther. Pat. 2021, 31, 223–238. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Hajjo, R.; Sweidan, K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Mahadevan, D. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. AntiCancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Nada, H.; Elkamhawy, A.; Lee, K. Structure Activity Relationship of Key Heterocyclic Anti-Angiogenic Leads of Promising Potential in the Fight against Cancer. Molecules 2021, 26, 553. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Paik, S.; Hassan, A.H.E.; Lee, Y.S.; Roh, E.J. Hit discovery of 4-amino-N-(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide: A novel EGFR inhibitor from a designed small library. Bioorg. Chem. 2017, 75, 393–405. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Farag, A.K.; Viswanath, A.N.; Bedair, T.M.; Leem, D.G.; Lee, K.T.; Pae, A.N.; Roh, E.J. Targeting EGFR/HER2 tyrosine kinases with a new potent series of 6-substituted 4-anilinoquinazoline hybrids: Design, synthesis, kinase assay, cell-based assay, and molecular docking. Bioorg. Med. Chem. Lett. 2015, 25, 5147–5154. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Al-Sanea, M.M.; Song, C.; Sim, T.; Roh, E.J. Design and Synthesis of New [1,2,3]Triazolo[4,5-d]pyrimidine Derivatives as Potential Antiproliferative Agents. Bull. Korean Chem. Soc. 2015, 36, 1863–1873. [Google Scholar] [CrossRef]
- Nakada, M.; Kita, D.; Teng, L.; Pyko, I.V.; Watanabe, T.; Hayashi, Y.; Hamada, J.I. Receptor Tyrosine Kinases: Principles and Functions in Glioma Invasion. Adv. Exp. Med. Biol. 2020, 1202, 151–178. [Google Scholar] [CrossRef]
- Bhanumathy, K.; Balagopal, A.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Freywald, A.; Giambra, V. Protein Tyrosine Kinases: Their Roles and Their Targeting in Leukemia. Cancers 2021, 13, 184. [Google Scholar] [CrossRef]
- Rix, U.; Hantschel, O.; Dürnberger, G.; Remsing Rix, L.L.; Planyavsky, M.; Fernbach, N.V.; Kaupe, I.; Bennett, K.L.; Valent, P.; Colinge, J.; et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 2007, 110, 4055–4063. [Google Scholar] [CrossRef]
- Haura, E.B.; Tanvetyanon, T.; Chiappori, A.; Williams, C.; Simon, G.; Antonia, S.; Gray, J.; Litschauer, S.; Tetteh, L.; Neuger, A.; et al. Phase I/II study of the Src inhibitor dasatinib in combination with erlotinib in advanced non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 1387–1394. [Google Scholar] [CrossRef] [Green Version]
- Johnson, F.M.; Bekele, B.N.; Feng, L.; Wistuba, I.; Tang, X.M.; Tran, H.T.; Erasmus, J.J.; Hwang, L.L.; Takebe, N.; Blumenschein, G.R.; et al. Phase II study of dasatinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 4609–4615. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.L.; Riely, G.J.; Rizvi, N.A.; Azzoli, C.G.; Kris, M.G.; Sima, C.S.; Ginsberg, M.S.; Pao, W.; Miller, V.A. Phase II trial of dasatinib for patients with acquired resistance to treatment with the epidermal growth factor receptor tyrosine kinase inhibitors erlotinib or gefitinib. J. Thor. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2011, 6, 1128–1131. [Google Scholar] [CrossRef] [Green Version]
