
The Impact of the Tumor Microenvironment on Macrophage Polarization in Cancer Metastatic Progression



Abstract
:
1. Introduction
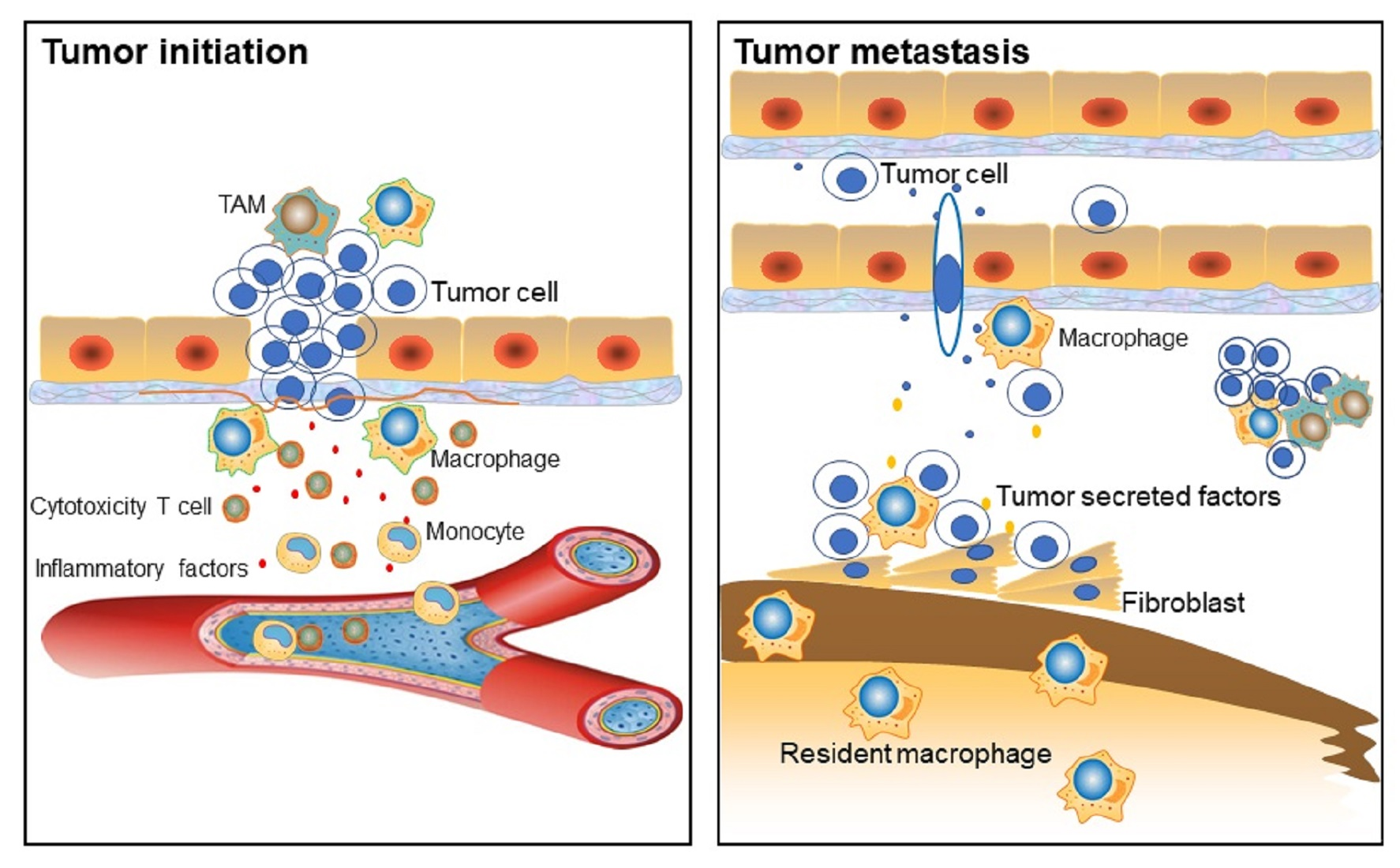
2. Macrophage Phenotypes
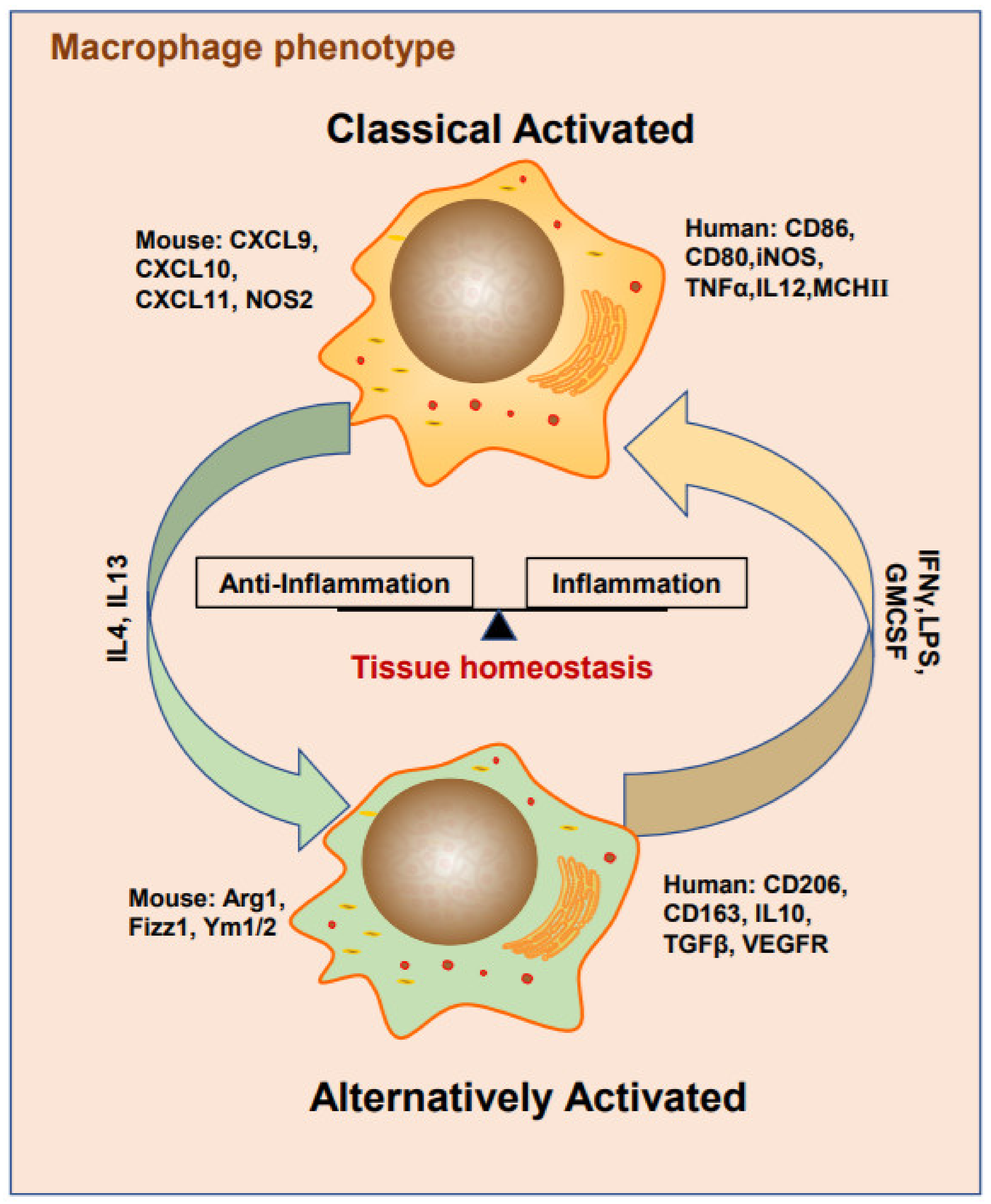
3. Heterogeneous TAM Polarization
3.1. Cytokines
3.2. Metabolites
3.3. Exosomes
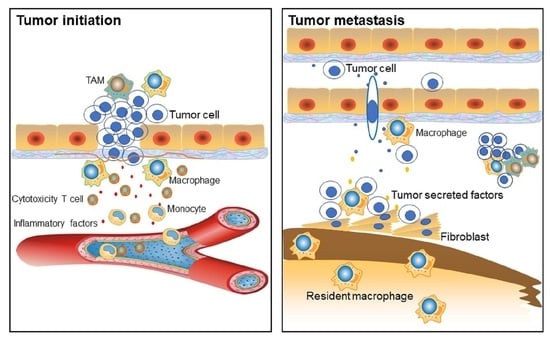
4. TAMs are Involved in Tumor Progression
4.1. Macrophages in Cancer Initiation
4.2. TAMs Facilitate Tumor Metastasis and Intra-tumoral Heterogeneity
5. Immunotherapy and TAM-targeted Therapy
5.1. Inhibition of TAMs Recruitment or Reprogramming of TAMs
5.2. Phagocytosis Checkpoints
5.3. cGAS-STING in TAMs
6. Conclusions, Perspectives, and Limitations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [Green Version]
- Loret, N.; Denys, H.; Tummers, P.; Berx, G. The role of epithelial-to-mesenchymal plasticity in ovarian cancer progression and therapy resistance. Cancers 2019, 11, 838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, M.; Shen, J.; Yu, S.; Fei, J.; Zhu, X.; Zhao, J.; Zhai, L.; Sadhukhan, A.; Zhou, J. Tumor-associated macrophages (TAMs): A critical activator in ovarian cancer metastasis. Onco. Targets Ther. 2019, 12, 8687–8699. [Google Scholar] [CrossRef] [Green Version]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hall mark of cancer revisited. Signal. Transduct. Target Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [Green Version]
- Janssen, L.M.E.; Ramsay, E.E.; Logsdon, C.D.; Overwijk, W.W. The immune system in cancer metastasis: Friend or foe? J. Immunother. Cancer 2017, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Lee, C.K.; Jeong, S.H.; Jang, C.; Bae, H.; Kim, Y.H.; Park, I.; Kim, S.K.; Koh, G.Y. Tumor metastasis to lymphnodes requiresYAP-dependent metabolic adaptation. Science 2019, 363, 644–649. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [Green Version]
- Komai, T.; Inoue, M.; Okamura, T.; Morita, K.; Iwasaki, Y.; Sumitomo, S.; Shoda, H.; Yamamoto, K.; Fujio, K. transforminggrowth factor-beta and inter leukin-10 synergistically regulate humoral immunity via modulating metabolic signals. Front. Immunol. 2018, 9, 1364. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-derived suppressor cells: Immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Hendry, S.; Salgado, R.; Gevaert, T.; Russell, P.A.; John, T.; Thapa, B.; Christie, M.; van deVijver, K.; Estrada, M.V.; Gonzalez-Ericsson, P.I.; et al. Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method from the International immuno-oncology biomarkers working group: Part 2: TILsin melanoma, gastrointestinal tract carcinomas, non-small cell lung carcinoma and mesothelioma, endometrial and ovarian carcinomas, squamous cell carcinoma of the head and neck, genitourinary carcinomas, and primary brain tumors. Adv. Anat. Pathol. 2017, 24, 311–335. [Google Scholar] [CrossRef]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. Ahigh M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian. Res. 2014, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-associated macrophages in human breast, colorectal, lung, ovarianand prostate cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gruosso, T.; Zuo, D.; Omeroglu, A.; Meterissian, S.; Guiot, M.C.; Salazar, A.; Park, M.; Levine, H. Infiltration of CD8(+) T cells in to tumor cell clusters in triple-negative breastcancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3678–3687. [Google Scholar] [CrossRef] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soncin, I.; Sheng, J.; Chen, Q.; Foo, S.; Duan, K.; Lum, J.; Poidinger, M.; Zolezzi, F.; Karjalainen, K.; Ruedl, C. The tumour micro environment create saniche for the self-renewal of tumour-promoting macrophages in colonadenoma. Nat. Commun. 