
Neurodegeneration in Niemann–Pick Type C Disease: An Updated Review on Pharmacological and Non-Pharmacological Approaches to Counteract Brain and Cognitive Impairment

 ,
, 
 , ,
, ,
Abstract
:1. Introduction
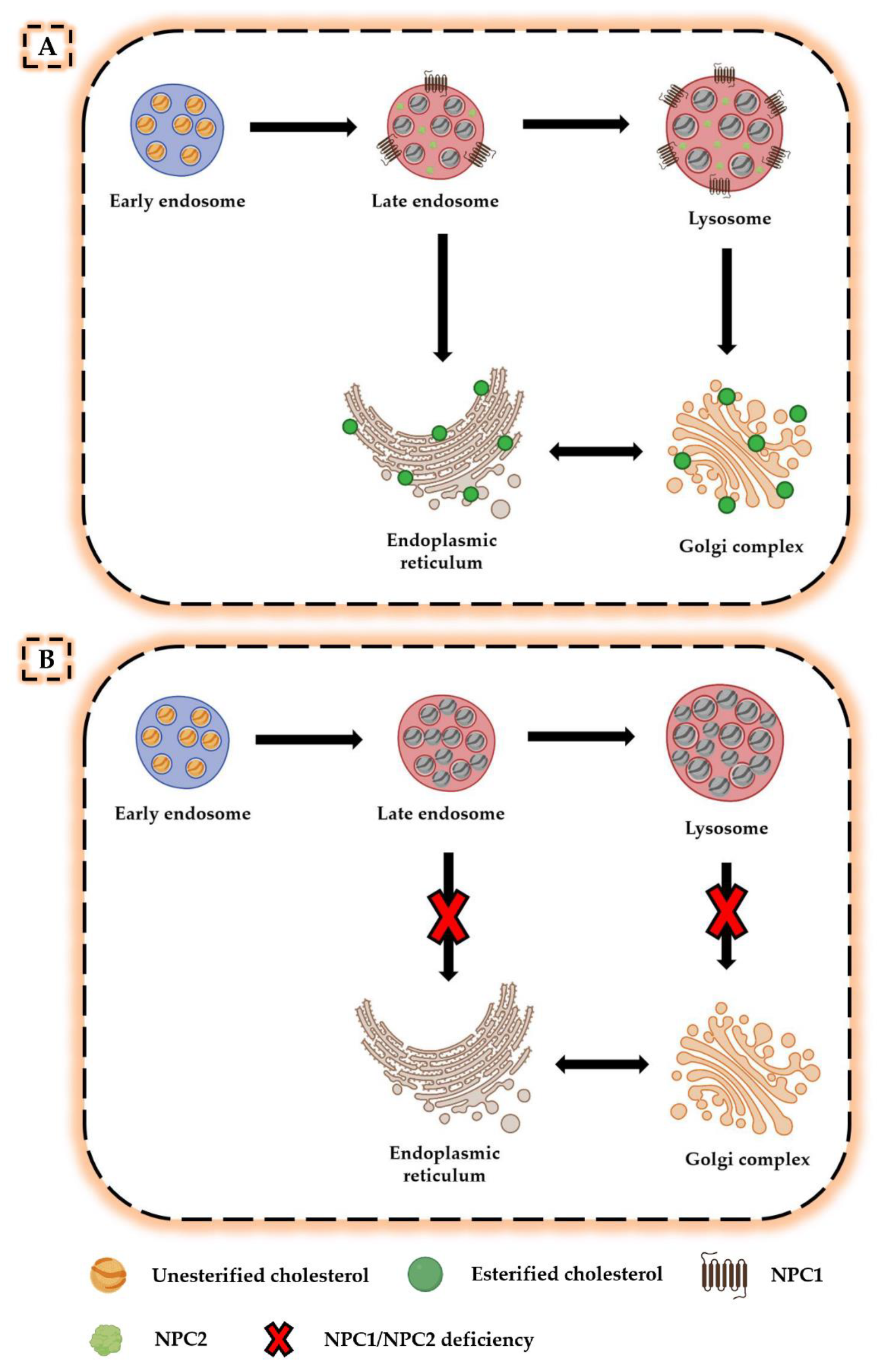
2. Neurodegeneration in NPC Disease
2.1. Molecular and Biochemical Events
2.2. Hyperexcitability and Altered Glutamatergic Neurotransmission in NPC Disease
3. Therapeutic Approaches Used to Counteract Cognitive Deficits
3.1. Miglustat Treatment
3.2. Cyclodextrin Treatment
4. Additional Non-Pharmacologic Approaches for Neurodegeneration
4.1. Physical Exercise
Effects of Physical Exercise on Synaptic Plasticity in Npc1 Model Mice
4.2. Nutrition
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newton, J.; Milstien, S.; Spiegel, S. Niemann-Pick type C disease: The atypical sphingolipidosis. Adv. Biol. Regul. 2018, 70, 82–88. [Google Scholar] [CrossRef]
- Wheeler, S.; Sillence, D.J. Niemann-Pick type C disease: Cellular pathology and pharmacotherapy. J. Neurochem. 2020, 153, 674–692. [Google Scholar] [CrossRef] [Green Version]
- Bi, X.; Liao, G. Cholesterol in Niemann-Pick Type C Disease. In Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2010; Volume 51, pp. 319–335. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Evans, E.; Platt, F.M. Lipids on trial: The search for the offending metabolite in Niemann-Pick type C disease. Traffic 2010, 11, 419–428. [Google Scholar] [CrossRef]
- Newton, J.; Hait, N.C.; Maceyka, M.; Colaco, A.; Maczis, M.; Wassif, C.A.; Cougnoux, A.; Porter, F.D.; Milstien, S.; Platt, N.; et al. FTY720/fingolimod increases NPC1 and NPC2 expression and reduces cholesterol and sphingolipid accumulation in Niemann-Pick type C mutant fibroblasts. FASEB J. 2017, 31, 1719–1730. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.L.; Gabrielides, C.; Davidson, R.K.; Swingler, T.E.; Clark, I.M.; Wallis, G.A.; Boot-Handford, R.P.; Kirkwood, T.B.L.; Taylor, R.W.; Young, D.A. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann. Rheum. Dis. 2010, 69, 1502–1510. [Google Scholar] [CrossRef] [Green Version]
- Héron, B.; Valayannopoulos, V.; Baruteau, J.; Chabrol, B.; Ogier, H.; Latour, P.; Dobbelaere, D.; Eyer, D.; Labarthe, F.; Maurey, H.; et al. Miglustat therapy in the French cohort of paediatric patients with Niemann-Pick disease type C. Orphanet J. Rare Dis. 2012, 7, 36. [Google Scholar] [CrossRef]
- Mauch, D.H.; Nägler, K.; Schumacher, S.; Göritz, C.; Müller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Pagano, R.E. Endocytic trafficking of glycosphingolipids in sphingolipid storage diseases. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2003, 358, 885–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.L.; Motamed, M.; Infante, R.E.; Abi-Mosleh, L.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 2010, 12, 166–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Brown, M.S.; Shelton, J.M.; Richardson, J.A.; Goldstein, J.L.; Liang, G. Amino acid substitution in NPC1 that abolishes cholesterol binding reproduces phenotype of complete NPC1 deficiency in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 15330–15335. [Google Scholar] [CrossRef] [Green Version]
- Wojtanik, K.M.; Liscum, L. The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem. 2003, 278, 14850–14856. [Google Scholar] [CrossRef] [Green Version]
- Berzina, Z.; Solanko, L.M.; Mehadi, A.S.; Jensen, M.L.V.; Lund, F.W.; Modzel, M.; Szomek, M.; Solanko, K.A.; Dupont, A.; Nielsen, G.K.; et al. Niemann-Pick C2 protein regulates sterol transport between plasma membrane and late endosomes in human fibroblasts. Chem. Phys. Lipids 2018, 213, 48–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodošček, M.; Elghobashi-Meinhardt, N. Simulations of NPC1(NTD):NPC2 Protein Complex Reveal Cholesterol Transfer Pathways. Int. J. Mol. Sci. 2018, 19, 2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, M.B.L.; Kidmose, R.T.; Szomek, M.; Thaysen, K.; Rawson, S.; Muench, S.P.; Wüstner, D.; Pedersen, B.P. Structural Insight into Eukaryotic Sterol Transport through Niemann-Pick Type C Proteins. Cell 2019, 179, 485–497.e18. [Google Scholar] [CrossRef]
