
Endothelial Progenitor Cells Dysfunctions and Cardiometabolic Disorders: From Mechanisms to Therapeutic Approaches

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Components of Metabolic Syndrome
2. Metabolic Syndrome and Endothelial Dysfunction
3. Endothelial Progenitor Cells
4. Endothelial Progenitor Cells Dysfunction in Cardiometabolic Disorders
4.1. Endothelial Progenitor Cells Dysfunction in Type-2 Diabetes
4.2. Endothelial Progenitor Cells Dysfunction in Hypertension and Cardiovascular Diseases
4.3. Endothelial Progenitor Cells Dysfunction in Obesity
4.4. Endothelial Progenitor Cells Dysfunction and Dyslipidemia
4.5. Endothelial Progenitor Cells Dysfunction and NAFLD
5. Mechanisms Potentially Associated with Impaired Functionality of Endothelial Progenitor Cells
5.1. Oxidative Stress
5.2. Cellular Senescence
5.3. Impaired Angiogenic Function
5.4. Inflammation-Induced EPCs Dysfunction
5.5. Epigenetic Regulation
5.5.1. MicroRNAs
5.5.2. DNA Methylation
5.5.3. Histone Modification
5.6. Hyperhomocysteinemia
6. Developmental Programming of Cardiometabolic Diseases
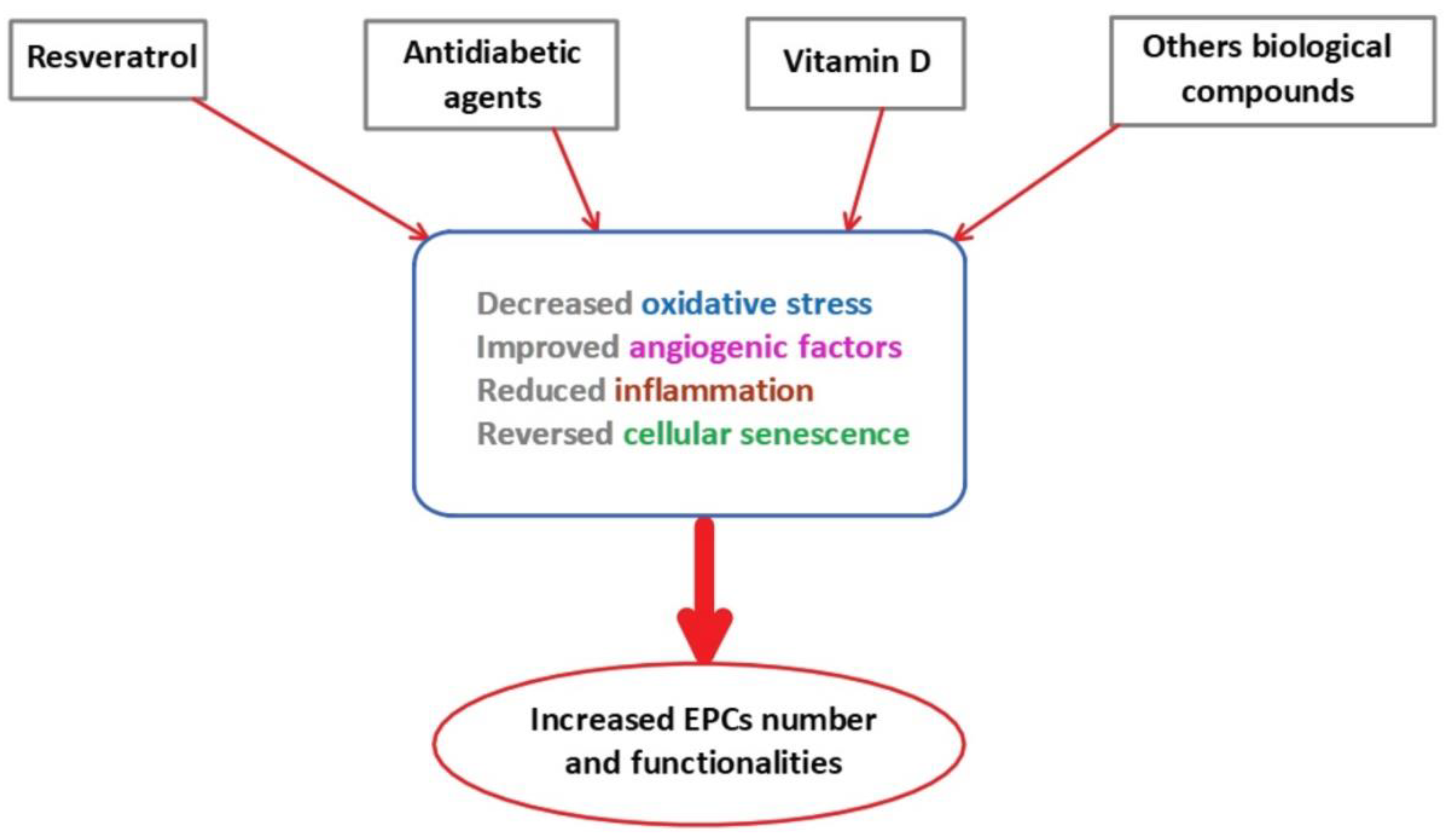
7. Reversibility of EPCs Dysfunction
7.1. Resveratrol
7.2. Oral Antidiabetic Agents
7.3. Vitamin D
7.4. Other Biological Therapies
7.4.1. Supplementation of EPCs with Proangiogenic Factors
7.4.2. The Renin–Angiotensin–Aldosterone System and Endothelin
7.4.3. Lifestyle Modifications
7.4.4. Treatment Using H2S Donors
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Spahis, S.; Borys, J.M.; Levy, E. Metabolic Syndrome as a Multifaceted Risk Factor for Oxidative Stress. Antioxid. Redox. Signal. 2017, 26, 445–461. [Google Scholar] [CrossRef]
- Bonomini, F.; Rodella, L.F.; Rezzani, R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015, 6, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C., Jr.; et al. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005, 112, 2735–2752. [Google Scholar] [CrossRef] [Green Version]
- Stefan, N.; Haring, H.U.; Cusi, K. Non-alcoholic fatty liver disease: Causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019, 7, 313–324. [Google Scholar] [CrossRef]
- Sperling, L.S.; Mechanick, J.I.; Neeland, I.J.; Herrick, C.J.; Despres, J.P.; Ndumele, C.E.; Vijayaraghavan, K.; Handelsman, Y.; Puckrein, G.A.; Araneta, M.R.; et al. The CardioMetabolic Health Alliance: Working Toward a New Care Model for the Metabolic Syndrome. J. Am. Coll. Cardiol. 2015, 66, 1050–1067. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, S.A.; Connell, J.M. The link between abdominal obesity, metabolic syndrome and cardiovascular disease. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Berwick, Z.C.; Dick, G.M.; Tune, J.D. Heart of the matter: Coronary dysfunction in metabolic syndrome. J. Mol. Cell. Cardiol. 2012, 52, 848–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luscher, T.F.; Barton, M. Biology of the endothelium. Clin. Cardiol. 1997, 20, II-3–10. [Google Scholar] [CrossRef]
- Martin, F.A.; Murphy, R.P.; Cummins, P.M. Thrombomodulin and the vascular endothelium: Insights into functional, regulatory, and therapeutic aspects. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1585-97. [Google Scholar] [CrossRef] [Green Version]
- Minshall, R.D.; Tiruppathi, C.; Vogel, S.M.; Malik, A.B. Vesicle formation and trafficking in endothelial cells and regulation of endothelial barrier function. Histochem. Cell. Biol. 2002, 117, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R.; Anggard, E.E.; Botting, R.M. Regulatory functions of the vascular endothelium. N. Engl. J. Med. 1990, 323, 27–36. [Google Scholar]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, N.A. Atherosclerosis or lipoprotein-induced endothelial dysfunction. Potential mechanisms underlying reduction in EDRF/nitric oxide activity. Circulation 1992, 85, 1927–1938. [Google Scholar] [CrossRef] [Green Version]
- Leeson, P.; Thorne, S.; Donald, A.; Mullen, M.; Clarkson, P.; Deanfield, J. Non-invasive measurement of endothelial function: Effect on brachial artery dilatation of graded endothelial dependent and independent stimuli. Heart 1997, 78, 22–27. [Google Scholar] [CrossRef]