2018, 9, 582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerriero, J.L. Macrophages: The road less traveled, changing anticancer therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Y.; Huang, T.W.; Hsieh, Y.T.; Wang, Y.F.; Yen, C.C.; Lee, G.L.; Yeh, C.C.; Peng, Y.J.; Kuo, Y.Y.; Wen, H.T.; et al. Cancer -derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol. Cell. 2020, 77, 213–227. [Google Scholar] [CrossRef]
- Wenes, M.; Shang, M.; DiMatteo, M.; Goveia, J.; Martin-Perez, R.; Serneels, J.; Prenen, H.; Ghesquiere, B.; Carmeliet, P.; Mazzone, M. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell. Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [Green Version]
- Marigo, I.; Trovato, R.; Hofer, F.; Ingangi, V.; Desantis, G.; Leone, K.; DeSanctis, F.; Ugel, S.; Cane, S.; Simonelli, A.; et al. Disabled homolog 2 controls prometastatic activity of tumor-associated macrophages. Cancer Discov. 2020, 10, 1758–1773. [Google Scholar] [CrossRef]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell 2019, 35, 588–602. [Google Scholar] [CrossRef] [Green Version]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Kumar, M.P.; Du, J.; Lagoudas, G.; Jiao, Y.; Sawyer, A.; Drummond, D.C.; Lauffenburger, D.A.; Raue, A. Analysis of single-cell RNA-Seq identifies cell-cell communication associated with tumor characteristics. Cell. Rep. 2018, 25, 1458–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.K.; Wang, M.; Sun, Y.; DiCostanzo, N.; Mitchell, C.; Achuthan, A.; Hamilton, J.A.; Busuttil, R.A.; Boussioutas, A. Macrophage spatial heterogeneity in gastric cancer defined by multiplex immunohisto chemistry. Nat. Commun. 2019, 10, 3928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000 Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage polarization: Different gene signatures in M1(LPS+) vs. classically and M2 (LPS-) vs. alternatively activated macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Kim, S.Y.; Nair, M.G. Macrophages in wound healing: Activation and plasticity. Immunol. Cell. Biol. 2019, 97, 258–267. [Google Scholar] [CrossRef]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [Green Version]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal. Transduct. Target Ther. 2021, 6, 75. [Google Scholar] [CrossRef]
- Takeya, M.; Komohara, Y. Role of tumor-associated macrophages in human malignancies: Friend or foe? Pathol. Int. 2016, 66, 491–505. [Google Scholar] [CrossRef]
- Ding, T.; Xu, J.; Wang, F.; Shi, M.; Zhang, Y.; Li, S.P.; Zheng, L. High tumor-infiltrating macrophage density predicts poor prognosis in patients with primary hepatocellular carcinoma after resection. Hum. Pathol. 2009, 40, 381–389. [Google Scholar] [CrossRef]
- Poh, A.R.; Ernst, M. Targeting macrophages in cancer: From bench to bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, W.; Fang, W.Y.; Ye, L.; Shen, L.Y.; Zhang, X.F.; Fei, X.C.; Chen, X.; Wang, W.Q.; Li, X.Y.; Xiao, J.C.; et al. Density of tumor-associated macrophages correlates with lymphno demetastasis in papillary thyroid carcinoma. Thyroid 2012, 22, 905–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumitomo, R.; Hirai, T.; Fujita, M.; Murakami, H.; Otake, Y.; Huang, C.L. M2 tumor-associated macrophages promote tumor progression in non-small-cell lung cancer. Exp. Ther. Med. 2019, 18, 4490–4498. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.C.; Zhao, J.L.; Lu, Y.T.; Gao, C.C.; Yang, Y.; Liang, S.Q.; Lu, Y.Y.; Wang, L.; Yue, S.Q.; Dou, K.F.; et al. NOTCH signaling via WNT regulates the proliferation of alternative, CCR2-independent tumor-associated macrophages in hepatocellular carcinoma. Cancer Res. 2019, 79, 4160–4172. [Google Scholar] [CrossRef]