- Elghobashi-Meinhardt, N. Computational Tools Unravel Putative Sterol Binding Sites in the Lysosomal NPC1 Protein. J. Chem. Inf. Model. 2019, 59, 2432–2441. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Coutavas, E.; Shi, H.; Hao, Q.; Blobel, G. Structure of human Niemann-Pick C1 protein. Proc. Natl. Acad. Sci. USA 2016, 113, 8212–8217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, S.; Schmid, R.; Sillence, D.J. Lipid–Protein Interactions in Niemann–Pick Type C Disease: Insights from Molecular Modeling. Int. J. Mol. Sci. 2019, 20, 717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanier, M.T.; Millat, G. Niemann-Pick disease type C. Clin. Genet. 2003, 64, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Reddy, J.V.; Ganley, I.G.; Pfeffer, S.R. Clues to neuro-degeneration in Niemann-Pick type C disease from global gene expression profiling. PLoS ONE 2006, 1, e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liscum, L.; Faust, J.R. Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J. Biol. Chem. 1987, 262, 17002–17008. [Google Scholar] [CrossRef]
- Olkkonen, V.M.; Levine, T.P. Oxysterol binding proteins: In more than one place at one time? Biochem. Cell Biol. 2004, 82, 87–98. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Platt, F.M. Lysosomal Ca(2+) homeostasis: Role in pathogenesis of lysosomal storage diseases. Cell Calcium 2011, 50, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Kågedal, K.; Zhao, M.; Svensson, I.; Brunk, U.T. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem. J. 2001, 359, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.C.; Hendriksz, C.J.; Walterfang, M.; Sedel, F.; Vanier, M.T.; Wijburg, F. Recommendations for the diagnosis and management of Niemann-Pick disease type C: An update. Mol. Genet. Metab. 2012, 106, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Raas-Rothschild, A.; Reish, O.; Regev, M.; Meiner, V.; Bargal, R.; Sury, V.; Meir, K.; Nadjari, M.; Hermann, G.; et al. The clinical spectrum of fetal Niemann-Pick type C. Am. J. Med. Genet. A 2009, 149A, 446–450. [Google Scholar] [CrossRef]
- Trendelenburg, G.; Vanier, M.T.; Maza, S.; Millat, G.; Bohner, G.; Munz, D.L.; Zschenderlein, R. Niemann-Pick type C disease in a 68-year-old patient. J. Neurol. Neurosurg. Psychiatry 2006, 77, 997–998. [Google Scholar] [CrossRef] [Green Version]
- Sévin, M.; Lesca, G.; Baumann, N.; Millat, G.; Lyon-Caen, O.; Vanier, M.T.; Sedel, F. The adult form of Niemann-Pick disease type C. Brain 2007, 130, 120–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garver, W.S.; Francis, G.A.; Jelinek, D.; Shepherd, G.; Flynn, J.; Castro, G.; Walsh Vockley, C.; Coppock, D.L.; Pettit, K.M.; Heidenreich, R.A.; et al. The National Niemann-Pick C1 disease database: Report of clinical features and health problems. Am. J. Med. Genet. A 2007, 143A, 1204–1211. [Google Scholar] [CrossRef]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassif, C.A.; Cross, J.L.; Iben, J.; Sanchez-Pulido, L.; Cougnoux, A.; Platt, F.M.; Ory, D.S.; Ponting, C.P.; Bailey-Wilson, J.E.; Biesecker, L.G.; et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet. Med. 2016, 18, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Arenas, F.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Intracellular Cholesterol Trafficking and Impact in Neurodegeneration. Front. Mol. Neurosci. 2017, 10, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, M.L.; Krus, K.L.; Lieberman, A.P. Lysosome and endoplasmic reticulum quality control pathways in Niemann-Pick type C disease. Brain Res. 2016, 1649, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malnar, M.; Hecimovic, S.; Mattsson, N.; Zetterberg, H. Bidirectional links between Alzheimer’s disease and Niemann-Pick type C disease. Neurobiol. Dis. 2014, 72, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Elrick, M.J.; Pacheco, C.D.; Yu, T.; Dadgar, N.; Shakkottai, V.G.; Ware, C.; Paulson, H.L.; Lieberman, A.P. Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum. Mol. Genet. 2010, 19, 837–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Shakkottai, V.G.; Chung, C.; Lieberman, A.P. Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum. Mol. Genet. 2011, 20, 4440–4451. [Google Scholar] [CrossRef]