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part I: Basic mechanisms and in vivo monitoring of ROS. Circulation 2003, 108, 1912–1916. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, E.J.; Larson, M.G.; Keyes, M.J.; Mitchell, G.F.; Vasan, R.S.; Keaney, J.F., Jr.; Lehman, B.T.; Fan, S.; Osypiuk, E.; Vita, J.A. Clinical correlates and heritability of flow-mediated dilation in the community: The Framingham Heart Study. Circulation 2004, 109, 613–619. [Google Scholar] [CrossRef] [Green Version]
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27. [Google Scholar] [CrossRef]
- Brook, R.D.; Bard, R.L.; Rubenfire, M.; Ridker, P.M.; Rajagopalan, S. Usefulness of visceral obesity (waist/hip ratio) in predicting vascular endothelial function in healthy overweight adults. Am. J. Cardiol. 2001, 88, 1264–1269. [Google Scholar] [CrossRef]
- Perticone, F.; Ceravolo, R.; Candigliota, M.; Ventura, G.; Iacopino, S.; Sinopoli, F.; Mattioli, P.L. Obesity and body fat distribution induce endothelial dysfunction by oxidative stress: Protective effect of vitamin C. Diabetes 2001, 50, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Arcaro, G.; Zamboni, M.; Rossi, L.; Turcato, E.; Covi, G.; Armellini, F.; Bosello, O.; Lechi, A. Body fat distribution predicts the degree of endothelial dysfunction in uncomplicated obesity. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 936–942. [Google Scholar] [CrossRef] [Green Version]
- McVeigh, G.E.; Brennan, G.M.; Johnston, G.D.; McDermott, B.J.; McGrath, L.T.; Henry, W.R.; Andrews, J.W.; Hayes, J.R. Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 1992, 35, 771–776. [Google Scholar] [PubMed]
- Williams, S.B.; Cusco, J.A.; Roddy, M.A.; Johnstone, M.T.; Creager, M.A. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J. Am. Coll. Cardiol. 1996, 27, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.A.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal relationships between insulin resistance and endothelial dysfunction: Molecular and pathophysiological mechanisms. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef]
- Fox, C.S. Cardiovascular disease risk factors, type 2 diabetes mellitus, and the Framingham Heart Study. Trends Cardiovasc. Med. 2010, 20, 90–95. [Google Scholar] [CrossRef] [Green Version]
- Avogaro, A.; de Kreutzenberg, S.V.; Fadini, G. Endothelial dysfunction: Causes and consequences in patients with diabetes mellitus. Diabetes Res. Clin. Pract. 2008, 82, S94–S101. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Tousoulis, D.; Papageorgiou, N.; Androulakis, E.; Siasos, G.; Latsios, G.; Tentolouris, K.; Stefanadis, C. Diabetes mellitus-associated vascular impairment: Novel circulating biomarkers and therapeutic approaches. J. Am. Coll. Cardiol. 2013, 62, 667–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arinc, H.; Sarli, B.; Baktir, A.O.; Saglam, H.; Demirci, E.; Dogan, Y.; Kurtul, S.; Karaman, H.; Erden, A.; Karaman, A. Serum gamma glutamyl transferase and alanine transaminase concentrations predict endothelial dysfunction in patients with non-alcoholic steatohepatitis. Ups. J. Med. Sci. 2013, 118, 228–234. [Google Scholar] [CrossRef]
- Villanova, N.; Moscatiello, S.; Ramilli, S.; Bugianesi, E.; Magalotti, D.; Vanni, E.; Zoli, M.; Marchesini, G. Endothelial dysfunction and cardiovascular risk profile in nonalcoholic fatty liver disease. Hepatology 2005, 42, 473–480. [Google Scholar] [CrossRef]
- Colak, Y.; Senates, E.; Yesil, A.; Yilmaz, Y.; Ozturk, O.; Doganay, L.; Coskunpinar, E.; Kahraman, O.T.; Mesci, B.; Ulasoglu, C.; et al. Assessment of endothelial function in patients with nonalcoholic fatty liver disease. Endocrine 2013, 43, 100–107. [Google Scholar] [CrossRef]
- Asahara, T.; Masuda, H.; Takahashi, T.; Kalka, C.; Pastore, C.; Silver, M.; Kearne, M.; Magner, M.; Isner, J.M. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ. Res. 1999, 85, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Murohara, T. Angiogenesis and vasculogenesis for therapeutic neovascularization. Nagoya J. Med. Sci. 2003, 66, 1–7. [Google Scholar]
