
SOS2 Comes to the Fore: Differential Functionalities in Physiology and Pathology



Abstract
:1. SOS2 vs. SOS1 Function: An Introductory Timeline Perspective
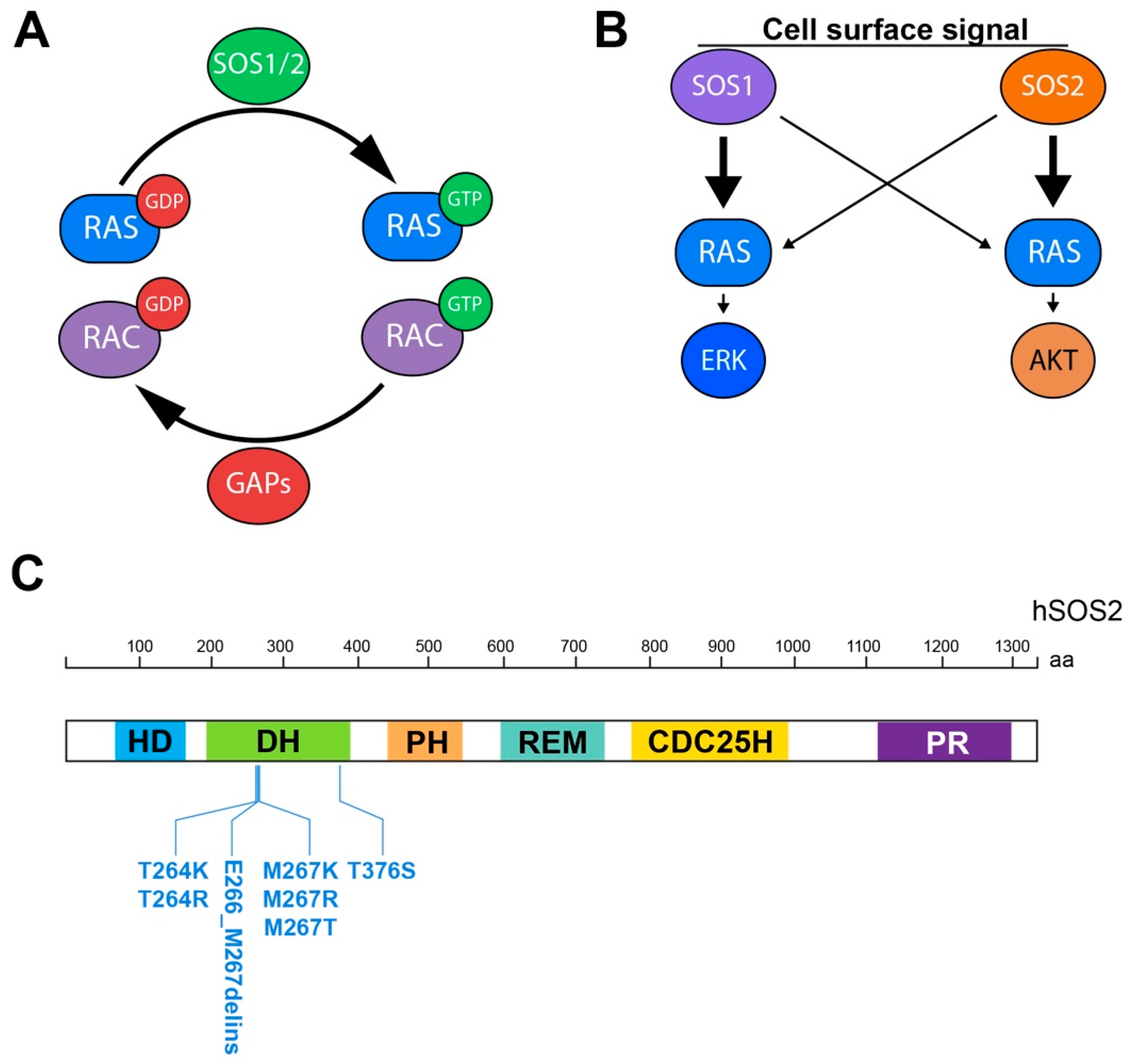
1.1. Ras GEFs and the SOS Family
1.2. Functional Redundancy/Specificity of SOS2 vs. SOS1
1.3. Hierarchy of Action of the SOS Family Members
1.4. Distinct Functional Roles of SOS2 and SOS1 in the Skin and Epidermal Cancers
1.5. Differential Involvement of SOS2 and SOS1 in Cellular Pathological Contexts
1.6. SOS1/2 Inhibitors in Pathological Settings

{kind=link}
| Compound | Mode of Action | Preclinical/Clinical Trial Identifier | Reference |
|---|---|---|---|
| Sotorasib (AMG510) | KRASG12C inhibitor | NCT04185883 NCT03600883 NCT04303780 | [43,44,45] |
| BAY-293 | SOS1 inhibitor | Preclinical | [30,47] |
| BI-3406 | SOS1 inhibitor | Preclinical | [23] |
| BI-1701963 | SOS1 inhibitor | NCT04111458 | https://clinicaltrials.gov/ct2/show/NCT04111458 (accessed on 20 June 2021) |