- Zheng, X.; Turkowski, K.; Mora, J.; Brune, B.; Seeger, W.; Weigert, A.; Savai, R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 2017, 8, 48436–48452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; Qin, M.; Song, Y.; Tang, Q.; Huang, Y.; Shen, P.; Lu, Y. M2polarization of tumor-associated macrophages is dependent on integrin beta 3 via peroxi some proliferator-activated receptor-gamma up-regulation in breast cancer. Immunology 2020, 160, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Sarode, P.; Zheng, X.; Giotopoulou, G.A.; Weigert, A.; Kuenne, C.; Gunther, S.; Friedrich, A.; Gattenlohner, S.; Stiewe, T.; Brune, B.; et al. Reprogramming of tumor-associated macrophages by targeting beta-catenin/FOSL2/ARID5A signaling: A potentialtreatment of lung cancer. Sci. Adv. 2020, 6, eaaz6105. [Google Scholar] [CrossRef]
- Xu, F.; Wei, Y.; Tang, Z.; Liu, B.; Dong, J. Tumor associated macrophages in lung cancer: Friend or foe? (Review). Mol. Med. Rep. 2020, 22, 4107–4115. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and metabolism in the tumor microenvironment. Cell. Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Sarode, P.; Schaefer, M.B.; Grimminger, F.; Seeger, W.; Savai, R. Macrophage and tumor cell cross-talk is fundamental for lung tumor progression: We need to talk. Front. Oncol. 2020, 10, 324. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: Anaccomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Cervantes-Villagrana, R.D.; Albores-Garcia, D.; Cervantes-Villagrana, A.R.; Garcia-Acevez, S.J. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal. Transduct. Target Ther. 2020, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.M.; Pierides, C.; Christodoulou, M.I.; Costeas, P.; Kyriakou, T.C.; Papageorgis, P. The role of tumor-associated myeloid cells in modulating cancer therapy. Front. Oncol. 2020, 10, 899. [Google Scholar] [CrossRef]
- Pass, H.I.; Lavilla, C.; Canino, C.; Goparaju, C.; Preiss, J.; Noreen, S.; Blandino, G.; Cioce, M. Inhibition of the colony-stimulating-factor-1receptor affects the resistance of lung cancer cells tocisplatin. Oncotarget 2016, 7, 56408–56421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug. Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef] [PubMed]
- Zabuawala, T.; Taffany, D.A.; Sharma, S.M.; Merchant, A.; Adair, B.; Srinivasan, R.; Rosol, T.J.; Fernandez, S.; Huang, K.; Leone, G.; et al. A nets2-driven transcriptional program in tumor-associated macrophages promotes tumor metastasis. Cancer Res. 2010, 70, 1323–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Cross talk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, X.; Li, Z.; Zhu, B. Metabolic regulatory cross talk between tumor micro environment and tumor-associated macrophages. Theranostics 2021, 11, 1016–1030. [Google Scholar] [CrossRef]
- Wang, J.; Li, D.; Cang, H.; Guo, B. Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019, 8, 4709–4721. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.; Huang, X.; Zhang, G.; Hong, Z.; Bai, X.; Liang, T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal. Transduct. Target Ther. 2021, 6, 72. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STAT sincancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Chen, R.R.; Yung, M.M.H.; Xuan, Y.; Zhan, S.; Leung, L.L.; Liang, R.R.; Leung, T.H.Y.; Yang, H.; Xu, D.; Sharma, R.; et al. Targeting of lipid metabolism with a metabolic inhibitor cocktaile radicates peritoneal metastases in ovarian cancer cells. Commun. Biol. 2019, 2, 281. [Google Scholar] [CrossRef]