- Werner, N.; Junk, S.; Laufs, U.; Link, A.; Walenta, K.; Bohm, M.; Nickenig, G. Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circ. Res. 2003, 93, e17–e24. [Google Scholar] [CrossRef] [Green Version]
- Ingram, D.A.; Mead, L.E.; Tanaka, H.; Meade, V.; Fenoglio, A.; Mortell, K.; Pollok, K.; Ferkowicz, M.J.; Gilley, D.; Yoder, M.C. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 2004, 104, 2752–2760. [Google Scholar] [CrossRef]
- Ingram, D.A.; Mead, L.E.; Moore, D.B.; Woodard, W.; Fenoglio, A.; Yoder, M.C. Vessel wall-derived endothelial cells rapidly proliferate because they contain a complete hierarchy of endothelial progenitor cells. Blood 2005, 105, 2783–2786. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Weisdorf, D.J.; Solovey, A.; Hebbel, R.P. Origins of circulating endothelial cells and endothelial outgrowth from blood. J. Clin. Investig. 2000, 105, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Duong, H.T.; Comhair, S.A.; Aldred, M.A.; Mavrakis, L.; Savasky, B.M.; Erzurum, S.C.; Asosingh, K. Pulmonary artery endothelium resident endothelial colony-forming cells in pulmonary arterial hypertension. Pulm. Circ. 2011, 1, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Alphonse, R.S.; Vadivel, A.; Zhong, S.; McConaghy, S.; Ohls, R.; Yoder, M.C.; Thebaud, B. The isolation and culture of endothelial colony-forming cells from human and rat lungs. Nat. Protoc. 2015, 10, 1697–1708. [Google Scholar] [CrossRef] [Green Version]
- Solomon, I.; O’Reilly, M.; Ionescu, L.; Alphonse, R.S.; Rajabali, S.; Zhong, S.; Vadivel, A.; Shelley, W.C.; Yoder, M.C.; Thebaud, B. Functional Differences Between Placental Micro- and Macrovascular Endothelial Colony-Forming Cells. Stem Cells Transl. Med. 2016, 5, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Prasain, N.; Lee, M.R.; Vemula, S.; Meador, J.L.; Yoshimoto, M.; Ferkowicz, M.J.; Fett, A.; Gupta, M.; Rapp, B.M.; Saadatzadeh, M.R.; et al. Differentiation of human pluripotent stem cells to cells similar to cord-blood endothelial colony-forming cells. Nat. Biotechnol. 2014, 32, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial Progenitors: A Consensus Statement on Nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Mund, J.A.; Estes, M.L.; Yoder, M.C.; Ingram, D.A., Jr.; Case, J. Flow cytometric identification and functional characterization of immature and mature circulating endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Estes, M.L.; Mund, J.A.; Ingram, D.A.; Case, J. Identification of endothelial cells and progenitor cell subsets in human peripheral blood. Curr. Protoc. Cytom. 2010, 33, 1–11. [Google Scholar] [CrossRef]
- Asahara, T.; Kawamoto, A.; Masuda, H. Concise review: Circulating endothelial progenitor cells for vascular medicine. Stem Cells 2011, 29, 1605–1650. [Google Scholar] [CrossRef]
- Fadini, G.P.; Baesso, I.; Albiero, M.; Sartore, S.; Agostini, C.; Avogaro, A. Technical notes on endothelial progenitor cells: Ways to escape from the knowledge plateau. Atherosclerosis 2008, 197, 496–503. [Google Scholar] [CrossRef]
- Boldicke, T.; Tesar, M.; Griesel, C.; Rohde, M.; Grone, H.J.; Waltenberger, J.; Kollet, O.; Lapidot, T.; Yayon, A.; Weich, H. Anti-VEGFR-2 scFvs for cell isolation. Single-chain antibodies recognizing the human vascular endothelial growth factor receptor-2 (VEGFR-2/flk-1) on the surface of primary endothelial cells and preselected CD34+ cells from cord blood. Stem Cells 2001, 19, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Kunz, G.A.; Liang, G.; Cuculi, F.; Gregg, D.; Vata, K.C.; Shaw, L.K.; Goldschmidt-Clermont, P.J.; Dong, C.; Taylor, D.A.; Peterson, E.D. Circulating endothelial progenitor cells predict coronary artery disease severity. Am. Heart J. 2006, 152, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Keymel, S.; Kalka, C.; Rassaf, T.; Yeghiazarians, Y.; Kelm, M.; Heiss, C. Impaired endothelial progenitor cell function predicts age-dependent carotid intimal thickening. Basic Res. Cardiol. 2008, 103, 582–586. [Google Scholar] [CrossRef]