- Lopez, M.E.; Klein, A.D.; Dimbil, U.J.; Scott, M.P. Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J. Neurosci. 2011, 31, 4367–4378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Gong, J.-S.; Ko, M.; Garver, W.S.; Yanagisawa, K.; Michikawa, M. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J. Biol. Chem. 2005, 280, 11731–11739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, A.; Llacuna, L.; Fernández-Checa, J.C.; Colell, A. Mitochondrial cholesterol loading exacerbates amyloid beta peptide-induced inflammation and neurotoxicity. J. Neurosci. 2009, 29, 6394–6405. [Google Scholar] [CrossRef] [Green Version]
- Benussi, A.; Cotelli, M.S.; Padovani, A.; Borroni, B. Recent neuroimaging, neurophysiological, and neuropathological advances for the understanding of NPC. F1000Research 2018, 7, 194. [Google Scholar] [CrossRef] [Green Version]
- Love, S.; Bridges, L.R.; Case, C.P. Neurofibrillary tangles in Niemann-Pick disease type C. Brain 1995, 118, 119–129. [Google Scholar] [CrossRef]
- Malnar, M.; Kosicek, M.; Mitterreiter, S.; Omerbasic, D.; Lichtenthaler, S.F.; Goate, A.; Hecimovic, S. Niemann-Pick type C cells show cholesterol dependent decrease of APP expression at the cell surface and its increased processing through the beta-secretase pathway. Biochim. Biophys. Acta 2010, 1802, 682–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, T.; Chang, T.Y.; Haass, C.; Ihara, Y. Accumulation and aggregation of amyloid beta-protein in late endosomes of Niemann-pick type C cells. J. Biol. Chem. 2001, 276, 4454–4460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runz, H.; Rietdorf, J.; Tomic, I.; de Bernard, M.; Beyreuther, K.; Pepperkok, R.; Hartmann, T. Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J. Neurosci. 2002, 22, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.-W.; Shie, F.-S.; Maezawa, I.; Vincent, I.; Bird, T. Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am. J. Pathol. 2004, 164, 975–985. [Google Scholar] [CrossRef]
- Burns, M.; Gaynor, K.; Olm, V.; Mercken, M.; LaFrancois, J.; Wang, L.; Mathews, P.M.; Noble, W.; Matsuoka, Y.; Duff, K. Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J. Neurosci. 2003, 23, 5645–5649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guénette, S.Y.; Chen, J.; Jondro, P.D.; Tanzi, R.E. Association of a novel human FE65-like protein with the cytoplasmic domain of the beta-amyloid precursor protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10832–10837. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Tesco, G.; Jeong, W.J.; Lindsley, L.; Eckman, E.A.; Eckman, C.B.; Tanzi, R.E.; Guénette, S.Y. Generation of the beta-amyloid peptide and the amyloid precursor protein C-terminal fragment gamma are potentiated by FE65L1. J. Biol. Chem. 2003, 278, 51100–51107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kågedal, K.; Kim, W.S.; Appelqvist, H.; Chan, S.; Cheng, D.; Agholme, L.; Barnham, K.; McCann, H.; Halliday, G.; Garner, B. Increased expression of the lysosomal cholesterol transporter NPC1 in Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 831–838. [Google Scholar] [CrossRef]
- Yu, T.; Lieberman, A.P. Npc1 acting in neurons and glia is essential for the formation and maintenance of CNS myelin. PLoS Genet. 2013, 9, e1003462. [Google Scholar] [CrossRef] [Green Version]
- Takikita, S.; Fukuda, T.; Mohri, I.; Yagi, T.; Suzuki, K. Perturbed myelination process of premyelinating oligodendrocyte in Niemann-Pick type C mouse. J. Neuropathol. Exp. Neurol. 2004, 63, 660–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, E.A.; Velakoulis, D.; Desmond, P.; Walterfang, M. Longitudinal Changes in White Matter Fractional Anisotropy in Adult-Onset Niemann-Pick Disease Type C Patients Treated with Miglustat. JIMD Rep. 2018, 39, 39–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masingue, M.; Adanyeguh, I.; Nadjar, Y.; Sedel, F.; Galanaud, D.; Mochel, F. Evolution of structural neuroimaging biomarkers in a series of adult patients with Niemann-Pick type C under treatment. Orphanet J. Rare Dis. 2017, 12, 22. [Google Scholar] [CrossRef] [Green Version]