2. SOS2 and SOS1: So Similar but So Functionally Different. Some Mechanistic Considerations
2.1. Is SOS2 a Bona Fide Rac-GEF In Vivo?
2.2. SOS2 as a Key Modulator of PI3K–AKT Signaling
3. SOS2 Functional Role(s) in Pathological Contexts
3.1. SOS2 in Noonan Syndrome
3.2. SOS2 in Sporadic Cancers
| SOS2 Disrution Strategy | Tumor Cell Line | Phenotypic Effect | Reference |
|---|---|---|---|
| miR-148a | BJAB and DG-75 and U2932 (B cell lymphoma and Burkitt lymphoma) | Reduction of ERK activation | [68] |
| miR-148a-3p | A549, HCC827 (lung cancer) | Reduction of proliferation and EM transition | [69] |
| miR-193a-3p | HEK293, SKOV3, and OVCA433 (ovarian cancer) | Suppression of MAPK–ERK signal transmission | [70] |
| SOS2 KO mice | MEFs expressing mutant RAS isoforms: HRASG12V, NRASG12V, or KRASG12V | Impairment of RTK-dependent AKT phosphorylation Dispensable for RTK-dependent ERK activation | [31] |
| CRISPR/Cas9 | H358 NSCLC cells (lung cancer) | Revert the transformed phenotype of KRAS mutant cells. SOS2 participates in anchorage-independent, but not in anchorage-dependent, growth. | [31,32] |
| CRISPR/Cas9 | H23 NSCLC cells (lung cancer) | SOS2 participates in anchorage-independent growth. Reduce cell viability. | [32] |
| CRISPR/Cas9 | SW620 (colorrectal cancer) | SOS2 participates in anchorage-independent growth. | [32] |
| CRISPR/Cas9 | NCI-H1299 NSCLC cells (lung cancer) | SOS2 participates in anchorage-independent growth. | [32] |
| CRISPR/Cas9 | YAPC cells (pancreatic cancer) | Revert the transformed phenotype of KRAS oncogenic cells. | [31] |
3.3. SOS2 in Non-Tumoral Pathologies
Author Contributions
Funding
Conflicts of Interest
References
- Buday, L.; Downward, J. Many faces of Ras activation. Biochim. Biophys. Acta 2008, 1786, 178–187. [Google Scholar] [CrossRef]
- Cherfils, J.; Zeghouf, M. Regulation of Small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [Green Version]
- Hennig, A.; Markwart, R.; Esparza-Franco, M.A.; Ladds, G.; Rubio, I. Ras activation revisited: Role of GEF and GAP systems. Biol. Chem. 2015, 396, 831–848. [Google Scholar] [CrossRef]
- Castellano, E.; Santos, E. Functional Specificity of Ras Isoforms: So Similar but So Different. Genes Cancer 2011, 2, 216–231. [Google Scholar] [CrossRef] [Green Version]
- Baltanás, F.C.; Zarich, N.; Rojas-Cabañeros, J.M.; Santos, E. SOS GEFs in health and disease. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188445. [Google Scholar] [CrossRef]
- Fernández-Medarde, A.; Santos, E. The RasGrf family of mammalian guanine nucleotide exchange factors. Biochim. Biophys. Acta Rev. Cancer 2011, 1815, 170–188. [Google Scholar] [CrossRef]
- Ksionda, O.; Limnander, A.; Roose, J.P. RasGRP Ras guanine nucleotide exchange factors in cancer. Front. Biol. 2013, 8, 508–532. [Google Scholar]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Tian, X.; Hartley, D.M.; Feig, L.A. Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J. Neurosci. 2006, 26, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Gómez, C.; Jimeno, D.; Fernández-Medarde, A.; García-Navas, R.; Calzada, N.; Santos, E. Ras-GRF2 regulates nestin-positive stem cell density and onset of differentiation during adult neurogenesis in the mouse dentate gyrus. Mol. Cell. Neurosci. 2017, 85, 127–147. [Google Scholar] [CrossRef]