- Iwasaki, H.; Kawamoto, A.; Ishikawa, M.; Oyamada, A.; Nakamori, S.; Nishimura, H.; Sadamoto, K.; Horii, M.; Matsumoto, T.; Murasawa, S.; et al. Dose-dependent contribution of CD34-positive cell transplantation to concurrent vasculogenesis and cardiomyogenesis for functional regenerative recovery after myocardial infarction. Circulation 2006, 113, 1311–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, H.K.; Chang, L.T.; Sun, C.K.; Sheu, J.J.; Chiang, C.H.; Youssef, A.A.; Lee, F.Y.; Wu, C.J.; Fu, M. Autologous transplantation of bone marrow-derived endothelial progenitor cells attenuates monocrotaline-induced pulmonary arterial hypertension in rats. Crit. Care Med. 2008, 36, 873–880. [Google Scholar] [CrossRef]
- Yip, H.K.; Chang, L.T.; Chang, W.N.; Lu, C.H.; Liou, C.W.; Lan, M.Y.; Liu, J.S.; Youssef, A.A.; Chang, H.W. Level and value of circulating endothelial progenitor cells in patients after acute ischemic stroke. Stroke 2008, 39, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A.E.; Kremzer, A.A.; Berezina, T.A.; Martovitskaya, Y.V. The pattern of circulating microparticles in patients with diabetes mellitus with asymptomatic atherosclerosis. Acta Clin. Belg. 2016, 71, 38–45. [Google Scholar] [CrossRef]
- Bakogiannis, C.; Tousoulis, D.; Androulakis, E.; Briasoulis, A.; Papageorgiou, N.; Vogiatzi, G.; Kampoli, A.M.; Charakida, M.; Siasos, G.; Latsios, G.; et al. Circulating endothelial progenitor cells as biomarkers for prediction of cardiovascular outcomes. Curr. Med. Chem. 2012, 19, 2597–2604. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A.E.; Kremzer, A.A. Circulating endothelial progenitor cells as markers for severity of ischemic chronic heart failure. J. Card. Fail. 2014, 20, 438–447. [Google Scholar] [CrossRef]
- Berezin, A.E.; Kremzer, A.A.; Samura, T.A.; Berezina, T.A.; Martovitskaya, Y.V. Serum uric Acid predicts declining of circulating proangiogenic mononuclear progenitor cells in chronic heart failure patients. J. Cardiovasc. Thorac. Res. 2014, 6, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, C.G.; Lavery, R.; Caporali, F.; Fondelli, C.; Laghi-Pasini, F.; Dotta, F.; Sorrentino, V. Generalised reduction of putative endothelial progenitors and CXCR4-positive peripheral blood cells in type 2 diabetes. Diabetologia 2008, 51, 1296–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadini, G.P. A reappraisal of the role of circulating (progenitor) cells in the pathobiology of diabetic complications. Diabetologia 2014, 57, 4–15. [Google Scholar] [CrossRef] [Green Version]
- Langford-Smith, A.W.W.; Hasan, A.; Weston, R.; Edwards, N.; Jones, A.M.; Boulton, A.J.M.; Bowling, F.L.; Rashid, S.T.; Wilkinson, F.L.; Alexander, M.Y. Diabetic endothelial colony forming cells have the potential for restoration with glycomimetics. Sci. Rep. 2019, 9, 2309. [Google Scholar] [CrossRef] [Green Version]
- Leicht, S.F.; Schwarz, T.M.; Hermann, P.C.; Seissler, J.; Aicher, A.; Heeschen, C. Adiponectin pretreatment counteracts the detrimental effect of a diabetic environment on endothelial progenitors. Diabetes 2011, 60, 652–661. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, S.A.; Bahlmann, F.H.; Besler, C.; Muller, M.; Schulz, S.; Kirchhoff, N.; Doerries, C.; Horvath, T.; Limbourg, A.; Limbourg, F.; et al. Oxidant stress impairs in vivo reendothelialization capacity of endothelial progenitor cells from patients with type 2 diabetes mellitus: Restoration by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation 2007, 116, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Tepper, O.M.; Galiano, R.D.; Capla, J.M.; Kalka, C.; Gagne, P.J.; Jacobowitz, G.R.; Levine, J.P.; Gurtner, G.C. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 2002, 106, 2781–2786. [Google Scholar] [CrossRef] [Green Version]
