
Integrated Insight into the Molecular Mechanisms of Spontaneous Abortion during Early Pregnancy in Pigs

 , , and
, , and 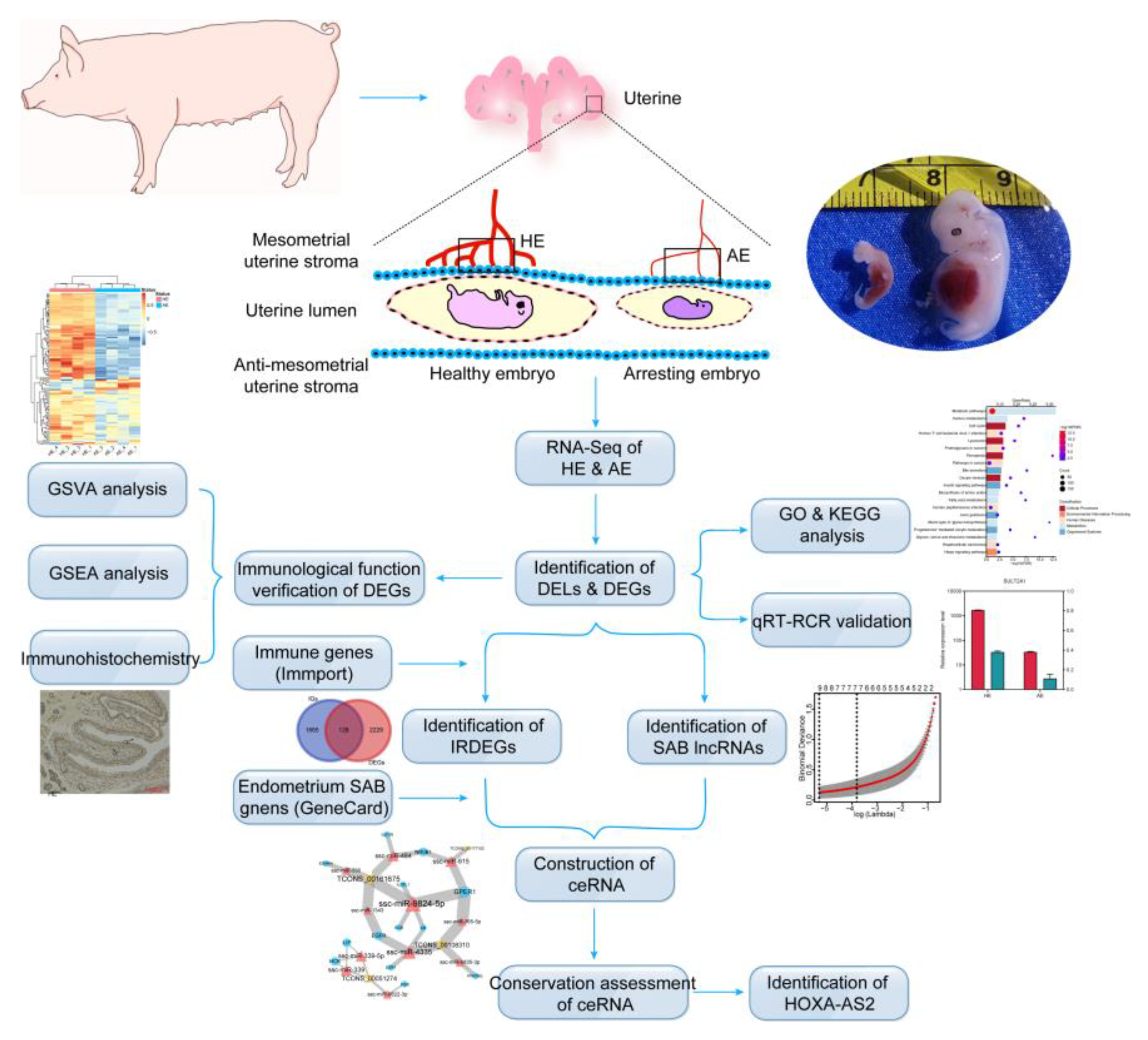{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
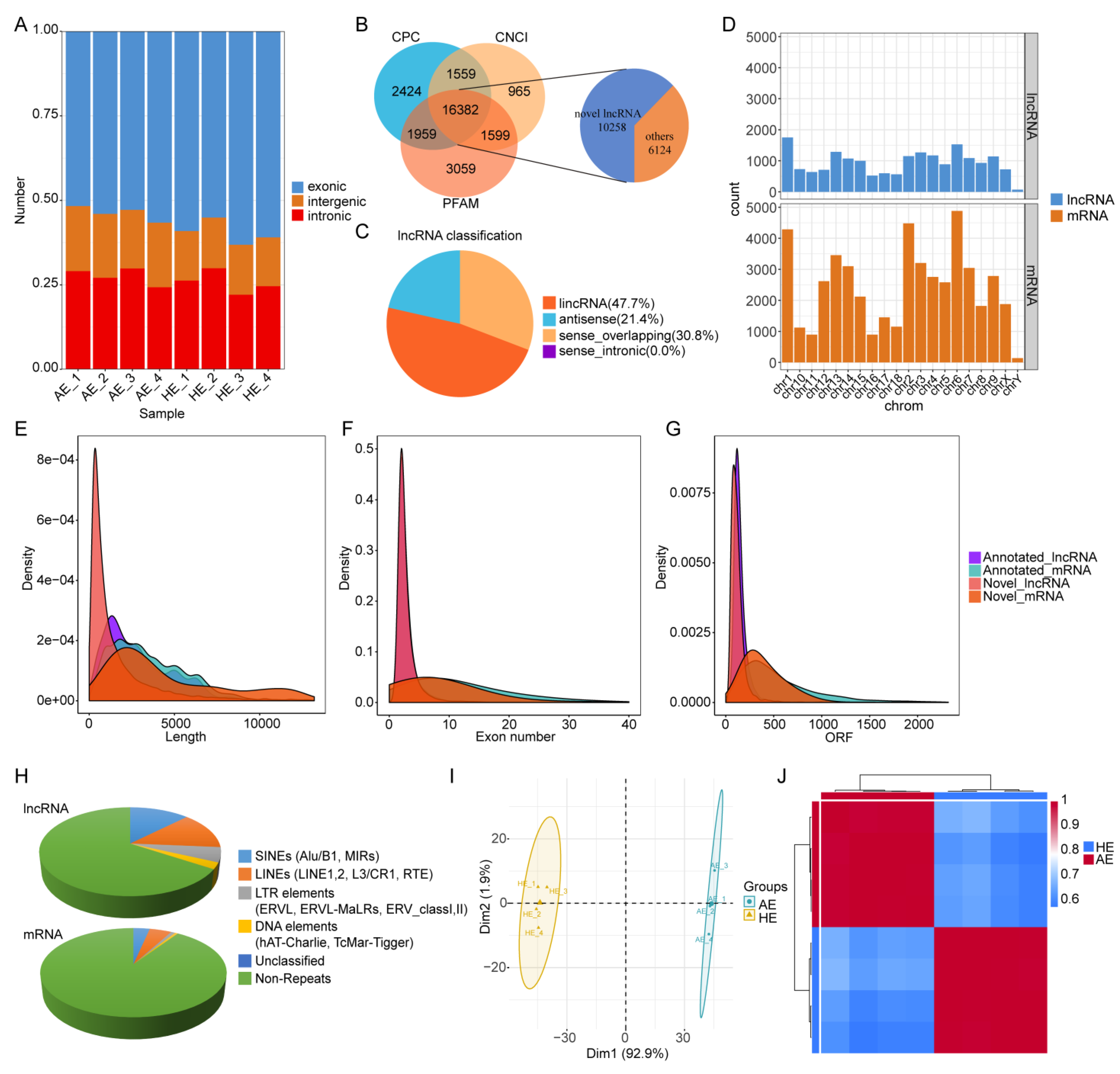
2.1. Genome-Wide Identification and Characterization of the lncRNAs and mRNAs
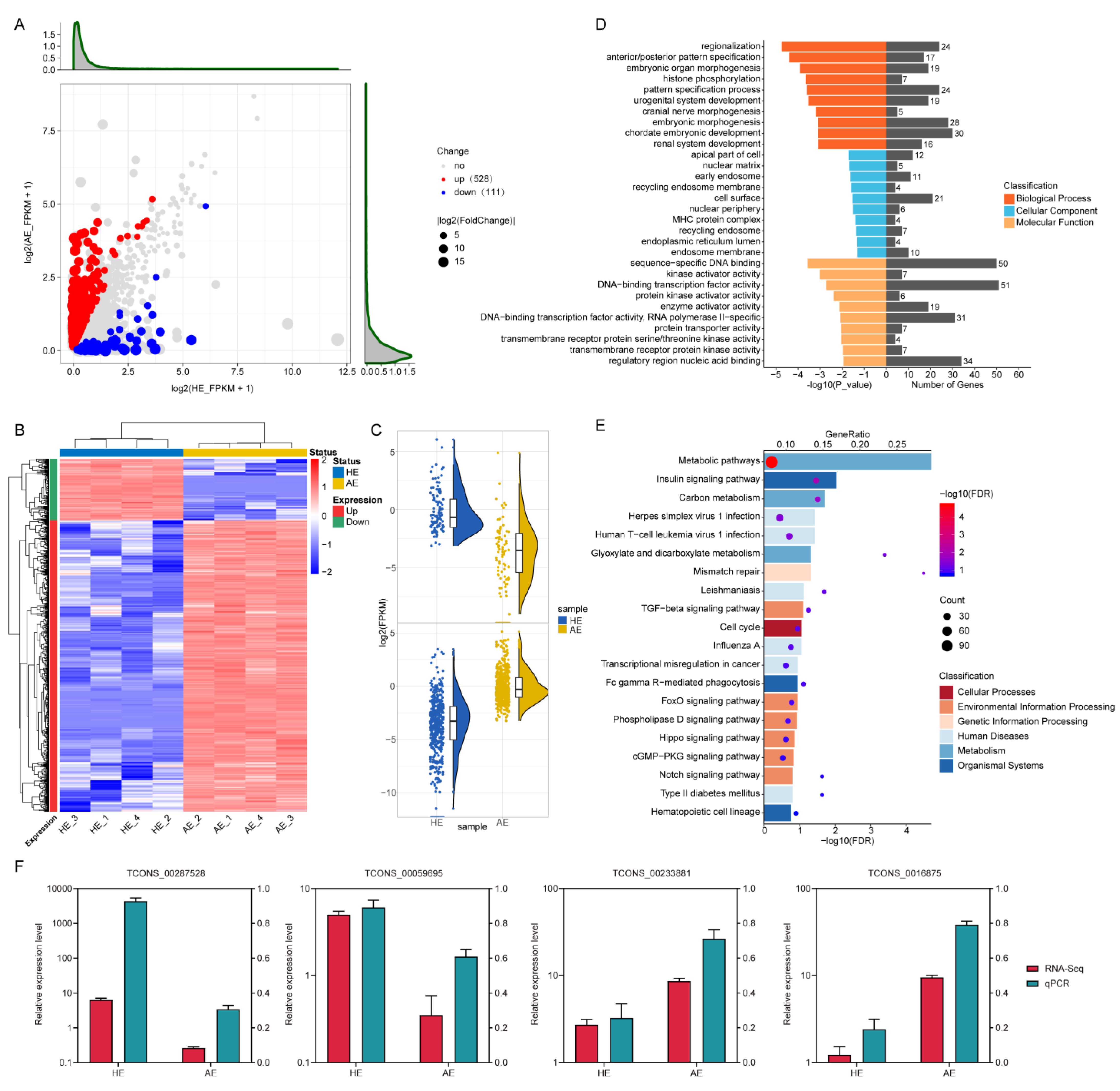
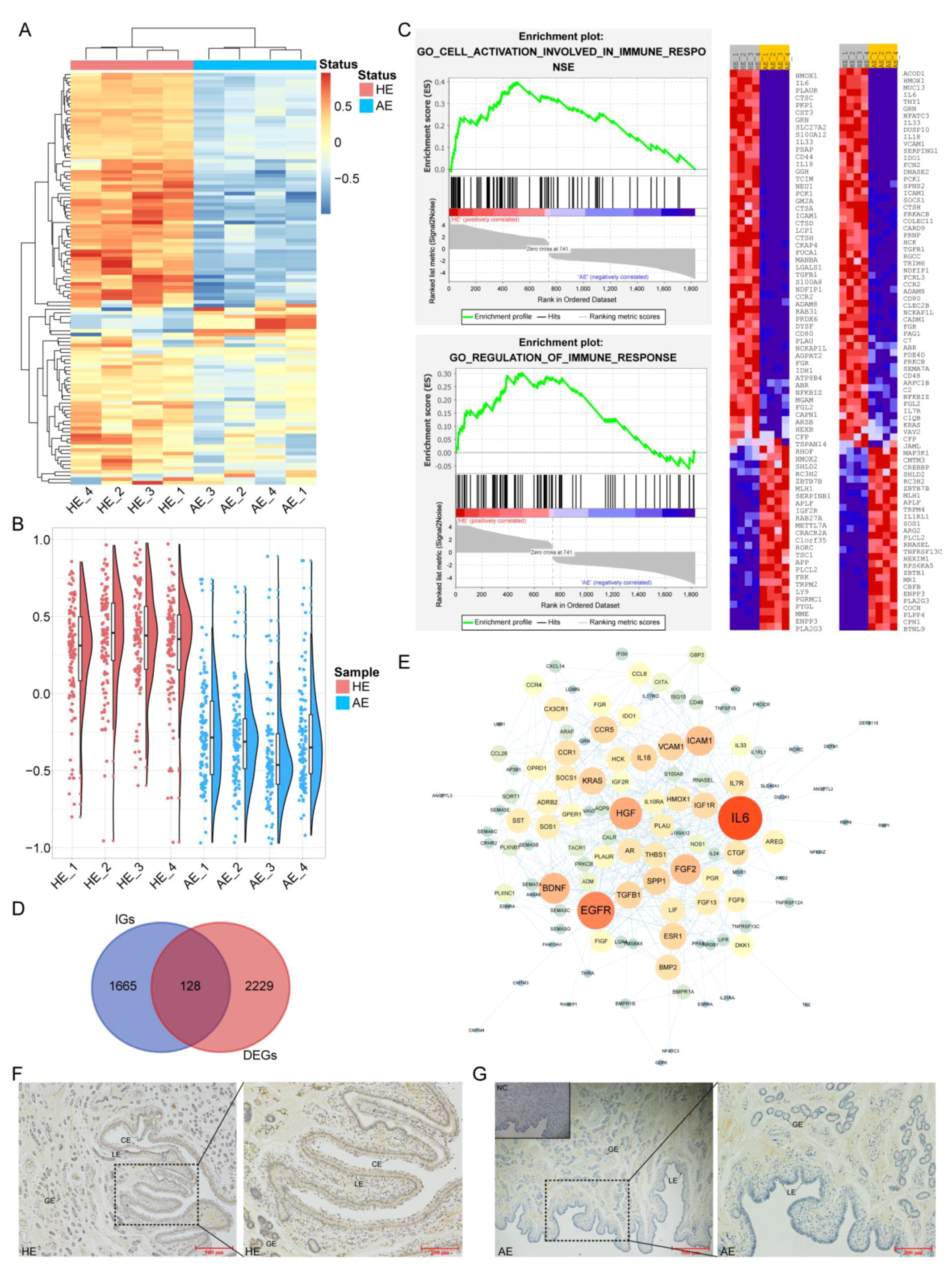
2.2. Differentially Expressed LncRNAs (DELs) and Their Functions
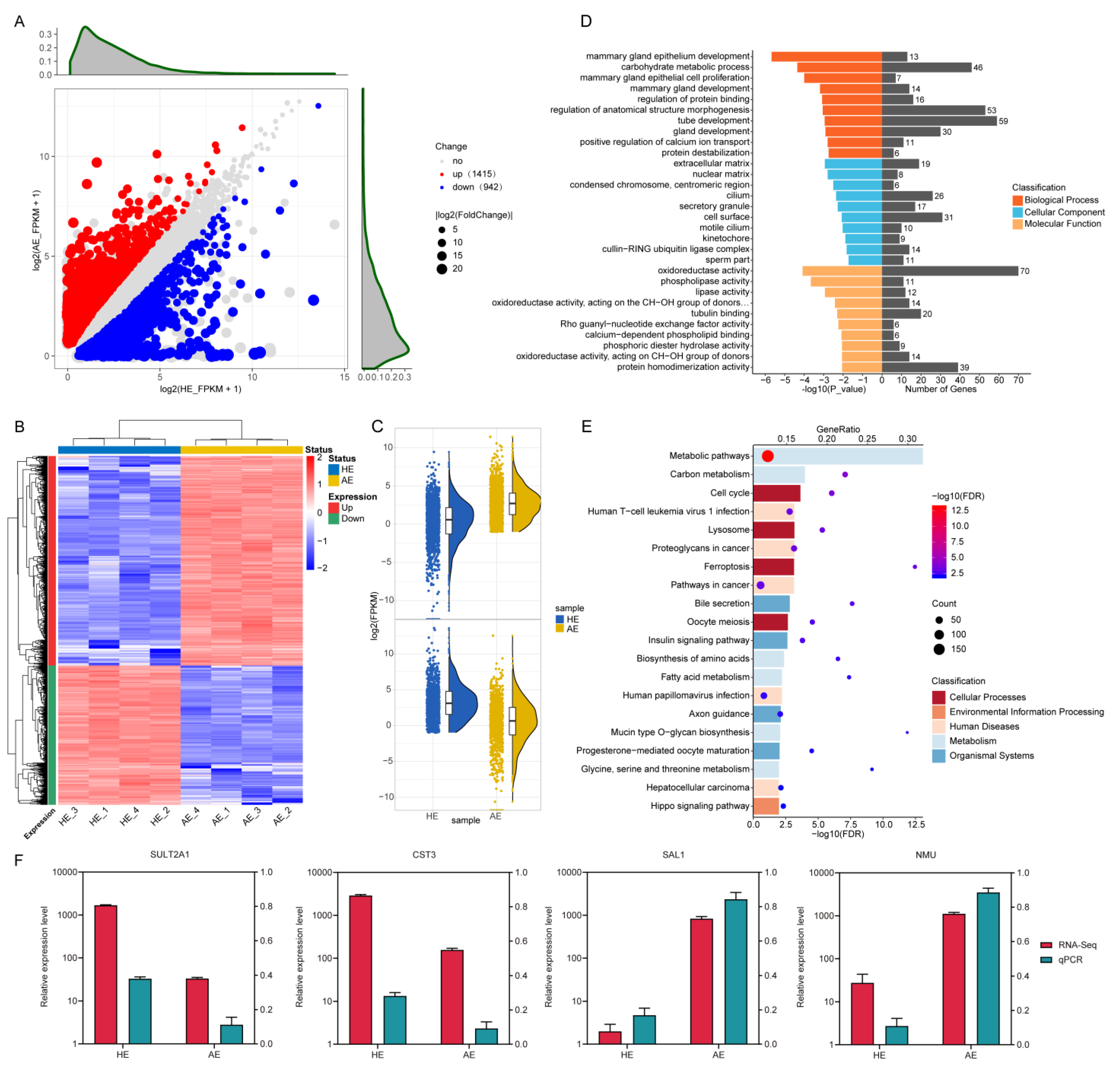
2.3. Analysis of Differentially Expressed Genes (DEGs) and Their Functions
2.4. Weakened Immunobiological Activities in AE
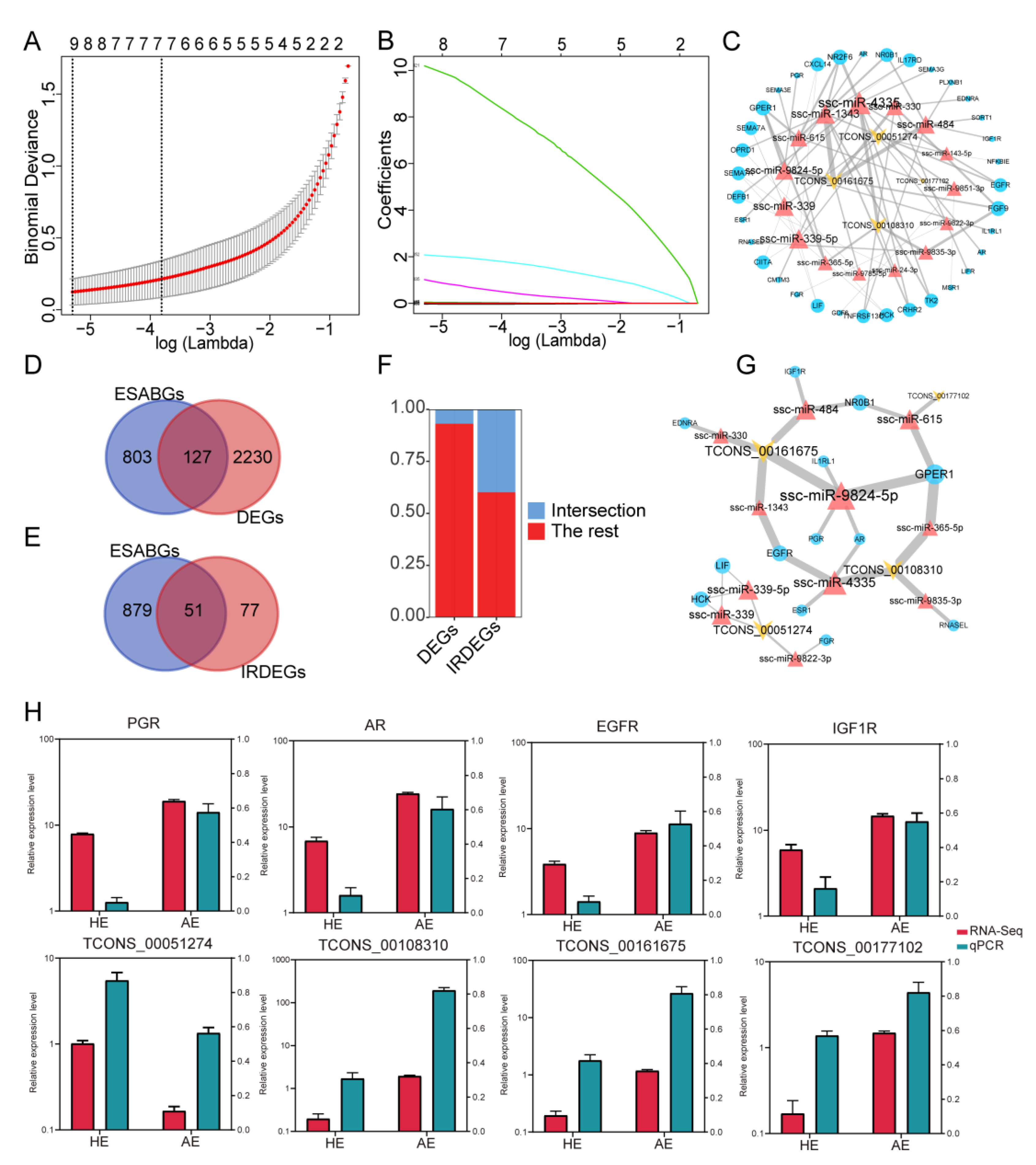
2.5. Construction of Immunological ceRNA Network Related to the SAB
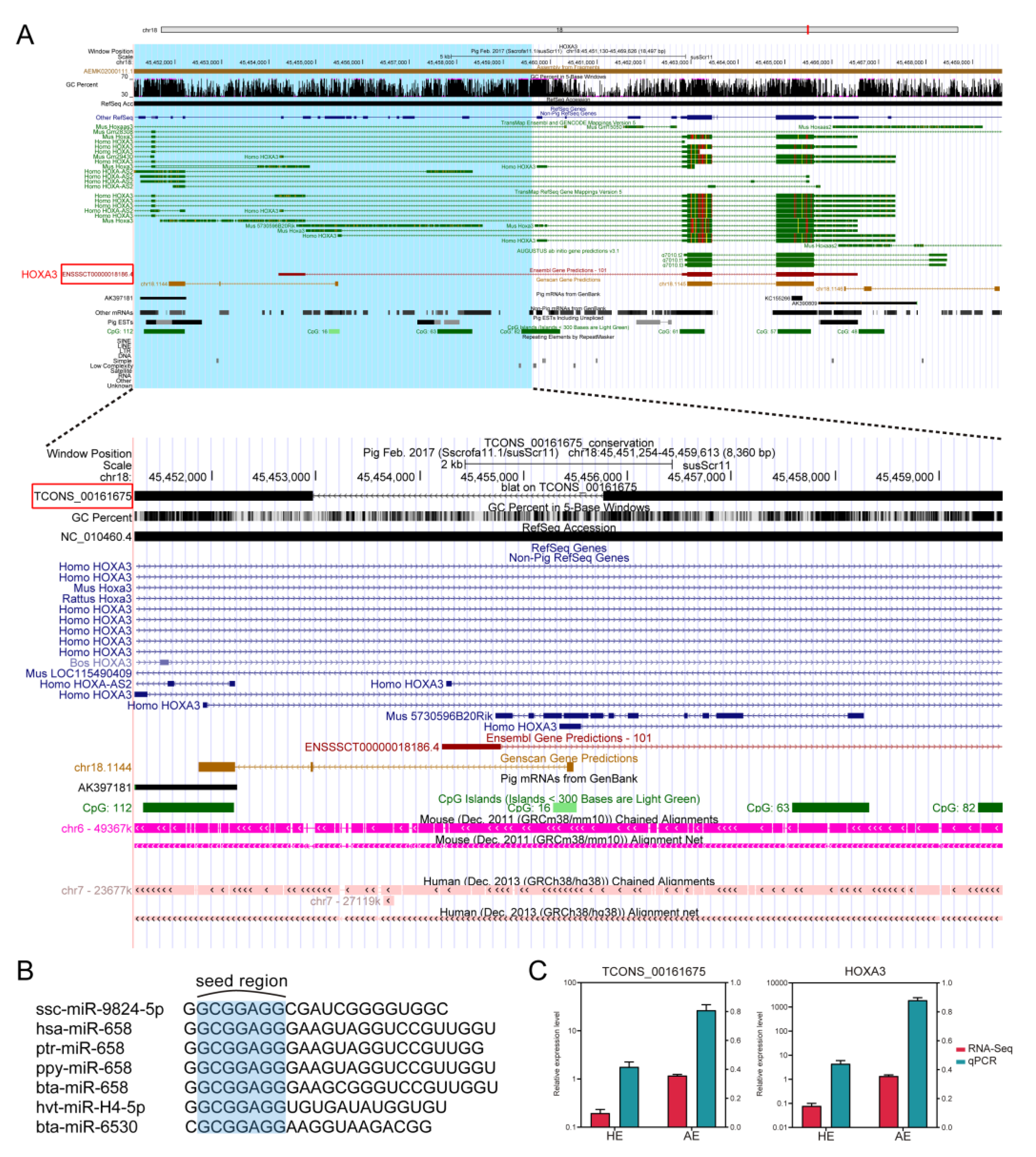
2.6. Evaluation of the Applicability of SAB ceRNA Network among Animals Species
3. Discussion
4. Materials and Methods
4.1. Ethics Statement and Sample Collection
4.2. RNA Isolation, Library Construction, and Sequencing
4.3. Quality Control and Transcriptome Assembly
4.4. LncRNA Identification and Characterization
4.5. Differential Expression Analysis and Function Enrichment Analysis
4.6. Quantitativereal-Time RT-PCR
4.7. Immunological Function Verification of DEGs
4.8. Immunohistochemistry
4.9. Construction of Competing Endogenous RNA (ceRNA) Network
4.10. Sequence Conservation Analysis of lncRNA and miRNA