- Stone, J.C. Regulation of Ras in lymphocytes: Get a GRP. Biochem. Soc. Trans. 2006, 34, 858–861. [Google Scholar] [CrossRef] [Green Version]
- Stone, J.C. Regulation and function of the rasGRP family of ras activators in blood cells. Genes Cancer 2011, 2, 320–334. [Google Scholar] [CrossRef]
- Innocenti, M.; Tenca, P.; Frittoli, E.; Faretta, M.; Tocchetti, A.; Di Fiore, P.P.; Scita, G. Mechanisms through which Sos-1 coordinates the activation of Ras and Rac. J. Cell Biol. 2002, 156, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Esteban, L.; Vass, W.C.; Upadhyaya, C.; Papageorge, A.G.; Yienger, K.; Ward, J.M.; Lowy, D.R.; Santos, E. The Sos1 and Sos2 Ras-specific exchange factors: Differences in placental expression and signaling properties. EMBO J. 2000, 19, 642–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.Z.M.; Hammond, V.E.; Abud, H.E.; Bertoncello, I.; McAvoy, J.W.; Bowtell, D.D.L. Mutation in Sos1 dominantly enhances a weak allele of the EGFR, demonstrating a requirement for sos1 in EGFR signaling and development. Genes Dev. 1997, 11, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Esteban, L.M.; Fernández-Medarde, A.; López, E.; Yienger, K.; Guerrero, C.; Ward, J.M.; Tessarollo, L.; Santos, E. Ras-guanine nucleotide exchange factor sos2 is dispensable for mouse growth and development. Mol. Cell. Biol. 2000, 20, 6410–6413. [Google Scholar] [CrossRef] [PubMed]
- Kessler, D.; Gerlach, D.; Kraut, N.; McConnell, D.B. Targeting Son of Sevenless 1: The pacemaker of KRAS. Curr. Opin. Chem. Biol. 2021, 62, 109–118. [Google Scholar] [CrossRef]
- Hong, S.H.; Yoo, D.Y.; Conway, L.; Richards-Corke, K.C.; Parker, C.G.; Arora, P.S. A Sos proteomimetic as a pan-Ras inhibitor. Proc. Natl. Acad. Sci. USA 2021, 118, e2101027118. [Google Scholar] [CrossRef]
- Baltanás, F.C.; Pérez-Andrés, M.; Ginel-Picardo, A.; Diaz, D.; Jimeno, D.; Liceras-Boillos, P.; Kortum, R.L.; Samelson, L.E.; Orfao, A.; Santos, E. Functional Redundancy of Sos1 and Sos2 for Lymphopoiesis and Organismal Homeostasis and Survival. Mol. Cell. Biol. 2013, 33, 4562–4578. [Google Scholar] [CrossRef] [Green Version]
- Liceras-Boillos, P.; García-Navas, R.; Ginel-Picardo, A.; Anta, B.; Pérez-Andrés, M.; Lillo, C.; Gómez, C.; Jimeno, D.; Fernández-Medarde, A.; Baltanás, F.C.; et al. Sos1 disruption impairs cellular proliferation and viability through an increase in mitochondrial oxidative stress in primary MEFs. Oncogene 2016, 1–14. [Google Scholar] [CrossRef]
- Liceras-Boillos, P.; Jimeno, D.; García-Navas, R.; Lorenzo-Martín, L.F.; Menacho-Marquez, M.; Segrelles, C.; Gómez, C.; Calzada, N.; Fuentes-Mateos, R.; Paramio, J.M.; et al. Differential Role of the RasGEFs Sos1 and Sos2 in Mouse Skin Homeostasis and Carcinogenesis. Mol. Cell. Biol. 2018, 38, e00049-18. [Google Scholar] [CrossRef] [Green Version]
- Suire, S.; Baltanas, F.C.; Segonds-Pichon, A.; Davidson, K.; Santos, E.; Hawkins, P.T.; Stephens, L.R. Frontline Science: TNF-α and GM-CSF1 priming augments the role of SOS1/2 in driving activation of Ras, PI3K-γ, and neutrophil proinflammatory responses. J. Leukoc. Biol. 2019, 106, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a potent and selective SOS1::KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov. 2020, CD-20-0142. [Google Scholar] [CrossRef]