- Ii, M.; Takenaka, H.; Asai, J.; Ibusuki, K.; Mizukami, Y.; Maruyama, K.; Yoon, Y.S.; Wecker, A.; Luedemann, C.; Eaton, E.; et al. Endothelial progenitor thrombospondin-1 mediates diabetes-induced delay in reendothelialization following arterial injury. Circ. Res. 2006, 98, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, A.; Siragusa, M.; Quaini, F.; Mangialardi, G.; Katare, R.G.; Caporali, A.; van Buul, J.D.; van Alphen, F.P.; Graiani, G.; Spinetti, G.; et al. Diabetes mellitus induces bone marrow microangiopathy. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Albiero, M.; Menegazzo, L.; Boscaro, E.; Agostini, C.; Avogaro, A.; Fadini, G.P. Defective recruitment, survival and proliferation of bone marrow-derived progenitor cells at sites of delayed diabetic wound healing in mice. Diabetologia 2011, 54, 945–953. [Google Scholar] [CrossRef] [Green Version]
- Kuliszewski, M.A.; Ward, M.R.; Kowalewski, J.W.; Smith, A.H.; Stewart, D.J.; Kutryk, M.J.; Leong-Poi, H. A direct comparison of endothelial progenitor cell dysfunction in rat metabolic syndrome and diabetes. Atherosclerosis 2013, 226, 58–66. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, W.S.; Lau, K.K.; Siu, C.W.; Wang, M.; Yan, G.H.; Yiu, K.H.; Tse, H.F. Impact of glycemic control on circulating endothelial progenitor cells and arterial stiffness in patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2011, 10, 113. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Miorin, M.; Facco, M.; Bonamico, S.; Baesso, I.; Grego, F.; Menegolo, M.; de Kreutzenberg, S.V.; Tiengo, A.; Agostini, C.; et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2005, 45, 1449–1457. [Google Scholar] [CrossRef]
- Fadini, G.P.; Sartore, S.; Albiero, M.; Baesso, I.; Murphy, E.; Menegolo, M.; Grego, F.; Vigili de Kreutzenberg, S.; Tiengo, A.; Agostini, C.; et al. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2140–2146. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.C.; Sheu, J.J.; Wang, P.W.; Chen, C.Y.; Kuo, M.C.; Hsieh, C.J.; Chen, J.F.; Chang, H.W. Complications impaired endothelial progenitor cell function in Type 2 diabetic patients with or without critical leg ischaemia: Implication for impaired neovascularization in diabetes. Diabet. Med. 2009, 26, 134–141. [Google Scholar] [CrossRef]
- Moon, J.H.; Chae, M.K.; Kim, K.J.; Kim, H.M.; Cha, B.S.; Lee, H.C.; Kim, Y.J.; Lee, B.W. Decreased endothelial progenitor cells and increased serum glycated albumin are independently correlated with plaque-forming carotid artery atherosclerosis in type 2 diabetes patients without documented ischemic disease. Circ. J. 2012, 76, 2273–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Lin, S.J.; Lin, F.Y.; Wu, T.C.; Tsao, C.R.; Huang, P.H.; Liu, P.L.; Chen, Y.L.; Chen, J.W. High glucose impairs early and late endothelial progenitor cells by modifying nitric oxide-related but not oxidative stress-mediated mechanisms. Diabetes 2007, 56, 1559–1568. [Google Scholar] [CrossRef] [Green Version]
- Werner, N.; Kosiol, S.; Schiegl, T.; Ahlers, P.; Walenta, K.; Link, A.; Bohm, M.; Nickenig, G. Circulating endothelial progenitor cells and cardiovascular outcomes. N. Engl. J. Med. 2005, 353, 999–1007. [Google Scholar] [CrossRef]
- MacEneaney, O.J.; DeSouza, C.A.; Weil, B.R.; Kushner, E.J.; Van Guilder, G.P.; Mestek, M.L.; Greiner, J.J.; Stauffer, B.L. Prehypertension and endothelial progenitor cell function. J. Hum. Hypertens. 2011, 25, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Mandraffino, G.; Imbalzano, E.; Sardo, M.A.; D’Ascola, A.; Mamone, F.; Lo Gullo, A.; Alibrandi, A.; Loddo, S.; Mormina, E.; David, A.; et al. Circulating progenitor cells in hypertensive patients with different degrees of cardiovascular involvement. J. Hum. Hypertens. 2014, 28, 543–550. [Google Scholar] [CrossRef]
- Skrzypkowska, M.W.; Ryba-Stanislawowska, M.E.; Slominski, B.; Gutknecht, P.G.; Siebert, J.; Mysliwska, J.M. Association of circulating progenitor cells with angiotensin II in newly diagnosed hypertensive patients. J. Hum. Hypertens. 2017, 32, 46–53. [Google Scholar] [CrossRef]
- Marketou, M.E.; Kalyva, A.; Parthenakis, F.I.; Pontikoglou, C.; Maragkoudakis, S.; Kontaraki, J.E.; Chlouverakis, G.; Zacharis, E.A.; Patrianakos, A.; Papadaki, H.A.; et al. Circulating endothelial progenitor cells in hypertensive patients with increased arterial stiffness. J. Clin. Hypertens. 2014, 16, 295–300. [Google Scholar] [CrossRef]