- Rego, T.; Farrand, S.; Goh, A.M.Y.; Eratne, D.; Kelso, W.; Mangelsdorf, S.; Velakoulis, D.; Walterfang, M. Psychiatric and Cognitive Symptoms Associated with Niemann-Pick Type C Disease: Neurobiology and Management. CNS Drugs 2019, 33, 125–142. [Google Scholar] [CrossRef]
- Peake, K.B.; Vance, J.E. Defective cholesterol trafficking in Niemann-Pick C-deficient cells. FEBS Lett. 2010, 584, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Burns, D.K.; Turley, S.D.; Dietschy, J.M. Cholesterol is sequestered in the brains of mice with Niemann-Pick type C disease but turnover is increased. J. Neuropathol. Exp. Neurol. 2000, 59, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Repa, J.J.; Valasek, M.A.; Beltroy, E.P.; Turley, S.D.; German, D.C.; Dietschy, J.M. Molecular, anatomical, and biochemical events associated with neurodegeneration in mice with Niemann-Pick type C disease. J. Neuropathol. Exp. Neurol. 2005, 64, 323–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Lund, E.G.; Turley, S.D.; Russell, D.W.; Dietschy, J.M. Quantitation of two pathways for cholesterol excretion from the brain in normal mice and mice with neurodegeneration. J. Lipid Res. 2003, 44, 1780–1789. [Google Scholar] [CrossRef] [Green Version]
- Lund, E.G.; Guileyardo, J.M.; Russell, D.W. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. USA 1999, 96, 7238–7243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repa, J.J.; Mangelsdorf, D.J. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Rev. Cell Dev. Biol. 2000, 16, 459–481. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Yan, Z.; Patel, R.C.; Patel, Y.C.; Patel, S.C. Cellular cholesterol storage in the Niemann-Pick disease type C mouse is associated with increased expression and defective processing of apolipoprotein D. J. Neurochem. 1998, 70, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Rall, S.C.J. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef]
- Wu, Y.-P.; Mizukami, H.; Matsuda, J.; Saito, Y.; Proia, R.L.; Suzuki, K. Apoptosis accompanied by up-regulation of TNF-alpha death pathway genes in the brain of Niemann-Pick type C disease. Mol. Genet. Metab. 2005, 84, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Vivas, O.; Tiscione, S.A.; Dixon, R.E.; Ory, D.S.; Dickson, E.J. Niemann-Pick Type C Disease Reveals a Link between Lysosomal Cholesterol and PtdIns(4,5)P(2) That Regulates Neuronal Excitability. Cell Rep. 2019, 27, 2636–2648.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuisset, J.-M.; Sukno, S.; Trauffler, A.; Latour, P.; Dobbelaere, D.; Michaud, L.; Vallée, L. Impact of miglustat on evolution of atypical presentation of late-infantile-onset Niemann-Pick disease type C with early cognitive impairment, behavioral dysfunction, epilepsy, ophthalmoplegia, and cerebellar involvement: A case report. J. Med. Case Rep. 2016, 10, 241. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett. 2006, 580, 5518–5524. [Google Scholar] [CrossRef] [Green Version]
- Grassi, S.; Giussani, P.; Mauri, L.; Prioni, S.; Sonnino, S.; Prinetti, A. Lipid rafts and neurodegeneration: Structural and functional roles in physiologic aging and neurodegenerative diseases. J. Lipid Res. 2020, 61, 636–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, C.; Rufini, S.; Tancredi, V.; Forcina, R.; Grossi, D.; D’Arcangelo, G. Cholesterol depletion inhibits synaptic transmission and synaptic plasticity in rat hippocampus. Exp. Neurol. 2008, 212, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, W.A.; Kurland, L.T. The epidemiology of epilepsy in Rochester, Minnesota, 1935 through 1967. Epilepsia 1975, 16, 1–66. [Google Scholar] [CrossRef] [PubMed]