- Gerboth, S.; Frittoli, E.; Palamidessi, A.; Baltanas, F.C.; Salek, M.; Rappsilber, J.; Giuliani, C.; Troglio, F.; Rolland, Y.; Pruneri, G.; et al. Phosphorylation of SOS1 on tyrosine 1196 promotes its RAC GEF activity and contributes to BCR-ABL leukemogenesis. Leukemia 2018, 32, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Baltanás, F.C.; Mucientes-Valdivieso, C.; Lorenzo-Martín, L.F.; Fernández-Parejo, N.; García-Navas, R.; Segrelles, C.; Calzada, N.; Fuentes-Mateos, R.; Paramio, J.M.; Bustelo, X.R.; et al. Functional Specificity of the Members of the Sos Family of Ras-GEF Activators: Novel Role of Sos2 in Control of Epidermal Stem Cell Homeostasis. Cancers 2021, 13, 2152. [Google Scholar] [CrossRef]
- Garcia-Navas, R.; Liceras-Boillos, P.; Gomez, C.; Baltanás, F.C.; Nuevo-Tapioles, C.; Cuezva, J.; Calzada, N.; Santos, E. Critical requirement of SOS1 RAS-GEF function for mitochondrial dynamics, metabolism and redox homeostasis. Oncogene 2021, in press. [Google Scholar] [CrossRef]
- You, X.; Kong, G.; Ranheim, E.A.; Yang, D.; Zhou, Y.; Zhang, J. Unique dependence on Sos1 in KrasG12D-induced leukemogenesis. Blood 2018, 132, 2575–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibilia, M.; Fleischmann, A.; Behrens, A.; Stingl, L.; Carroll, J.; Watt, F.M.; Schlessinger, J.; Wagner, E.F. The EGF Receptor Provides an Essential Survival Signal for SOS-Dependent Skin Tumor Development. Cell 2000, 102, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Wong, G.S.; Zhou, J.; Liu, J.B.; Wu, Z.; Xu, X.; Li, T.; Xu, D.; Schumacher, S.E.; Puschhof, J.; McFarland, J.; et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 2018, 24, 968–977. [Google Scholar] [CrossRef]
- Theard, P.L.; Sheffels, E.; Sealover, N.E.; Linke, A.J.; Pratico, D.J.; Kortum, R.L. Marked synergy by vertical inhibition of EGFR signaling in NSCLC spheroids shows SOS1 is a therapeutic target in EGFR-mutated cancer. eLife 2020, 9. [Google Scholar] [CrossRef]
- Sheffels, E.; Sealover, N.E.; Wang, C.; Kim, D.H.; Vazirani, I.A.; Lee, E.; Terrell, E.M.; Morrison, D.K.; Luo, J.; Kortum, R.L. Oncogenic RAS isoforms show a hierarchical requirement for the guanine nucleotide exchange factor SOS2 to mediate cell transformation. Sci. Signal. 2018, 11, eaar8371. [Google Scholar] [CrossRef] [Green Version]
- Sheffels, E.; Sealover, N.E.; Theard, P.L.; Kortum, R.L. Anchorage-independent growth conditions reveal a differential SOS2 dependence for transformation and survival in RAS -mutant cancer cells. Small GTPases 2019, 12, 67–78. [Google Scholar] [CrossRef]
- Drosten, M.; Lechuga, C.G.; Barbacid, M. Ras signaling is essential for skin development. Oncogene 2014, 33, 2857–2865. [Google Scholar] [CrossRef]
- Kern, F.; Niault, T.; Baccarini, M. Ras and Raf pathways in epidermis development and carcinogenesis. Br. J. Cancer 2011, 104, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Doma, E.; Rupp, C.; Baccarini, M. EGFR-Ras-Raf Signaling in Epidermal Stem Cells: Roles in Hair Follicle Development, Regeneration, Tissue Remodeling and Epidermal Cancers. Int. J. Mol. Sci. 2013, 14, 19361–19384. [Google Scholar] [CrossRef]