- Lewington, S.; Clarke, R.; Qizilbash, N.; Peto, R.; Collins, R.; Prospective Studies, C. Age-specific relevance of usual blood pressure to vascular mortality: A meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002, 360, 1903–1913. [Google Scholar]
- Britton, K.A.; Gaziano, J.M.; Djousse, L. Normal systolic blood pressure and risk of heart failure in US male physicians. Eur. J. Heart Fail. 2009, 11, 1129–1134. [Google Scholar] [CrossRef]
- Jahangiry, L.; Farhangi, M.A.; Rezaei, F. Framingham risk score for estimation of 10-years of cardiovascular diseases risk in patients with metabolic syndrome. J. Health Popul. Nutr. 2017, 36, 36. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.M.; Zalos, G.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef]
- Vasa, M.; Fichtlscherer, S.; Aicher, A.; Adler, K.; Urbich, C.; Martin, H.; Zeiher, A.M.; Dimmeler, S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res. 2001, 89, E1–E7. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.W.; Huang, P.H.; Huang, S.S.; Leu, H.B.; Huang, C.C.; Wu, T.C.; Chen, J.W.; Lin, S.J. Decreased circulating endothelial progenitor cell levels and function in essential hypertensive patients with electrocardiographic left ventricular hypertrophy. Hypertens. Res. 2011, 34, 999–1003. [Google Scholar] [CrossRef]
- Bitterli, L.; Afan, S.; Buhler, S.; DiSanto, S.; Zwahlen, M.; Schmidlin, K.; Yang, Z.; Baumgartner, I.; Diehm, N.; Kalka, C. Endothelial progenitor cells as a biological marker of peripheral artery disease. Vasc. Med. 2016, 21, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Sartore, S.; Baesso, I.; Lenzi, M.; Agostini, C.; Tiengo, A.; Avogaro, A. Endothelial progenitor cells and the diabetic paradox. Diabetes Care 2006, 29, 714–716. [Google Scholar] [CrossRef] [Green Version]
- Jialal, I.; Devaraj, S.; Singh, U.; Huet, B.A. Decreased number and impaired functionality of endothelial progenitor cells in subjects with metabolic syndrome: Implications for increased cardiovascular risk. Atherosclerosis 2010, 211, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Muller-Ehmsen, J.; Braun, D.; Schneider, T.; Pfister, R.; Worm, N.; Wielckens, K.; Scheid, C.; Frommolt, P.; Flesch, M. Decreased number of circulating progenitor cells in obesity: Beneficial effects of weight reduction. Eur. Heart J. 2008, 29, 1560–1568. [Google Scholar] [CrossRef] [Green Version]
- Heida, N.M.; Muller, J.P.; Cheng, I.F.; Leifheit-Nestler, M.; Faustin, V.; Riggert, J.; Hasenfuss, G.; Konstantinides, S.; Schafer, K. Effects of obesity and weight loss on the functional properties of early outgrowth endothelial progenitor cells. J. Am. Coll. Cardiol. 2010, 55, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.L.; Chang, C.L.; Sun, C.K.; Wu, C.J.; Tsai, T.H.; Chung, S.Y.; Chua, S.; Yeh, K.H.; Leu, S.; Sheu, J.J.; et al. Impact of obesity control on circulating level of endothelial progenitor cells and angiogenesis in response to ischemic stimulation. J. Transl. Med. 2012, 10, 86. [Google Scholar] [CrossRef] [Green Version]
- Tsai, T.H.; Chai, H.T.; Sun, C.K.; Yen, C.H.; Leu, S.; Chen, Y.L.; Chung, S.Y.; Ko, S.F.; Chang, H.W.; Wu, C.J.; et al. Obesity suppresses circulating level and function of endothelial progenitor cells and heart function. J. Transl. Med. 2012, 10, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Z.; Zhang, F.R.; Tao, Q.M.; Wang, X.X.; Zhu, J.H.; Zhu, J.H. Number and activity of endothelial progenitor cells from peripheral blood in patients with hypercholesterolaemia. Clin. Sci. 2004, 107, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Rossi, F.; Bertone, C.; Montanile, F.; Miglietta, F.; Lubrano, C.; Gandini, L.; Santiemma, V. HDL cholesterol is a strong determinant of endothelial progenitor cells in hypercholesterolemic subjects. Microvasc. Res. 2010, 80, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, T.; Hano, T.; Sawamura, T.; Nishio, I. Oxidized low-density lipoprotein induces endothelial progenitor cell senescence, leading to cellular dysfunction. Clin. Exp. Pharmacol. Physiol. 2004, 31, 407–413. [Google Scholar] [CrossRef]