- Cariati, I.; Bonanni, R.; Pallone, G.; Annino, G.; Tancredi, V.; D’Arcangelo, G. Modulation of Synaptic Plasticity by Vibratory Training in Young and Old Mice. Brain Sci. 2021, 11, 82. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Grossi, D.; De Chiara, G.; de Stefano, M.C.; Cortese, G.; Citro, G.; Rufini, S.; Tancredi, V.; Merlo, D.; Frank, C. Glutamatergic neurotransmission in a mouse model of Niemann-Pick type C disease. Brain Res. 2011, 1396, 11–19. [Google Scholar] [CrossRef]
- Calabresi, P.; Centonze, D.; Pisani, A.; Cupini, L.; Bernardi, G. Synaptic plasticity in the ischaemic brain. Lancet. Neurol. 2003, 2, 622–629. [Google Scholar] [CrossRef]
- Calabresi, P.; Saulle, E.; Centonze, D.; Pisani, A.; Marfia, G.A.; Bernardi, G. Post-ischaemic long-term synaptic potentiation in the striatum: A putative mechanism for cell type-specific vulnerability. Brain 2002, 125, 844–860. [Google Scholar] [CrossRef] [Green Version]
- Lo Castro, A.; Murdocca, M.; Pucci, S.; Zaratti, A.; Greggi, C.; Sangiuolo, F.; Tancredi, V.; Frank, C.; D’Arcangelo, G. Early Hippocampal i-LTP and LOX-1 Overexpression Induced by Anoxia: A Potential Role in Neurodegeneration in NPC Mouse Model. Int. J. Mol. Sci. 2017, 18, 1442. [Google Scholar] [CrossRef] [Green Version]
- Crocker, A.C.; Farber, S. Niemann-Pick disease: A review of eighteen patients. Medicine (Baltimore) 1958, 37, 1–95. [Google Scholar] [CrossRef]
- Sylvain, M.; Arnold, D.L.; Scriver, C.R.; Schreiber, R.; Shevell, M.I. Magnetic resonance spectroscopy in Niemann-Pick disease type C: Correlation with diagnosis and clinical response to cholestyramine and lovastatin. Pediatr. Neurol. 1994, 10, 228–232. [Google Scholar] [CrossRef]
- Patterson, M.C.; Di Bisceglie, A.M.; Higgins, J.J.; Abel, R.B.; Schiffmann, R.; Parker, C.C.; Argoff, C.E.; Grewal, R.P.; Yu, K.; Pentchev, P.G. The effect of cholesterol-lowering agents on hepatic and plasma cholesterol in Niemann-Pick disease type C. Neurology 1993, 43, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare Dis. 2018, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Erickson, R.P.; Garver, W.S.; Camargo, F.; Hossain, G.S.; Heidenreich, R.A. Pharmacological and genetic modifications of somatic cholesterol do not substantially alter the course of CNS disease in Niemann-Pick C mice. J. Inherit. Metab. Dis. 2000, 23, 54–62. [Google Scholar] [CrossRef]
- Beheregaray, A.P.C.; Souza, F.T.S.; Coelho, J.C. Effect of a peroxysomal proliferator agent on intracellular cholesterol accumulation in cultured fibroblasts from Niemann-Pick type C disease patients. Clin. Chim. Acta 2003, 336, 137–142. [Google Scholar] [CrossRef]
- Wraith, J.E.; Baumgartner, M.R.; Bembi, B.; Covanis, A.; Levade, T.; Mengel, E.; Pineda, M.; Sedel, F.; Topçu, M.; Vanier, M.T.; et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol. Genet. Metab. 2009, 98, 152–165. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Miglustat: A review of its use in Niemann-Pick disease type C. Drugs 2014, 74, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Vecchio, D.; Jacklin, E.; Abel, L.; Chadha-Boreham, H.; Luzy, C.; Giorgino, R.; Patterson, M.C. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: Long-term data from a clinical trial. Mol. Genet. Metab. 2010, 99, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.E.; Pastores, G.M.; Gianutsos, J.; Luzy, C.; Kolodny, E.H. Miglustat in late-onset Tay-Sachs disease: A 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genet. Med. 2009, 11, 425–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, A.; Bagel, J.; Sampson, M.; Farhat, N.; Ding, W.; Swain, G.; Prociuk, M.; O’Donnell, P.; Drobatz, K.; Gurda, B.; et al. Cerebrospinal Fluid Calbindin D Concentration as a Biomarker of Cerebellar Disease Progression in Niemann-Pick Type C1 Disease. J. Pharmacol. Exp. Ther. 2016, 358, 254–261. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Evans, E.; Pelled, D.; Riebeling, C.; Bodennec, J.; de-Morgan, A.; Waller, H.; Schiffmann, R.; Futerman, A.H. Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. J. Biol. Chem. 2003, 278, 23594–23599. [Google Scholar] [CrossRef] [Green Version]