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ross, J.W.; Ashworth, M.D.; Stein, D.R.; Couture, O.P.; Tuggle, C.K.; Geisert, R.D. Identification of differential gene expression during porcine conceptus rapid trophoblastic elongation and attachment to uterine luminal epithelium. Physiol. Genom. 2009, 36, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Enders, A.C.; Blankenship, T.N. Comparative placental structure. Adv. Drug Deliv. Rev. 1999, 38, 3–15. [Google Scholar] [CrossRef]
- Geisert, R.D.; Lucy, M.C.; Whyte, J.J.; Ross, J.W.; Mathew, D.J. Cytokines from the pig conceptus: Roles in conceptus development in pigs. J. Anim. Sci. Biotechnol. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and Chemokine Receptors: Positioning Cells for Host Defense and Immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidarimath, M. Immune-Angiogenesis Mechanisms Associated with Porcine Pregnancy Success and Failure. Ph.D. Thesis, Queen’s University, Kingston, ON, Canada, 2016. [Google Scholar]
- Engelhardt, H.; Croy, B.A.; King, G.J. Conceptus influences the distribution of uterine leukocytes during early porcine pregnancy. Biol. Reprod. 2002, 66, 1875–1880. [Google Scholar] [CrossRef] [Green Version]
- Winther, H.; Ahmed, A.; Dantzer, V. Immunohistochemical Localization of Vascular Endothelial Growth Factor (VEGF) and its Two Specific Receptors, Flt-1 and KDR, in the Porcine Placenta and Non-pregnant Uterus. Placenta 1999, 20, 35–43. [Google Scholar] [CrossRef]