- Tarantino, G.; Saldalamacchia, G.; Conca, P.; Arena, A. Non-alcoholic fatty liver disease: Further expression of the metabolic syndrome. J. Gastroenterol. Hepatol. 2007, 22, 293–303. [Google Scholar] [CrossRef]
- Kotronen, A.; Yki-Jarvinen, H. Fatty liver: A novel component of the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 27–38. [Google Scholar] [CrossRef]
- Despres, J.P.; Lemieux, I.; Bergeron, J.; Pibarot, P.; Mathieu, P.; Larose, E.; Rodes-Cabau, J.; Bertrand, O.F.; Poirier, P. Abdominal obesity and the metabolic syndrome: Contribution to global cardiometabolic risk. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1039–1049. [Google Scholar] [CrossRef]
- Vlachopoulos, C.; Manesis, E.; Baou, K.; Papatheodoridis, G.; Koskinas, J.; Tiniakos, D.; Aznaouridis, K.; Archimandritis, A.; Stefanadis, C. Increased arterial stiffness and impaired endothelial function in nonalcoholic Fatty liver disease: A pilot study. Am. J. Hypertens. 2010, 23, 1183–1189. [Google Scholar] [CrossRef]
- Chiang, C.H.; Huang, P.H.; Chung, F.P.; Chen, Z.Y.; Leu, H.B.; Huang, C.C.; Wu, T.C.; Chen, J.W.; Lin, S.J. Decreased circulating endothelial progenitor cell levels and function in patients with nonalcoholic fatty liver disease. PLoS ONE 2012, 7, e31799. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Grobe, Y.; Gavilanes-Espinar, J.G.; Masso-Rojas, F.A.; Sanchez-Valle, V.; Paez-Arenas, A.; Ponciano-Rodriguez, G.; Chavez-Tapia, N.C.; Uribe, M.; Mendez-Sanchez, N. Metabolic syndrome and nonalcoholic fatty liver disease. The role of endothelial progenitor cells. Ann. Hepatol. 2013, 12, 908–914. [Google Scholar] [CrossRef]
- Dean, R.T.; Fu, S.; Stocker, R.; Davies, M.J. Biochemistry and pathology of radical-mediated protein oxidation. Biochem. J. 1997, 324, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef]
- Versari, D.; Daghini, E.; Virdis, A.; Ghiadoni, L.; Taddei, S. Endothelium-dependent contractions and endothelial dysfunction in human hypertension. Br. J. Pharmacol. 2009, 157, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Watson, T.; Goon, P.K.; Lip, G.Y. Endothelial progenitor cells, endothelial dysfunction, inflammation, and oxidative stress in hypertension. Antioxid. Redox Signal. 2008, 10, 1079–1088. [Google Scholar] [CrossRef]
- Schatteman, G.C.; Hanlon, H.D.; Jiao, C.; Dodds, S.G.; Christy, B.A. Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J. Clin. Investig. 2000, 106, 571–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thum, T.; Fraccarollo, D.; Galuppo, P.; Tsikas, D.; Frantz, S.; Ertl, G.; Bauersachs, J. Bone marrow molecular alterations after myocardial infarction: Impact on endothelial progenitor cells. Cardiovasc. Res. 2006, 70, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Dernbach, E.; Urbich, C.; Brandes, R.P.; Hofmann, W.K.; Zeiher, A.M.; Dimmeler, S. Antioxidative stress-associated genes in circulating progenitor cells: Evidence for enhanced resistance against oxidative stress. Blood 2004, 104, 3591–3597. [Google Scholar] [CrossRef] [PubMed]
- Collett, J.A.; Mehrotra, P.; Crone, A.; Shelley, W.C.; Yoder, M.C.; Basile, D.P. Endothelial colony-forming cells ameliorate endothelial dysfunction via secreted factors following ischemia-reperfusion injury. Am. J. Physiol. Renal. Physiol. 2017, 312, F897–F907. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Yang, Z.; Wang, J.M.; Wang, L.C.; Luo, C.F.; Tang, A.L.; Dong, Y.G.; Ma, H. Shear stress increases Cu/Zn SOD activity and mRNA expression in human endothelial progenitor cells. J. Hum. Hypertens. 2007, 21, 353–358. [Google Scholar] [CrossRef]
- He, T.; Peterson, T.E.; Holmuhamedov, E.L.; Terzic, A.; Caplice, N.M.; Oberley, L.W.; Katusic, Z.S. Human endothelial progenitor cells tolerate oxidative stress due to intrinsically high expression of manganese superoxide dismutase. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2021–2027. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.Y.; Tsao, N.W.; Shih, C.M.; Lin, Y.W.; Yeh, J.S.; Chen, J.W.; Nakagami, H.; Morishita, R.; Sawamura, T.; Huang, C.Y. The biphasic effects of oxidized-low density lipoprotein on the vasculogenic function of endothelial progenitor cells. PLoS ONE 2015, 10, e0123971. [Google Scholar] [CrossRef]