- Puthenveetil, A.; Dubey, S. Metabolic reprograming of tumor-associated macrophages. Ann. Transl. Med. 2020, 8, 1030. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Verdeguer, F.; Aouadi, M. Macrophage heterogeneity and energy metabolism. Exp. Cell Res. 2017, 360, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The metabolic signature of macrophage responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, M.; Muller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via arginase or nitric oxide synthase: Two competing arginine pathways in macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Xian, M.; Xiang, S.; Xiang, D.; Shao, X.; Wang, J.; Cao, J.; Yang, X.; Yang, B.; Ying, M.; et al. All-trans retinoic acid prevents osteosarcoma metastasis by inhibiting M2 polarization of tumor-associated macrophages. Cancer Immunol. Res. 2017, 5, 547–559. [Google Scholar] [CrossRef] [Green Version]
- Etzerodt, A.; Tsalkitzi, K.; Maniecki, M.; Damsky, W.; Delfini, M.; Baudoin, E.; Moulin, M.; Bosenberg, M.; Graversen, J.H.; Auphan-Anezin, N.; et al. Specific targeting of CD163 (+) TAMs mobilizes inflammatory monocytes and promotes Tcell-mediated tumor regression. J. Exp. Med. 2019, 216, 2394–2411. [Google Scholar] [CrossRef] [PubMed]
- Poznanski, S.M.; Singh, K.; Ritchie, T.M.; Aguiar, J.A.; Fan, I.Y.; Portillo, A.L.; Rojas, E.A.; Vahedi, F.; El-Sayes, A.; Xing, S.; et al. Metabolic flexibility determines human NKcell functional fate in the tumor microenvironment. Cell Metab. 2021. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Ma, J.; Li, J.; Gu, Y.; Yin, L.; Wang, Y.; Zhou, X.; Wang, J.; Ji, H.; Zhang, Q. Sodium/glucoseco transporter 1-dependent metabolic alterations induce tamoxifen resistance in breast cancer by promoting macrophage M2 polarization. Cell Death Dis. 2021, 12, 509. [Google Scholar] [CrossRef]
- Su, P.; Wang, Q.; Bi, E.; Ma, X.; Liu, L.; Yang, M.; Qian, J.; Yi, Q. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res. 2020, 80, 1438–1450. [Google Scholar] [CrossRef]
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; van Eck, M.; Auphan-Anezin, N.; et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab. 2019, 29, 1376–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Bai, X.; Ni, J.; Zhang, H.; Duan, W.; Graham, P.; Li, Y. Exosomes and breast cancer drug resistance. Cell Death Dis. 2020, 11, 987. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Zheng, S.; Zhang, T.; Wu, J.; Sun, Y.; Zhang, J.; Liu, G. Role of exosomes in the immune microenvironment of ovarian cancer. Oncol. Lett. 2021, 21, 377. [Google Scholar] [CrossRef]
- Li, B.; Song, T.N.; Wang, F.R.; Yin, C.; Li, Z.; Lin, J.P.; Meng, Y.Q.; Feng, H.M.; Jing, T. Tumor-derived exosomal HMGB1 promotes esophageal squamous cell carcinoma progression through inducing PD1 (+) TAM expansion. Oncogenesis 2019, 8, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, P.; Luo, Q.; Wang, W.; Li, J.; Wang, T.; Wang, P.; Chen, L.; Zhang, P.; Chen, H.; Liu, Y.; et al. Tumor-associated macrophages-derived exosomes promote the migration of gastric cancer cells by transfer of functional Apolipoprotein, E. Cell Death Dis. 2018, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Bunggulawa, E.J.; Wang, W.; Yin, T.; Wang, N.; Durkan, C.; Wang, Y.; Wang, G. Recent advancements in the use of exosomes as drug delivery systems. J. Nanobiotechnology 2018, 16, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Li, Y.; Pang, W.; Shen, H. Exosomes: A potential therapeutic tool targeting communications between tumorcells and macrophages. Mol. Ther. 2020, 28, 1953–1964. [Google Scholar] [CrossRef]