- Tayade, C.; Fang, Y.; Hilchie, D.; Croy, B.A. Lymphocyte contributions to altered endometrial angiogenesis during early and midgestation fetal loss. J. Leukoc. Biol. 2007, 82, 877–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffett, A.; Loke, C. Immunology of placentation in eutherian mammals. Nat. Rev. Immunol. 2006, 6, 584–594. [Google Scholar] [CrossRef]
- Tayade, C.; Fang, Y.; Croy, B.A. A review of gene expression in porcine endometrial lymphocytes, endothelium and trophoblast during pregnancy success and failure. J. Reprod. Dev. 2007, 53, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, S.; Jones, S.P. RNA editing: Unexplored opportunities in the cardiovascular system. Circul. Res. 2018, 122, 399–401. [Google Scholar] [CrossRef] [Green Version]
- Zimmer-Bensch, G. Emerging Roles of Long Non-Coding RNAs as Drivers of Brain Evolution. Cells 2019, 8, 1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Diao, Z.; Yue, X.; Chen, Y.; Zhao, H.; Cheng, L.; Sun, J. Construction and analysis of dysregulated lncRNA-associated ceRNA network identified novel lncRNA biomarkers for early diagnosis of human pancreatic cancer. Oncotarget 2016, 7, 56383–56394. [Google Scholar] [CrossRef] [Green Version]
- Swarr, D.; Herriges, M.; Li, S.; Morley, M.; Fernandes, S.; Sridharan, A.; Zhou, S.; Garcia, B.A.; Stewart, K.; Morrisey, E.E. The long noncoding RNA Falcor regulates Foxa2 expression to maintain lung epithelial homeostasis and promote regeneration. Genes Dev. 2019, 33, 656–668. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.; Tong, M.; Xia, B.; Feng, G.; Wang, L.; Li, A.; Luo, G.; Wan, H.; Zhang, Z.; Zhang, H. Long noncoding RNA lnc-NAP sponges mmu-miR-139-5p to modulate Nanog functions in mouse ESCs and embryos. RNA Biol. 2020, 18, 875–887. [Google Scholar] [CrossRef]