- Patterson, M.C.; Mengel, E.; Vanier, M.T.; Schwierin, B.; Muller, A.; Cornelisse, P.; Pineda, M. Stable or improved neurological manifestations during miglustat therapy in patients from the international disease registry for Niemann-Pick disease type C: An observational cohort study. Orphanet J. Rare Dis. 2015, 10, 65. [Google Scholar] [CrossRef] [Green Version]
- Pineda, M.; Wraith, J.E.; Mengel, E.; Sedel, F.; Hwu, W.-L.; Rohrbach, M.; Bembi, B.; Walterfang, M.; Korenke, G.C.; Marquardt, T.; et al. Miglustat in patients with Niemann-Pick disease Type C (NP-C): A multicenter observational retrospective cohort study. Mol. Genet. Metab. 2009, 98, 243–249. [Google Scholar] [CrossRef]
- Fecarotta, S.; Romano, A.; Della Casa, R.; Del Giudice, E.; Bruschini, D.; Mansi, G.; Bembi, B.; Dardis, A.; Fiumara, A.; Di Rocco, M.; et al. Long term follow-up to evaluate the efficacy of miglustat treatment in Italian patients with Niemann-Pick disease type C. Orphanet J. Rare Dis. 2015, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Zervas, M.; Somers, K.L.; Thrall, M.A.; Walkley, S.U. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr. Biol. 2001, 11, 1283–1287. [Google Scholar] [CrossRef] [Green Version]
- Zervas, M.; Dobrenis, K.; Walkley, S.U. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J. Neuropathol. Exp. Neurol. 2001, 60, 49–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, V.M.; Crooks, A.; Ding, W.; Prociuk, M.; O’Donnell, P.; Bryan, C.; Sikora, T.; Dingemanse, J.; Vanier, M.T.; Walkley, S.U.; et al. Miglustat improves purkinje cell survival and alters microglial phenotype in feline Niemann-Pick disease type C. J. Neuropathol. Exp. Neurol. 2012, 71, 434–448. [Google Scholar] [CrossRef] [Green Version]
- D’Arcangelo, G.; Grossi, D.; Racaniello, M.; Cardinale, A.; Zaratti, A.; Rufini, S.; Cutarelli, A.; Tancredi, V.; Merlo, D.; Frank, C. Miglustat Reverts the Impairment of Synaptic Plasticity in a Mouse Model of NPC Disease. Neural Plast. 2016, 2016, 3830424. [Google Scholar] [CrossRef] [Green Version]
- D’Arcangelo, G.; Triossi, T.; Buglione, A.; Melchiorri, G.; Tancredi, V. Modulation of synaptic plasticity by short-term aerobic exercise in adult mice. Behav. Brain Res. 2017, 332, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Roch Santed, M.; Cabañas Poy, M.J.; Del Toro Riera, M.; Cañete Ramírez, C.; Fernández Polo, A.; Clemente Bautista, S. Intrathecal cyclodextrin in the treatment of Niemann-Pick disease type C. Eur. J. Hosp. Pharm. Sci. Pract. 2017, 24, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Matencio, A.; Navarro-Orcajada, S.; González-Ramón, A.; García-Carmona, F.; López-Nicolás, J.M. Recent advances in the treatment of Niemann pick disease type C: A mini-review. Int. J. Pharm. 2020, 584, 119440. [Google Scholar] [CrossRef] [PubMed]
- Ory, D.S.; Ottinger, E.A.; Farhat, N.Y.; King, K.A.; Jiang, X.; Weissfeld, L.; Berry-Kravis, E.; Davidson, C.D.; Bianconi, S.; Keener, L.A.; et al. Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: A non-randomised, open-label, phase 1-2 trial. Lancet (London, UK) 2017, 390, 1758–1768. [Google Scholar] [CrossRef] [Green Version]
- Grobin, A.C.; Heenan, E.J.; Lieberman, J.A.; Morrow, A.L. Perinatal neurosteroid levels influence GABAergic interneuron localization in adult rat prefrontal cortex. J. Neurosci. 2003, 23, 1832–1839. [Google Scholar] [CrossRef] [Green Version]
- Concas, A.; Mostallino, M.C.; Porcu, P.; Follesa, P.; Barbaccia, M.L.; Trabucchi, M.; Purdy, R.H.; Grisenti, P.; Biggio, G. Role of brain allopregnanolone in the plasticity of gamma-aminobutyric acid type A receptor in rat brain during pregnancy and after delivery. Proc. Natl. Acad. Sci. USA 1998, 95, 13284–13289. [Google Scholar] [CrossRef] [Green Version]
- Griffin, L.D.; Gong, W.; Verot, L.; Mellon, S.H. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat. Med. 2004, 10, 704–711. [Google Scholar] [CrossRef]