- Bantscheff, M.; Eberhard, D.; Abraham, Y.; Bastuck, S.; Boesche, M.; Hobson, S.; Mathieson, T.; Perrin, J.; Raida, M.; Rau, C.; et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007, 25, 1035–1044. [Google Scholar] [CrossRef]
- Rix, U.; Remsing Rix, L.L.; Terker, A.S.; Fernbach, N.V.; Hantschel, O.; Planyavsky, M.; Breitwieser, F.P.; Herrmann, H.; Colinge, J.; Bennett, K.L.; et al. A comprehensive target selectivity survey of the BCR-ABL kinase inhibitor INNO-406 by kinase profiling and chemical proteomics in chronic myeloid leukemia cells. Leukemia 2010, 24, 44–50. [Google Scholar] [CrossRef]
- Ren, X.; Pan, X.; Zhang, Z.; Wang, D.; Lu, X.; Li, Y.; Wen, D.; Long, H.; Luo, J.; Feng, Y.; et al. Identification of GZD824 as an orally bioavailable inhibitor that targets phosphorylated and nonphosphorylated breakpoint cluster region-Abelson (Bcr-Abl) kinase and overcomes clinically acquired mutation-induced resistance against imatinib. J. Med. Chem. 2013, 56, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.J.; Hebron, M.; Missner, A.A.; Wang, R.; Gao, X.; Kurd-Misto, B.T.; Liu, X.; Moussa, C.E. Multikinase Abl/DDR/Src Inhibition Produces Optimal Effects for Tyrosine Kinase Inhibition in Neurodegeneration. Drugs R D 2019, 19, 149–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Sun, X.; Phan, T.N.; Jung, S.H.; Kim, S.Y.; Cho, J.U.; Lee, H.; Woo, S.H.; Park, T.K.; Yang, B.S. LCB 03-0110, a novel pan-discoidin domain receptor/c-Src family tyrosine kinase inhibitor, suppresses scar formation by inhibiting fibroblast and macrophage activation. J. Pharmacol. Exp. Ther. 2012, 340, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Okram, B.; Nagle, A.; Adrián, F.J.; Lee, C.; Ren, P.; Wang, X.; Sim, T.; Xie, Y.; Wang, X.; Xia, G.; et al. A general strategy for creating “inactive-conformation” abl inhibitors. Chem. Biol. 2006, 13, 779–786. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.G.; Tan, L.; Weisberg, E.L.; Liu, F.; Canning, P.; Choi, H.G.; Ezell, S.A.; Wu, H.; Zhao, Z.; Wang, J.; et al. Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem. Biol. 2013, 8, 2145–2150. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-G.; Tan, L.; Weisberg, E.L.; Liu, F.; Canning, P.; Choi, H.-G.; Ezell, S.; Zhao, Z.; Wu, H.; Wang, J.; et al. Correction to Discovery of a Potent and Selective DDR1 Receptor Tyrosine Kinase Inhibitor. ACS Chem. Biol. 2014, 9, 840. [Google Scholar] [CrossRef]
- Murata, T.; Kawada, H.; Niizuma, S.; Hara, S.; Hada, K.; Shimada, H.; Tanaka, H.; Mio, T. Preparation of quinazolinedione derivatives as discoidin domain receptor 1 (DDR1) inhibitors. Patent No. WO201316 1853, 2013. [Google Scholar]
- Murata, T.; Niizuma, S.; Hara, S.; Kawada, H.; Hada, K.; Shimada, H.; Tanaka, H.; Nakanishi, Y. Preparation of benzamide derivatives as discoidin domain receptor 1 (DDR1) inhibitors. Patent No. WO201316 1851, 2013. [Google Scholar]