- Shyu, K.; Wang, B.; Fang, W.; Pan, C.; Lin, C. Hyperbaric oxygen-induced long non-coding RNA MALAT1 exosomes suppress MicroRNA-92a expression in a rat model of acute myocardial infarction. J. Cell. Mol. Med. 2020, 24, 12945–12954. [Google Scholar] [CrossRef] [PubMed]
- Smolinska, N.; Kiezun, M.; Dobrzyn, K.; Szeszko, K.; Maleszka, A.; Kaminski, T. Adiponectin, orexin A and orexin B concentrations in the serum and uterine luminal fluid during early pregnancy of pigs. Anim. Reprod. Sci. 2017, 178, 1–8. [Google Scholar] [CrossRef]
- Croy, B.; Waterfield, A.; Wood, W.; King, G.J. Normal murine and porcine embryos recruit NK cells to the uterus. Cell. Immunol. 1988, 115, 471–480. [Google Scholar] [CrossRef]
- Johnson, R.; Guigó, R. The RIDL hypothesis: Transposable elements as functional domains of long noncoding RNAs. RNA 2014, 20, 959–976. [Google Scholar] [CrossRef] [Green Version]
- Lim, L.P.; Lau, N.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nat. Cell Biol. 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.C.; Hentzel, M.D.; Dziuk, P.J. Relationships Between Uterine Length and Number of Fetuses and Prenatal Mortality in Pigs. J. Anim. Sci. 1987, 65, 762–770. [Google Scholar] [CrossRef]
- Zang, X.; Gu, T.; Hu, Q.; Xu, Z.; Xie, Y.; Zhou, C.; Zheng, E.; Huang, S.; Xu, Z.; Meng, F.; et al. Global Transcriptomic Analyses Reveal Genes Involved in Conceptus Development During the Implantation Stages in Pigs. Front. Genet. 2021, 12, 247. [Google Scholar] [CrossRef]