- Davidson, C.D.; Ali, N.F.; Micsenyi, M.C.; Stephney, G.; Renault, S.; Dobrenis, K.; Ory, D.S.; Vanier, M.T.; Walkley, S.U. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE 2009, 4, e6951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Li, H.; Repa, J.J.; Turley, S.D.; Dietschy, J.M. Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid Res. 2008, 49, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, M.; Shraishi, K.; Wada, K.; Ishitsuka, Y.; Doi, H.; Maeda, M.; Mizoguchi, T.; Eto, J.; Mochinaga, S.; Arima, H.; et al. Effects of intracerebroventricular administration of 2-hydroxypropyl-β-cyclodextrin in a patient with Niemann-Pick Type C disease. Mol. Genet. Metab. Rep. 2014, 1, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Caporali, P.; Dolas, A.; Johny, S.; Goyal, S.; Dragotto, J.; Macone, A.; Jayaraman, R.; Fiorenza, M.T. Linear Cyclodextrin Polymer Prodrugs as Novel Therapeutics for Niemann-Pick Type C1 Disorder. Sci. Rep. 2018, 8, 9547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontikis, C.C.; Davidson, C.D.; Walkley, S.U.; Platt, F.M.; Begley, D.J. Cyclodextrin alleviates neuronal storage of cholesterol in Niemann-Pick C disease without evidence of detectable blood-brain barrier permeability. J. Inherit. Metab. Dis. 2013, 36, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crumling, M.A.; Liu, L.; Thomas, P.V.; Benson, J.; Kanicki, A.; Kabara, L.; Hälsey, K.; Dolan, D.; Duncan, R.K. Hearing loss and hair cell death in mice given the cholesterol-chelating agent hydroxypropyl-β-cyclodextrin. PLoS ONE 2012, 7, e53280. [Google Scholar] [CrossRef]
- Ward, S.; O’Donnell, P.; Fernandez, S.; Vite, C.H. 2-hydroxypropyl-beta-cyclodextrin raises hearing threshold in normal cats and in cats with Niemann-Pick type C disease. Pediatr. Res. 2010, 68, 52–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, K.I.; Hillman, C.; Stillman, C.M.; Ballard, R.M.; Bloodgood, B.; Conroy, D.E.; Macko, R.; Marquez, D.X.; Petruzzello, S.J.; Powell, K.E. Physical Activity, Cognition, and Brain Outcomes: A Review of the 2018 Physical Activity Guidelines. Med. Sci. Sports Exerc. 2019, 51, 1242–1251. [Google Scholar] [CrossRef]
- Hamer, M.; Chida, Y. Physical activity and risk of neurodegenerative disease: A systematic review of prospective evidence. Psychol. Med. 2009, 39, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yan, T.; Chu, J.M.-T.; Chen, Y.; Dunnett, S.; Ho, Y.-S.; Wong, G.T.-C.; Chang, R.C.-C. The beneficial effects of physical exercise in the brain and related pathophysiological mechanisms in neurodegenerative diseases. Lab. Invest. 2019, 99, 943–957. [Google Scholar] [CrossRef]
- Di Liegro, C.M.; Schiera, G.; Proia, P.; Di Liegro, I. Physical Activity and Brain Health. Genes (Basel) 2019, 10, 720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, B.K. Physical activity and muscle-brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Dahlin, E.; Andersson, M.; Thorén, A.; Hanse, E.; Seth, H. Effects of physical exercise and stress on hippocampal CA1 and dentate gyrus synaptic transmission and long-term potentiation in adolescent and adult Wistar rats. Neuroscience 2019, 408, 22–30. [Google Scholar] [CrossRef]
- Brown, J.; Cooper-Kuhn, C.M.; Kempermann, G.; Van Praag, H.; Winkler, J.; Gage, F.H.; Kuhn, H.G. Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur. J. Neurosci. 2003, 17, 2042–2046. [Google Scholar] [CrossRef] [PubMed]
- Farmer, J.; Zhao, X.; van Praag, H.; Wodtke, K.; Gage, F.H.; Christie, B.R. Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague-Dawley rats in vivo. Neuroscience 2004, 124, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Van Praag, H.; Shubert, T.; Zhao, C.; Gage, F.H. Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 2005, 25, 8680–8685. [Google Scholar] [CrossRef]
- Erickson, K.I.; Voss, M.W.; Prakash, R.S.; Basak, C.; Szabo, A.; Chaddock, L.; Kim, J.S.; Heo, S.; Alves, H.; White, S.M.; et al. Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. USA 2011, 108, 3017–3022. [Google Scholar] [CrossRef] [Green Version]
- Firth, J.; Stubbs, B.; Vancampfort, D.; Schuch, F.; Lagopoulos, J.; Rosenbaum, S.; Ward, P.B. Effect of aerobic exercise on hippocampal volume in humans: A systematic review and meta-analysis. Neuroimage 2018, 166, 230–238. [Google Scholar] [CrossRef]