- Gao, M.; Duan, L.; Luo, J.; Zhang, L.; Lu, X.; Zhang, Y.; Zhang, Z.; Tu, Z.; Xu, Y.; Ren, X.; et al. Discovery and Optimization of 3-(2-(Pyrazolo[1,5-a]pyrimidin-6-yl)ethynyl)benzamides as Novel Selective and Orally Bioavailable Discoidin Domain Receptor 1 (DDR1) Inhibitors. J. Med. Chem. 2013, 56, 3281–3295. [Google Scholar] [CrossRef]
- Lu, Q.P.; Chen, W.D.; Peng, J.R.; Xu, Y.D.; Cai, Q.; Feng, G.K.; Ding, K.; Zhu, X.F.; Guan, Z. Antitumor activity of 7RH, a discoidin domain receptor 1 inhibitor, alone or in combination with dasatinib exhibits antitumor effects in nasopharyngeal carcinoma cells. Oncol. Lett. 2016, 12, 3598–3608. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Bian, H.; Bartual, S.G.; Du, W.; Luo, J.; Zhao, H.; Zhang, S.; Mo, C.; Zhou, Y.; Xu, Y.; et al. Structure-Based Design of Tetrahydroisoquinoline-7-carboxamides as Selective Discoidin Domain Receptor 1 (DDR1) Inhibitors. J. Med. Chem. 2016, 59, 5911–5916. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, Y.; Bartual, S.G.; Luo, J.; Xu, T.; Du, W.; Xun, Q.; Tu, Z.; Brekken, R.A.; Ren, X.; et al. Tetrahydroisoquinoline-7-carboxamide Derivatives as New Selective Discoidin Domain Receptor 1 (DDR1) Inhibitors. ACS Med. Chem. Lett. 2017, 8, 327–332. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.; Pinkas, D.M.; Fox, A.E.; Luo, J.; Huang, H.; Cui, S.; Xiang, Q.; Xu, T.; Xun, Q.; et al. Design, Synthesis, and Biological Evaluation of 3-(Imidazo[1,2-a]pyrazin-3-ylethynyl)-4-isopropyl-N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide as a Dual Inhibitor of Discoidin Domain Receptors 1 and 2. J. Med. Chem. 2018, 61, 7977–7990. [Google Scholar] [CrossRef]
- Richters, A.; Nguyen, H.D.; Phan, T.; Simard, J.R.; Grütter, C.; Engel, J.; Rauh, D. Identification of Type II and III DDR2 Inhibitors. J. Med. Chem. 2014, 57, 4252–4262. [Google Scholar] [CrossRef]
- Murray, C.W.; Berdini, V.; Buck, I.M.; Carr, M.E.; Cleasby, A.; Coyle, J.E.; Curry, J.E.; Day, J.E.H.; Day, P.J.; Hearn, K.; et al. Fragment-Based Discovery of Potent and Selective DDR1/2 Inhibitors. ACS Med. Chem. Lett. 2015, 6, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Elkamhawy, A.; Park, J.-e.; Cho, N.-C.; Sim, T.; Pae, A.N.; Roh, E.J. Discovery of a broad spectrum antiproliferative agent with selectivity for DDR1 kinase: Cell line-based assay, kinase panel, molecular docking, and toxicity studies. J. Enzym. Inhib. Med. Chem. 2016, 31, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Damasy, A.K.; Seo, S.H.; Cho, N.C.; Kang, S.B.; Pae, A.N.; Kim, K.S.; Keum, G. Design, synthesis, in-vitro antiproliferative activity and kinase profile of new picolinamide based 2-amido and ureido quinoline derivatives. Eur. J. Med. Chem. 2015, 101, 754–768. [Google Scholar] [CrossRef] [PubMed]
- El-Damasy, A.K.; Lee, J.H.; Seo, S.H.; Cho, N.C.; Pae, A.N.; Keum, G. Design and synthesis of new potent anticancer benzothiazole amides and ureas featuring pyridylamide moiety and possessing dual B-Raf(V600E) and C-Raf kinase inhibitory activities. Eur. J. Med. Chem. 2016, 115, 201–216. [Google Scholar] [CrossRef] [PubMed]