- Dantzer, V.; Leiser, R. Initial vascularisation in the pig placenta: I. Demonstration of nonglandular areas by histology and corrosion casts. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 1994, 238, 177–190. [Google Scholar] [CrossRef]
- Tayade, C.; Black, G.P.; Fang, Y.; Croy, B.A. Differential Gene Expression in Endometrium, Endometrial Lymphocytes, and Trophoblasts during Successful and Abortive Embryo Implantation. J. Immunol. 2005, 176, 148–156. [Google Scholar] [CrossRef] [Green Version]
- King, G.J. Comparative placentation in ungulates. J. Exp. Zool. 1993, 266, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Whyte, A.; Binns, R.M. Adhesion molecule expression and infiltrating maternal leucocyte phenotypes during blastocyst implantation in the pig. Cell Biol. Int. 1994, 18, 759–766. [Google Scholar] [CrossRef]
- Sallam, T.; Sandhu, J.; Tontonoz, P. Long noncoding RNA discovery in cardiovascular disease: Decoding form to function. Circul. Res. 2018, 122, 155–166. [Google Scholar] [CrossRef]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Währisch, S.; Beisaw, A.; Macura, K.; Bläss, G.; Kellis, M.; Werber, M.; et al. The Tissue-Specific lncRNA Fendrr Is an Essential Regulator of Heart and Body Wall Development in the Mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Sun, X.; Cai, H.; Sun, Y.; Plath, M.; Li, C.; Lan, X.; Lei, C.; Lin, F.; Bai, Y.; et