- Cordeddu, V.; Yin, J.C.; Gunnarsson, C.; Virtanen, C.; Drunat, S.; Lepri, F.; De Luca, A.; Rossi, C.; Ciolfi, A.; Pugh, T.J.; et al. Activating Mutations Affecting the Dbl Homology Domain of SOS2 Cause Noonan Syndrome. Hum. Mutat. 2015, 36, 1080–1087. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, G.L.; Aguena, M.; Gos, M.; Hung, C.; Pilch, J.; Fahiminiya, S.; Abramowicz, A.; Cristian, I.; Buscarilli, M.; Naslavsky, M.S.; et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J. Med. Genet. 2015, 52, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Zhao, W.; Yang, Z.; Wang, Y.M.; Dai, Y.; Chen, L.A. Novel resistance mechanisms to osimertinib analysed by whole-exome sequencing in non-small cell lung cancer. Cancer Manag. Res. 2021, 13, 2025–2032. [Google Scholar] [CrossRef]
- Lissewski, C.; Chune, V.; Pantaleoni, F.; De Luca, A.; Capri, Y.; Brinkmann, J.; Lepri, F.; Daniele, P.; Leenders, E.; Mazzanti, L.; et al. Variants of SOS2 are a rare cause of Noonan syndrome with particular predisposition for lymphatic complications. Eur. J. Hum. Genet. 2021, 29, 51–60. [Google Scholar] [CrossRef]
- Gentile, M.; Fanelli, T.; Lepri, F.R.; Gentile, A.; Orsini, P.; Volpe, P.; Novelli, A.; Ficarella, R. First prenatal case of Noonan syndrome with SOS2 mutation: Implications of early diagnosis for genetic counseling. Am. J. Med. Genet. Part A 2021. [Google Scholar] [CrossRef]
- Indini, A.; Rijavec, E.; Ghidini, M.; Cortellini, A.; Grossi, F. Targeting KRAS in Solid Tumors: Current Challenges and Future Opportunities of Novel KRAS Inhibitors. Pharmaceutics 2021, 13, 653. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS G12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, NEJMoa2103695. [Google Scholar] [CrossRef]
- Reck, M.; Carbone, D.P.; Garassino, M.; Barlesi, F. Targeting KRAS in non-small cell lung cancer: Recent progress and new approaches. Ann. Oncol. 2021. [Google Scholar] [CrossRef]
- Sheffels, E.; Kortum, R.L. Breaking Oncogene Addiction: Getting RTK/RAS-Mutated Cancers off the SOS. J. Med. Chem. 2021, 64, 6566–6568. [Google Scholar] [CrossRef]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J.; et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 2551–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, T.; Suda, K.; Fujino, T.; Ohara, S.; Hamada, A.; Nishino, M.; Chiba, M.; Shimoji, M.; Takemoto, T.; Arita, T.; et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From the In Vitro Experiments. J. Thorac. Oncol. 2021. [Google Scholar] [CrossRef]
- Shi, T.; Niepel, M.; McDermott, J.E.; Gao, Y.; Nicora, C.D.; Chrisler, W.B.; Markillie, L.M.; Petyuk, V.A.; Smith, R.D.; Rodland, K.D.; et al. Conservation of protein abundance patterns reveals the regulatory architecture of the EGFR-MAPK pathway. Sci. Signal. 2016, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, D.; Choi, P.S.; Gelbard, M.; Meyerson, M. Identification and characterization of oncogenic SOS1 mutations in lung adenocarcinoma. Mol. Cancer Res. 2019, 17, 1002–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minella, D.; Wannenes, F.; Biancolella, M.; Amati, F.; Testa, B.; Nardone, A.; Bueno, S.; Fabbri, A.; Lauro, D.; Novelli, G.; et al. SOS1 over-expression in genital skin fibroblasts from hirsute women: A putative role of the SOS1/RAS pathway in the pathogenesis of hirsutism. J. Biol. Regul. Homeost. Agents 2011, 25, 615–626. [Google Scholar]
- Yang, S.S.; Van Aelst, L.; Bar-Sagi, D. Differential interactions of human Sos1 and Sos2 with Grb2. J. Biol. Chem. 1995, 270, 18212–18215. [Google Scholar] [CrossRef] [Green Version]