- Wang, M.; Yu, F.; Li, P.; Wang, K. Emerging function and clinical significance of exosomalcirc RNAs in cancer. Mol. Ther. Nucleic Acids 2020, 21, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, R.; Li, J.; Tang, S.; Li, S.; Tong, Q.; Li, S. Down regulation of hsa_circ_0074854 suppresses the migration and invasion in hepatocellular carcinomavia interacting with Hu Rand via suppressing exosomes-mediated macrophage M2 polarization. Int. J. Nanomedicine 2021, 16, 2803–2818. [Google Scholar] [CrossRef]
- Chen, X.; Ying, X.; Wang, X.; Wu, X.; Zhu, Q.; Wang, X. Exosomes derived from hypoxicepithelial ovarian cancer deliver microRNA-940 to induce macrophage M2 polarization. Oncol. Rep. 2017, 38, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Ying, X.; Wu, Q.; Wu, X.; Zhu, Q.; Wang, X.; Jiang, L.; Chen, X.; Wang, X. Epithelial ovarian cancer-secreted exosomal miR-222-3p induces polarization of tumor-associated macrophages. Oncotarget 2016, 7, 43076–43087. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Han, Q.; Xu, D.; Zheng, B.; Zhao, X.; Zhang, J. SALL4-mediated up regulation of exosomal miR-146a-5p drives T-cell exhaustion b yM2tumor-associated macrophages in HCC. Oncoimmunology 2019, 8, 1601479. [Google Scholar] [CrossRef]
- Papait, A.; Stefani, F.R.; Cargnoni, A.; Magatti, M.; Parolini, O.; Silini, A.R. The multifaceted roles of MSCs in the tumor microenvironment: Interactions with immune cells and exploitation for therapy. Front. Cell Dev. Biol. 2020, 8, 447. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Li, W. Crosstalk between mesenchymal stromal cells and tumor-associated macrophages in gastric cancer. Front. Oncol. 2020, 10, 571516. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.; Caja, S.; Strano Moraes, M.C.; Couto, N.; Costa-Silva, B. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front. Cell Dev. Biol. 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Olejarz, W.; Dominiak, A.; Zolnierzak, A.; Kubiak-Tomaszewska, G.; Lorenc, T. Tumor-derived exosomes in immunosuppression and immunotherapy. J. Immunol. Res. 2020, 2020, 6272498. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. macrophage-based approaches for cancer immunotherapy. Cancer Res. 2021, 81, 1201–1208. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning cold into hot: Firing up the tumor microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.S.; Zhang, X.H. One microenvironment does not fit all: Heterogeneity beyond cancer cells. Cancer Metastasis Rev. 2016, 35, 601–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comen, E.A.; Bowman, R.L.; Kleppe, M. Underlying causes and therapeutic targeting of the inflammatory tumor microenvironment. Front. Cell Dev. Biol. 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Smirnova, T.; Kedrin, D.; Wyckoff, J.; Zhu, L.; Stanley, E.R.; Cox, D.; Muller, W.J.; Pollard, J.W.; van Rooijen, N.; et al. The EGF/CSF-1paracrine invasion loop can be triggered by heregulin beta1 and CXCL12. Cancer Res 2009, 69, 3221–3227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metal loproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.E.; Harney, A.S.; Pollard, J.W. The multifaceted role of perivascular macrophagesin tumors. Cancer Cell 2016, 30, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Investig. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, D.M.; Hastings, P.J.; Rosenberg, S.M. Stress-induced mutagenesis: Implications in cancer and drug resistance. Annu. Rev. Cancer Biol. 2017, 1, 119–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matic, I. Mutation rate heterogeneity increases odds of survival in unpredictable environments. Mol. Cell 2019, 75, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Gast, C.E.; Silk, A.D.; Zarour, L.; Riegler, L.; Burkhart, J.G.; Gustafson, K.T.; Parappilly, M.S.; Roh-Johnson, M.; Goodman, J.R.; Olson, B.; et al. Cell fusion potentialtes tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci. Adv. 2018, 4, 7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belgiovine, C.; Digifico, E.; Anfray, C.; Ummarino, A.; TorresAndon, F. Targeting tumor-associated macrophages in anti-cancer therapies: Convincing the traitors to do the right thing. J. Clin. Med. 2020, 9, 3226. [Google Scholar] [CrossRef]
- Castro, F.; Cardoso, A.P.; Goncalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [Green Version]
- Gun, S.Y.; Lee, S.W.L.; Sieow, J.L.; Wong, S.C. Targeting immune cells for cancer therapy. Redox Biol. 2019, 25, 101174. [Google Scholar] [CrossRef] [PubMed]
- Marigo, I.; Zilio, S.; Desantis, G.; Mlecnik, B.; Agnellini, A.H.R.; Ugel, S.; Sasso, M.S.; Qualls, J.E.; Kratochvill, F.; Zanovello, P.; et al. T cell cancer therapy requires CD40-CD40L activation of tumor necrosis factor and inducible nitric-oxide-synthase-producing dendritic cells. Cancer Cell 2016, 30, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guerin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anfray, C.; Ummarino, A.; Andon, F.T.; Allavena, P. Current strategies to target tumor-associated-macrophages to improve anti-tumor immune responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argyle, D.; Kitamura, T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front. Immunol. 2018, 9, 2629. [Google Scholar] [CrossRef]
- Zhou, W.; Guo, S.; Liu, M.; Burow, M.E.; Wang, G. TargetingCXCL12/CXCR4Axis in tumor immunotherapy. Curr. Med. Chem. 2019, 26, 3026–3041. [Google Scholar] [CrossRef]
- Li, X.; Bu, W.; Meng, L.; Liu, X.; Wang, S.; Jiang, L.; Ren, M.; Fan, Y.; Sun, H. CXCL12/CXCR4 path way orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp. Cell Res. 2019, 378, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.B.; Danielsen, A.V.; Hamilton-Dutoit, S.J.; Bendix, K.; Norgaard, P.; Moller, M.B.; Steiniche, T.; d’Amore, F. High intra tumor almacrophage content is an adverse prognostic feature in anaplastic large cell lymphoma. Histopathology 2014, 65, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Lenzo, J.C.; Turner, A.L.; Cook, A.D.; Vlahos, R.; Anderson, G.P.; Reynolds, E.C.; Hamilton, J.A. Control of macrophage lineage populations by CSF-1 receptor and GM-CSF in homeostasis and inflammation. Immunol. Cell Biol. 2012, 90, 429–440. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Ruttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Benner, B.; Good, L.; Quiroga, D.; Schultz, T.E.; Kassem, M.; Carson, W.E.; Cherian, M.A.; Sardesai, S.; Wesolowski, R. Pexidartinib, a novel small molecule CSF-1R inhibitor in use for tenosynovial giant cell tumor: A systematic review of pre-clinical and clinical development. Drug Des. Devel. Ther. 2020, 14, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Opperman, K.S.; Vandyke, K.; Clark, K.C.; Coulter, E.A.; Hewett, D.R.; Mrozik, K.M.; Schwarz, N.; Evdokiou, A.; Croucher, P.I.; Psaltis, P.J.; et al. Clodronate-liposome mediated macrophage depletion abrogates multiple myeloma tumor establishment in vivo. Neoplasia 2019, 21, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, R.; Su, X.; Pan, Y.; Han, X.; Shao, C.; Shi, Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer 2019, 18, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosowski, E.E. Determining macrophage versus neutrophil contributions to in nateimmunity using larval zebrafish. Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Su, Y.; Kang, B.H.; Fan, Z.; Dong, T.; Brown, D.R.; Cheah, J.; Wittrup, K.D.; Chen, J. High-through put phenotypic screen and transcriptional analysis identify new compound sand targets for macrophage reprogramming. Nat. Commun. 2021, 12, 773. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigenreceptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Shields, C.W.t.; Evans, M.A.; Wang, L.L.; Baugh, N.; Iyer, S.; Wu, D.; Zhao, Z.; Pusuluri, A.; Ukidve, A.; Pan, D.C.; et al. Cellular backpacks for macrophage immunotherapy. Sci. Adv. 2020, 6, eaaz6579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Parayath, N.N.; Ene, C.I.; Stephan, S.B.; Koehne, A.L.; Coon, M.E.; Holland, E.C.; Stephan, M.T. Genetic programming of macrophages to performanti-tumorfunctions using targeted mRNA nanocarriers. Nat. Commun. 2019, 10, 3974. [Google Scholar] [CrossRef]
- Wei, Z.; Zhang, X.; Yong, T.; Bie, N.; Zhan, G.; Li, X.; Liang, Q.; Li, J.; Yu, J.; Huang, G.; et al. Boosting anti-PD-1 therapy with metformin-loaded macrophage-derived microparticles. Nat. Commun. 2021, 12, 440. [Google Scholar] [CrossRef]
- Eiro, N.; Sendon-Lago, J.; Seoane, S.; Bermudez, M.A.; Lamelas, M.L.; Garcia-Caballero, T.; Schneider, J.; Perez-Fernandez, R.; Vizoso, F.J. Potential therapeutic effect of the secretome from human uterine cervical stem cells against both cancer and stromal cells compared with a dipose tissue stem cells. Oncotarget 2014, 5, 10692–10708. [Google Scholar] [CrossRef] [Green Version]
- Silini, A.R.; Magatti, M.; Cargnoni, A.; Parolini, O. Is immune modulation the mechanism underlying the beneficial effects of amniotic cells and their derivatives in regenerative medicine? Cell Transplant. 2017, 26, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, N.; Watkins-Schulz, R.; Junkins, R.D.; David, C.N.; Johnson, B.M.; Montgomery, S.A.; Peine, K.J.; Darr, D.B.; Yuan, H.; McKinnon, K.P.; et al. Ananoparticle-incorporated STING activator enhances antitumorimmunity in PD-L1-insensitive models of triple-negative breast cancer. J. CI Insight 2018, 3. [Google Scholar] [CrossRef]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of a poptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.X.; Weissman, I.L. Phagocytosis check points as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Yoon, K.W. Dead cell phagocytosis and in nateimmune check point. BMB Rep. 2017, 50, 496–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in anti-tumor treatments targeting the CD47/SIR palpha axis. Front. Immunol. 2020, 11, 18. [Google Scholar] [CrossRef] [Green Version]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Zou, S.S.; Qiao, Y.; Zhu, S.; Gao, B.; Yang, N.; Liu, Y.J.; Chen, J. Intrinsic strategies for the evasion of cGAS-STING signaling -mediated immune surveillance in human cancer: How therapy can overcome them. Pharmacol. Res. 2021, 166, 105514. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Hornung, V. Molecular mechanism sand cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Qi, J.; Zhao, Q.; Wu, Q.N.; Wei, D.L.; Wei, X.L.; Liu, J.; Chen, J.; Zeng, Z.L.; Ju, H.Q.; et al. Targeting the STING pathway in tumor-associated macrophages regulates in nateimmune sensing of gastric cancer cells. Theranostics 2020, 10, 498–515. [Google Scholar] [CrossRef]
- Lv, M.; Chen, M.; Zhang, R.; Zhang, W.; Wang, C.; Zhang, Y.; Wei, X.; Guan, Y.; Liu, J.; Feng, K.; et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. 2020, 30, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through acytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TAM Targeted Strategies | Compound | Targets | Therapy | Tumor Type | Phase | References |