al. Long non-coding RNA ADNCR suppresses adipogenic differentiation by targeting miR-204. Biochim. Biophys. Acta (BBA)-Bioenergy 2016, 1859, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Sheng, F.; Sun, N.; Ji, Y.; Ma, Y.; Ding, H.; Zhang, Q.; Yang, F.; Li, W. Aberrant expression of imprinted lncRNA MEG8 causes trophoblast dysfunction and abortion. J. Cell. Biochem. 2019, 120, 17378–17390. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinger, M.; Amaral, P.D.P.; Mercer, T.R.; Pang, K.C.; Bruce, S.J.; Gardiner, B.B.; Askarian-Amiri, M.E.; Ru, K.; Soldà, G.; Simons, C.; et al. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 2008, 18, 1433–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothman, A.M.; Arnold, N.D.; Pickworth, J.A.; Iremonger, J.; Ciuclan, L.; Allen, R.M.; Guth-Gundel, S.; Southwood, M.; Morrell, N.W.; Thomas, M. MicroRNA-140-5p and SMURF1 regulate pulmonary arterial hypertension. J. Clin. Investig. 2016, 126, 2495–2508. [Google Scholar] [CrossRef] [Green Version]
- Chojnowski, J.L.; Masuda, K.; Trau, H.A.; Thomas, K.; Capecchi, M.; Manley, N.R. Multiple roles for HOXA3 in regulating thymus and parathyroid differentiation and morphogenesis in mouse. Development 2014, 141, 3697–3708. [Google Scholar] [CrossRef] [Green Version]
- Chojnowski, J.L.; Trau, H.A.; Masuda, K.; Manley, N.R. Temporal and spatial requirements for Hoxa3 in mouse embryonic development. Dev. Biol. 2016, 415, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhao, S.; Li, J.; Zhang, H.; Qian, C.; Wang, H.; Liu, J.; Zhao, Y. TCF7L2 activated HOXA-AS2 decreased the glucocorticoid sensitivity in acute lymphoblastic leukemia through regulating HOXA3/EGFR/Ras/Raf/MEK/ERK pathway. Biomed. Pharmacother. 