- Dubiel, D.; Rockel, B.; Naumann, M.; Dubiel, W. Diversity of COP9 signalosome structures and functional consequences. FEBS Lett. 2015, 589, 2507–2513. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, K.H.; Papageorge, A.G.; Vass, W.C.; Willumsen, B.M.; Lowy, D.R. The Ras-specific exchange factors mouse Sos1 (mSos1) and mSos2 are regulated differently: MSos2 contains ubiquitination signals absent in mSos1. Mol. Cell. Biol. 1997, 17, 7132–7138. [Google Scholar] [CrossRef] [Green Version]
- Zarich, N.; Anta, B.; Fernández-Medarde, A.; Ballester, A.; de Lucas, M.P.; Cámara, A.B.; Anta, B.; Oliva, J.L.; Rojas-Cabañeros, J.M.; Santos, E. The CSN3 subunit of the COP9 signalosome interacts with the HD region of Sos1 regulating stability of this GEF protein. Oncogenesis 2019, 8, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, K.K.; Bar-Sagi, D. Allosteric gating of Son of sevenless activity by the histone domain. Proc. Natl. Acad. Sci. USA 2010, 107, 3436–3440. [Google Scholar] [CrossRef] [Green Version]
- Sheffels, E.; Kortum, R.L. The Role of Wild-Type RAS in Oncogenic RAS Transformation. Genes 2021, 12, 662. [Google Scholar] [CrossRef] [PubMed]
- Scita, G.; Nordstrom, J.; Carbone, R.; Tenca, P.; Giardina, G.; Gutkind, S.; Bjarnegård, M.; Betsholtz, C.; Di Fiore, P.P. EPS8 and E3B1 transduce signals from Ras to Rac. Nature 1999, 401, 290–293. [Google Scholar] [CrossRef]
- Fan, P.-D.; Goff, S.P. Abl Interactor 1 Binds to Sos and Inhibits Epidermal Growth Factor- and v-Abl-Induced Activation of Extracellular Signal-Regulated Kinases. Mol. Cell. Biol. 2000, 20, 7591–7601. [Google Scholar] [CrossRef] [Green Version]
- Castellano, E.; Downward, J. Ras interaction with PI3K: More than just another effector pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Medarde, A.; Santos, E. Ras in Cancer and Developmental Diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [Green Version]
- Noonan, J.A. Noonan syndrome and related disorders: Alterations in growth and puberty. Rev. Endocr. Metab. Disord. 2006, 7, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, M.; Pennacchio, L.A.; Zhao, C.; Yadav, K.K.; Fodale, V.; Sarkozy, A.; Pandit, B.; Oishi, K.; Martinelli, S.; Schackwitz, W.; et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat. Genet. 2007, 39, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Tcheandjieu, C.; Aguirre, M.; Gustafsson, S.; Saha, P.; Potiny, P.; Haendel, M.; Ingelsson, E.; Rivas, M.A.; Priest, J.R. A phenome-wide association study of 26 mendelian genes reveals phenotypic expressivity of common and rare variants within the general population. PLoS Genet. 2020, 16. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Z.; Li, X.; Ye, J.; Wu, X.; Tan, Z.; Liu, C.; Shen, B.; Wang, X.-A.; Wu, W.; et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat. Genet. 2014, 46, 872–876. [Google Scholar] [CrossRef]
- Shain, A.H.; Garrido, M.; Botton, T.; Talevich, E.; Yeh, I.; Sanborn, J.Z.; Chung, J.; Wang, N.J.; Kakavand, H.; Mann, G.J.; et al. Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat. Genet. 2015, 47, 1194–1199. [Google Scholar] [CrossRef]
- Shin, J.; Choi, J.-H.; Jung, S.; Jeong, S.; Oh, J.; Yoon, D.-Y.; Rhee, M.H.; Ahn, J.; Kim, S.-H.; Oh, J.-W. MUDENG Expression Profiling in Cohorts and Brain Tumor Biospecimens to Evaluate Its Role in Cancer. Front. Genet. 2019, 10, 884. [Google Scholar] [CrossRef] [Green Version]