|---|---|---|---|---|---|---|
| Inhibit the recruitment | BMS-813160 | CCR2/5-inhibitor | Combination | Non-small Cell Lung Cancer or Hepatocellular Carcinoma, Pancreatic Ductal Adenocarcinoma | 1,2 | NCT04123379, NCT03496662 |
| Carlumab | anti-CCL2 antibodies | Single agent | Prostate Cancer | 2 | NCT00992186 | |
| Plerixafor | CXCR4/CXCL12 inhibitor | Combination | Metastatic Pancreatic Cancer | 2 | NCT04177810 | |
| BL-8040 | CXCR4 antagonist | Combination | Metastatic Pancreatic Adenocarcinoma | 2 | NCT02907099 | |
| DCC-3014 | CSF-1R inhibitor | Single agent | Advanced Malignant Neoplasm | 1, 2 | NCT03069469 | |
| SNDX-6352 | CSF-1R inhibitor | Combination | Solid Tumor, Metastatic Tumor, Unresectable Intrahepatic Cholangiocarcinoma | 1, 2 | NCT03238027, NCT04301778 | |
| TPX-0022 | MET/CSF1/SRC inhibitor | Single agent | Advanced Solid Tumor Metastatic Solid Tumors MET Gene Alterations | 1 | NCT03993873 | |
| LY3022855 | CSF-1R inhibitor | Combination | Melanoma | 1, 2 | NCT03101254 | |
| IMC-CS4 | CSF-1 R mAb | Combination | Pancreatic Cancer | 1 | NCT03153410 | |
| Cabiralizumab | CSF-1 R mAb | Combination | Advanced Melanoma, Non-small Cell Lung Cancer, Renal Cell Carcinoma | 1 | NCT03502330 | |
| Regorafenib | CSF-1R | Combination or Single agent | Hepatocellular Carcinoma | 1, 2 | NCT04170556 | |
| Active phagocytic checkpoint | STI-6643 | Anti-CD47 mAb | Single agent | Solid Tumor | 1 | NCT04900519 |
| Hu5F9-G4 | Anti-CD47 mAb | Combination | Hematological Malignancies | 1 | NCT03248479 | |
| TTI-621 | Anti SIRPαFc | Combination or Single agent | Hematologic Malignancies or Solid Tumor | 1 | NCT02663518 |
| TAM Targeted Strategies | Method | Therapy | References |
|---|---|---|---|
| Reprogramming M2-like into M1-like phenotypes | Thiostrepton | M1-activating compound could reprogram M2-like into M1-like phenotypes | [119] |
| CAR-M | Chimeric adenoviral vector transfer macrophage to proinflammation status (M1-like) | [120] | |
| Nanoparticle | Nanocarrier delivery interferon regulatory factor 5 and IKKβ to polarize macrophage to M1-like phenotypes | [122] | |
| EVs | Isolate normal MSCs derived Evs or conditional medium to switch M2-like to M1-like phnotypes | [124,125] | |
| Regulation of cGAS-STING pathway in TAMs | Nanoparticle | Liposomal nanoparticle-delivered cGAMP to TAMs to promote M1-like polarization | [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Yung, M.M.H.; Ngan, H.Y.S.; Chan, K.K.L.; Chan, D.W. The Impact of the Tumor Microenvironment on Macrophage Polarization in Cancer Metastatic Progression. Int. J. Mol. Sci. 2021, 22, 6560. https://doi.org/10.3390/ijms22126560
Wang H, Yung MMH, Ngan HYS, Chan KKL, Chan DW. The Impact of the Tumor Microenvironment on Macrophage Polarization in Cancer Metastatic Progression. International Journal of Molecular Sciences. 2021; 22(12):6560. https://doi.org/10.3390/ijms22126560
Chicago/Turabian StyleWang, Huogang, Mingo M. H. Yung, Hextan Y. S. Ngan, Karen K. L. Chan, and David W. Chan. 2021. "The Impact of the Tumor Microenvironment on Macrophage Polarization in Cancer Metastatic Progression" International Journal of Molecular Sciences 22, no. 12: 6560. https://doi.org/10.3390/ijms22126560
APA StyleWang, H., Yung, M. M. H., Ngan, H. Y. S., Chan, K. K. L., & Chan, D. W. (2021). The Impact of the Tumor Microenvironment on Macrophage Polarization in Cancer Metastatic Progression. International Journal of Molecular Sciences, 22(12), 6560. https://doi.org/10.3390/ijms22126560