- El-Damasy, A.K.; Cho, N.C.; Nam, G.; Pae, A.N.; Keum, G. Discovery of a Nanomolar Multikinase Inhibitor (KST016366): A New Benzothiazole Derivative with Remarkable Broad-Spectrum Antiproliferative Activity. ChemMedChem 2016, 11, 1587–1595. [Google Scholar] [CrossRef]
- Liu, L.; Hussain, M.; Luo, J.; Duan, A.; Chen, C.; Tu, Z.; Zhang, J. Synthesis and biological evaluation of novel dasatinib analogues as potent DDR1 and DDR2 kinase inhibitors. Chem. Biol. Drug Des. 2017, 89, 420–427. [Google Scholar] [CrossRef]
- Jeffries, D.E.; Borza, C.M.; Blobaum, A.L.; Pozzi, A.; Lindsley, C.W. Discovery of VU6015929: A Selective Discoidin Domain Receptor 1/2 (DDR1/2) Inhibitor to Explore the Role of DDR1 in Antifibrotic Therapy. ACS Med. Chem. Lett. 2020, 11, 29–33. [Google Scholar] [CrossRef]
- Terai, H.; Tan, L.; Beauchamp, E.M.; Hatcher, J.M.; Liu, Q.; Meyerson, M.; Gray, N.S.; Hammerman, P.S. Characterization of DDR2 Inhibitors for the Treatment of DDR2 Mutated Nonsmall Cell Lung Cancer. ACS Chem. Biol. 2015, 10, 2687–2696. [Google Scholar] [CrossRef]
- Wang, Q.; Dai, Y.; Ji, Y.; Shi, H.; Guo, Z.; Chen, D.; Chen, Y.; Peng, X.; Gao, Y.; Wang, X.; et al. Discovery and optimization of a series of 3-substituted indazole derivatives as multi-target kinase inhibitors for the treatment of lung squamous cell carcinoma. Eur. J. Med. Chem. 2019, 163, 671–689. [Google Scholar] [CrossRef]
- Zhu, W.; Chen, H.; Wang, Y.; Wang, J.; Peng, X.; Chen, X.; Gao, Y.; Li, C.; He, Y.; Ai, J.; et al. Design, Synthesis, and Pharmacological Evaluation of Novel Multisubstituted Pyridin-3-amine Derivatives as Multitargeted Protein Kinase Inhibitors for the Treatment of Non-Small Cell Lung Cancer. J. Med. Chem. 2017, 60, 6018–6035. [Google Scholar] [CrossRef]
- Hu, Y.; Potts, M.B.; Colosimo, D.; Herrera-Herrera, M.L.; Legako, A.G.; Yousufuddin, M.; White, M.A.; MacMillan, J.B. Discoipyrroles A–D: Isolation, Structure Determination, and Synthesis of Potent Migration Inhibitors from Bacillus hunanensis. J. Am. Chem. Soc. 2013, 135, 13387–13392. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Banwell, M.G.; Carr, P.D.; Willis, A.C. Modular Total Syntheses of the Alkaloids Discoipyrroles A and B, Potent Inhibitors of the DDR2 Signaling Pathway. Org. Lett. 2016, 18, 704–707. [Google Scholar] [CrossRef]
- Zhu, D.; Huang, H.; Pinkas, D.M.; Luo, J.; Ganguly, D.; Fox, A.E.; Arner, E.; Xiang, Q.; Tu, Z.-C.; Bullock, A.N.; et al. 2-Amino-2,3-dihydro-1H-indene-5-carboxamide-Based Discoidin Domain Receptor 1 (DDR1) Inhibitors: Design, Synthesis, and in Vivo Antipancreatic Cancer Efficacy. J. Med. Chem. 2019, 62, 7431–7444. [Google Scholar] [CrossRef]
- Mo, C.; Zhang, Z.; Li, Y.; Huang, M.; Zou, J.; Luo, J.; Tu, Z.-C.; Xu, Y.; Ren, X.; Ding, K.; et al. Design and Optimization of 3′-(Imidazo[1,2-a]pyrazin-3-yl)-[1,1′-biphenyl]-3-carboxamides as Selective DDR1 Inhibitors. ACS Med. Chem. Lett. 2020, 11, 379–384. [Google Scholar] [CrossRef]
- Dou, X.; Huang, H.; Jiang, L.; Zhu, G.; Jin, H.; Jiao, N.; Zhang, L.; Liu, Z.; Zhang, L. Rational modification, synthesis and biological evaluation of 3,4-dihydroquinoxalin-2(1H)-one derivatives as potent and selective c-Jun N-terminal kinase 3 (JNK3) inhibitors. Eur. J. Med. Chem. 2020, 201, 112445. [Google Scholar] [CrossRef]