2019, 109, 1640–1649. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wu, Z.; Meng, X.; Chu, X.; Huang, H.; Xu, C. LncRNA HOXA-AS2 Facilitates Tumorigenesis and Progression of Papillary Thyroid Cancer by Modulating the miR-15a-5p/HOXA3 Axis. Hum. Gene Ther. 2019, 30, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Parkhomchuk, D.; Borodina, T.; Amstislavskiy, V.; Banaru, M.; Hallen, L.; Krobitsch, S.; Lehrach, H.; Soldatov, A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009, 37, e123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef] [PubMed]
- Thissen, D.; Steinberg, L.; Kuang, D. Quick and Easy Implementation of the Benjamini-Hochberg Procedure for Controlling the False Positive Rate in Multiple Comparisons. J. Educ. Behav. Stat. 2002, 27, 77–83. [Google Scholar] [CrossRef]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. Clusterprofiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS: A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, F.A.; Binns, R.M.; Husmann, R.; Yang, H.; Carr, M.M.; Kim, Y.B.; Davis, W.C.; Misfeldt, M.; Lunney, J.K. Analyses of monoclonal antibodies reactive with porcine CD44 and CD45. Veter-Immunol. Immunopathol. 1994, 43, 293–305. [Google Scholar] [CrossRef]