- Palmieri, M.; Cariati, I.; Scimeca, M.; Pallone, G.; Bonanno, E.; Tancredi, V.; D’Arcangelo, G.; Frank, C. Effects of short-term aerobic exercise in a mouse model of Niemann-Pick type C disease on synaptic and muscle plasticity. Ann. Ist. Super. Sanita 2019, 55. [Google Scholar] [CrossRef]
- Cariati, I.; Scimeca, M.; Tancredi, V.; D’Amico, A.G.; Pallone, G.; Palmieri, M.; Frank, C.; D’Arcangelo, G. Effects of different continuous aerobic training protocols in a heterozygous mouse model of Niemann-Pick type C disease. J. Funct. Morphol. Kinesiol. 2020, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Pallone, G.; Palmieri, M.; Cariati, I.; Bei, R.; Masuelli, L.; D’Arcangelo, G.; Tancredi, V. Different continuous training modalities result in distinctive effects on muscle structure, plasticity and function. Biomed. Rep. 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Scholl-Bürgi, S.; Höller, A.; Pichler, K.; Michel, M.; Haberlandt, E.; Karall, D. Ketogenic diets in patients with inherited metabolic disorders. J. Inherit. Metab. Dis. 2015, 38, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Höller, A.; Albrecht, U.; Baumgartner Sigl, S.; Zöggeler, T.; Ramoser, G.; Bernar, B.; Karall, D.; Scholl-Bürgi, S. Successful implementation of classical ketogenic dietary therapy in a patient with Niemann-Pick disease type C. Mol. Genet. Metab. Rep. 2021, 27, 100723. [Google Scholar] [CrossRef]
- Marín, T.; Contreras, P.; Castro, J.F.; Chamorro, D.; Balboa, E.; Bosch-Morató, M.; Muñoz, F.J.; Alvarez, A.R.; Zanlungo, S. Vitamin E dietary supplementation improves neurological symptoms and decreases c-Abl/p73 activation in Niemann-Pick C mice. Nutrients 2014, 6, 3000–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, C.A.; Borbon, I.A.; Erickson, R.P. Relative efficacy of nicotinamide treatment of a mouse model of infantile Niemann-Pick C1 disease. J. Appl. Genet. 2017, 58, 99–102. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UC Protocol | PC Protocol | VC Protocol | |
|---|---|---|---|
| Main features | Single session training at 9 RPM, without speed changes | Incremental speed changes with gradually increasing exercise intensity. Intensity increases in 2 RPM intervals from 10 to 32 RPM, with 12 speed changes | Two series of 8-min bi-pyramidal inversion exercise, with a 2-min active recovery at 10 RPM between series |
| Training session duration | 30 min | 18 min | 18 min |
| Weekly frequency | 3 times a week | 3 times a week | 3 times (VCt) or 2 times (VCb) a week |
| Training period | 3 weeks | 12 weeks | 12 weeks |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cariati, I.; Masuelli, L.; Bei, R.; Tancredi, V.; Frank, C.; D’Arcangelo, G. Neurodegeneration in Niemann–Pick Type C Disease: An Updated Review on Pharmacological and Non-Pharmacological Approaches to Counteract Brain and Cognitive Impairment. Int. J. Mol. Sci. 2021, 22, 6600. https://doi.org/10.3390/ijms22126600
Cariati I, Masuelli L, Bei R, Tancredi V, Frank C, D’Arcangelo G. Neurodegeneration in Niemann–Pick Type C Disease: An Updated Review on Pharmacological and Non-Pharmacological Approaches to Counteract Brain and Cognitive Impairment. International Journal of Molecular Sciences. 2021; 22(12):6600. https://doi.org/10.3390/ijms22126600
Chicago/Turabian StyleCariati, Ida, Laura Masuelli, Roberto Bei, Virginia Tancredi, Claudio Frank, and Giovanna D’Arcangelo. 2021. "Neurodegeneration in Niemann–Pick Type C Disease: An Updated Review on Pharmacological and Non-Pharmacological Approaches to Counteract Brain and Cognitive Impairment" International Journal of Molecular Sciences 22, no. 12: 6600. https://doi.org/10.3390/ijms22126600
APA StyleCariati, I., Masuelli, L., Bei, R., Tancredi, V., Frank, C., & D’Arcangelo, G. (2021). Neurodegeneration in Niemann–Pick Type C Disease: An Updated Review on Pharmacological and Non-Pharmacological Approaches to Counteract Brain and Cognitive Impairment. International Journal of Molecular Sciences, 22(12), 6600. https://doi.org/10.3390/ijms22126600