- Alles, J.; Ludwig, N.; Rheinheimer, S.; Leidinger, P.; Grässer, F.A.; Keller, A.; Meese, E. MiR-148a impairs Ras/ERK signaling in B lymphocytes by targeting SOS proteins. Oncotarget 2017, 8, 56417–56427. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Yu, Z.; Lu, Y.; Fan, J.; Ni, Y.; Ma, L. microRNA-148a-3p inhibited the proliferation and epithelial–mesenchymal transition progression of non-small-cell lung cancer via modulating Ras/MAPK/Erk signaling. J. Cell. Physiol. 2019, 234, 12786–12799. [Google Scholar] [CrossRef]
- Chen, K.; Liu, M.X.; Mak, C.S.L.; Yung, M.M.H.; Leung, T.H.Y.; Xu, D.; Ngu, S.F.; Chan, K.K.L.; Yang, H.; Ngan, H.Y.S.; et al. Methylation-associated silencing of miR-193a-3p promotes ovarian cancer aggressiveness by targeting GRB7 and MAPK/ERK pathways. Theranostics 2018, 8, 423–436. [Google Scholar] [CrossRef]
- Jeng, H.-H.; Taylor, L.J.; Bar-Sagi, D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat. Commun. 2012, 3, 1168. [Google Scholar] [CrossRef] [Green Version]
- Nickerson, S.; Joy, S.T.; Arora, P.S.; Bar-Sagi, D. An Orthosteric Inhibitor of the RAS?SOS Interaction. Enzymes 2013, 34 Pt B, 25–39. [Google Scholar]
- Hamilton, G.; Proitsi, P.; Jehu, L.; Morgan, A.; Williams, J.; O’Donovan, M.C.; Owen, M.J.; Powell, J.F.; Lovestone, S. Candidate gene association study of insulin signaling genes and Alzheimer’s disease: Evidence for SOS2, PCK1, and PPARγ as susceptibility loci. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2007, 144, 508–516. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Riley, L.G.; Bratkovic, D.; Ketteridge, D.; Manton, N.; Cowley, M.J.; Gayevskiy, V.; Roscioli, T.; Mohamed, M.; Gardeitchik, T.; et al. Unique presentation of cutis laxa with Leigh-like syndrome due to ECHS1 deficiency. J. Inherit. Metab. Dis. 2017, 40, 745–747. [Google Scholar] [CrossRef]
- Shaffer, J.R.; Polk, D.E.; Wang, X.; Feingold, E.; Weeks, D.E.; Lee, M.K.; Cuenco, K.T.; Weyant, R.J.; Crout, R.J.; McNeil, D.W.; et al. Genome-wide association study of periodontal health measured by probing depth in adults ages 18–49 years. G3 Genes Genomes Genet. 2014, 4, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.R.; Huang, H.; Nannini, D.R.; Fan, F.; Kim, H. Genome-wide association analyses identify new loci influencing intraocular pressure. Hum. Mol. Genet. 2018, 27, 2205–2213. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baltanás, F.C.; García-Navas, R.; Santos, E. SOS2 Comes to the Fore: Differential Functionalities in Physiology and Pathology. Int. J. Mol. Sci. 2021, 22, 6613. https://doi.org/10.3390/ijms22126613
Baltanás FC, García-Navas R, Santos E. SOS2 Comes to the Fore: Differential Functionalities in Physiology and Pathology. International Journal of Molecular Sciences. 2021; 22(12):6613. https://doi.org/10.3390/ijms22126613
Chicago/Turabian StyleBaltanás, Fernando C., Rósula García-Navas, and Eugenio Santos. 2021. "SOS2 Comes to the Fore: Differential Functionalities in Physiology and Pathology" International Journal of Molecular Sciences 22, no. 12: 6613. https://doi.org/10.3390/ijms22126613
APA StyleBaltanás, F. C., García-Navas, R., & Santos, E. (2021). SOS2 Comes to the Fore: Differential Functionalities in Physiology and Pathology. International Journal of Molecular Sciences, 22(12), 6613. https://doi.org/10.3390/ijms22126613