- Dong, R.; Zhou, X.; Wang, M.; Li, W.; Zhang, J.-Y.; Zheng, X.; Tang, K.-X.; Sun, L.-P. Discovery of 4-amino-1H-pyrazolo[3,4-d]pyrimidin derivatives as novel discoidin domain receptor 1 (DDR1) inhibitors. Bioorg. Med. Chem. 2021, 29, 115876. [Google Scholar] [CrossRef]
- Richter, H.; Satz, A.L.; Bedoucha, M.; Buettelmann, B.; Petersen, A.C.; Harmeier, A.; Hermosilla, R.; Hochstrasser, R.; Burger, D.; Gsell, B.; et al. DNA-Encoded Library-Derived DDR1 Inhibitor Prevents Fibrosis and Renal Function Loss in a Genetic Mouse Model of Alport Syndrome. ACS Chem. Biol. 2019, 14, 37–49. [Google Scholar] [CrossRef]
- Vanajothi, R.; Hemamalini, V.; Jeyakanthan, J.; Premkumar, K. Ligand-based pharmacophore mapping and virtual screening for identification of potential discoidin domain receptor 1 inhibitors. J. Biomol. Struc. Dyn. 2020, 38, 2800–2808. [Google Scholar] [CrossRef]
- Chen, C.; Deng, J.; Yu, X.; Wu, F.; Men, K.; Yang, Q.; Zhu, Y.; Liu, X.; Jiang, Q. Identification of novel inhibitors of DDR1 against idiopathic pulmonary fibrosis by integrative transcriptome meta-analysis, computational and experimental screening. Mol. BioSyst. 2016, 12, 1540–1551. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Crunkhorn, S. Deep learning identifies DDR1 kinase inhibitors. Nat. Rev. Drug Discov. 2019, 18, 826. [Google Scholar] [CrossRef] [PubMed]
- Yoshimori, A.; Asawa, Y.; Kawasaki, E.; Tasaka, T.; Matsuda, S.; Sekikawa, T.; Tanabe, S.; Neya, M.; Natsugari, H.; Kanai, C. Design and Synthesis of DDR1 Inhibitors with a Desired Pharmacophore Using Deep Generative Models. ChemMedChem 2021, 16, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Jeon, W.; Kim, D. Autonomous molecule generation using reinforcement learning and docking to develop potential novel inhibitors. Sci. Rep. 2020, 10, 22104. [Google Scholar] [CrossRef] [PubMed]


























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkamhawy, A.; Lu, Q.; Nada, H.; Woo, J.; Quan, G.; Lee, K. The Journey of DDR1 and DDR2 Kinase Inhibitors as Rising Stars in the Fight Against Cancer. Int. J. Mol. Sci. 2021, 22, 6535. https://doi.org/10.3390/ijms22126535
Elkamhawy A, Lu Q, Nada H, Woo J, Quan G, Lee K. The Journey of DDR1 and DDR2 Kinase Inhibitors as Rising Stars in the Fight Against Cancer. International Journal of Molecular Sciences. 2021; 22(12):6535. https://doi.org/10.3390/ijms22126535
Chicago/Turabian StyleElkamhawy, Ahmed, Qili Lu, Hossam Nada, Jiyu Woo, Guofeng Quan, and Kyeong Lee. 2021. "The Journey of DDR1 and DDR2 Kinase Inhibitors as Rising Stars in the Fight Against Cancer" International Journal of Molecular Sciences 22, no. 12: 6535. https://doi.org/10.3390/ijms22126535
APA StyleElkamhawy, A., Lu, Q., Nada, H., Woo, J., Quan, G., & Lee, K. (2021). The Journey of DDR1 and DDR2 Kinase Inhibitors as Rising Stars in the Fight Against Cancer. International Journal of Molecular Sciences, 22(12), 6535. https://doi.org/10.3390/ijms22126535