- Hong, L.; Hou, C.; Li, X.; Li, C.; Zhao, S.; Yu, M. Expression of Heparanase Is Associated with Breed-Specific Morphological Characters of Placental Folded Bilayer Between Yorkshire and Meishan Pigs1. Biol. Reprod. 2014, 90, 56. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: MicroRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zang, X.; Gu, T.; Wang, W.; Zhou, C.; Ding, Y.; Gu, S.; Xu, Z.; Xie, Y.; Li, Z.; Cai, G.; et al. Integrated Insight into the Molecular Mechanisms of Spontaneous Abortion during Early Pregnancy in Pigs. Int. J. Mol. Sci. 2021, 22, 6644. https://doi.org/10.3390/ijms22126644
Zang X, Gu T, Wang W, Zhou C, Ding Y, Gu S, Xu Z, Xie Y, Li Z, Cai G, et al. Integrated Insight into the Molecular Mechanisms of Spontaneous Abortion during Early Pregnancy in Pigs. International Journal of Molecular Sciences. 2021; 22(12):6644. https://doi.org/10.3390/ijms22126644
Chicago/Turabian StyleZang, Xupeng, Ting Gu, Wenjing Wang, Chen Zhou, Yue Ding, Shengchen Gu, Zhiqian Xu, Yanshe Xie, Zicong Li, Gengyuan Cai, and et al. 2021. "Integrated Insight into the Molecular Mechanisms of Spontaneous Abortion during Early Pregnancy in Pigs" International Journal of Molecular Sciences 22, no. 12: 6644. https://doi.org/10.3390/ijms22126644
APA StyleZang, X., Gu, T., Wang, W., Zhou, C., Ding, Y., Gu, S., Xu, Z., Xie, Y., Li, Z., Cai, G., Hu, B., Hong, L., & Wu, Z. (2021). Integrated Insight into the Molecular Mechanisms of Spontaneous Abortion during Early Pregnancy in Pigs. International Journal of Molecular Sciences, 22(12), 6644. https://doi.org/10.3390/ijms22126644