- Tso, C.; Martinic, G.; Fan, W.H.; Rogers, C.; Rye, K.A.; Barter, P.J. High-density lipoproteins enhance progenitor-mediated endothelium repair in mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1144–1149. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. Endogenous oxidative damage of mtDNA. Mutat. Res. 1999, 424, 51–58. [Google Scholar] [CrossRef]
- Murphy, M.P. Induction of mitochondrial ROS production by electrophilic lipids: A new pathway of redox signaling? Am. J. Physiol. Heart. Circ. Physiol. 2006, 290, H1754–H1755. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.W.; Son, S.M.; Mook-Jung, I.; Moon, Y.J.; Lee, J.Y.; Wang, K.C.; Kang, H.S.; Phi, J.H.; Choi, S.A.; Chong, S.; et al. Mitochondrial abnormalities related to the dysfunction of circulating endothelial colony-forming cells in moyamoya disease. J. Neurosurg. 2018, 129, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- Lyons, C.J.; O’Brien, T. The Functionality of Endothelial-Colony-Forming Cells from Patients with Diabetes Mellitus. Cells 2020, 9, 1731. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2013, 5, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell. Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Martin-Ruiz, C.; Saretzki, G.; Petrie, J.; Ladhoff, J.; Jeyapalan, J.; Wei, W.; Sedivy, J.; von Zglinicki, T. Stochastic variation in telomere shortening rate causes heterogeneity of human fibroblast replicative life span. J. Biol. Chem. 2004, 279, 17826–17833. [Google Scholar] [CrossRef] [Green Version]
- Vassallo, P.F.; Simoncini, S.; Ligi, I.; Chateau, A.L.; Bachelier, R.; Robert, S.; Morere, J.; Fernandez, S.; Guillet, B.; Marcelli, M.; et al. Accelerated senescence of cord blood endothelial progenitor cells in premature neonates is driven by SIRT1 decreased expression. Blood 2014, 123, 2116–2126. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Ingram, D.A.; Lien, I.Z.; Mead, L.E.; Estes, M.; Prater, D.N.; Derr-Yellin, E.; DiMeglio, L.A.; Haneline, L.S. In vitro hyperglycemia or a diabetic intrauterine environment reduces neonatal endothelial colony-forming cell numbers and function. Diabetes 2008, 57, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, J.; Merino, A.; Briceno, C.; Soriano, S.; Buendia, P.; Calleros, L.; Rodriguez, M.; Martin-Malo, A.; Aljama, P.; Ramirez, R. Carbamylated low-density lipoprotein induces oxidative stress and accelerated senescence in human endothelial progenitor cells. FASEB J. 2011, 25, 1314–1322. [Google Scholar] [CrossRef]
- Satoh, M.; Ishikawa, Y.; Takahashi, Y.; Itoh, T.; Minami, Y.; Nakamura, M. Association between oxidative DNA damage and telomere shortening in circulating endothelial progenitor cells obtained from metabolic syndrome patients with coronary artery disease. Atherosclerosis 2008, 198, 347–353. [Google Scholar] [CrossRef]
- Alphonse, R.S.; Vadivel, A.; Fung, M.; Shelley, W.C.; Critser, P.J.; Ionescu, L.; O’Reilly, M.; Ohls, R.K.; McConaghy, S.; Eaton, F.; et al. Existence, functional impairment, and lung repair potential of endothelial colony-forming cells in oxygen-induced arrested alveolar growth. Circulation 2014, 129, 2144–2157. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Lee, J.H.; Han, Y.S.; Ryu, J.M.; Yoon, Y.M.; Han, H.J. Hypoxia accelerates vascular repair of endothelial colony-forming cells on ischemic injury via STAT3-BCL3 axis. Stem Cell Res. Ther. 2015, 6, 139. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, S.; Kwon, S.M.; Matsuda, T.; Jung, S.Y.; Lee, J.H.; Lee, S.H.; Masuda, H.; Asahara, T. Identification of mouse colony-forming endothelial progenitor cells for postnatal neovascularization: A novel insight highlighted by new mouse colony-forming assay. Stem Cell Res. Ther. 2013, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Luque Contreras, D.; Vargas Robles, H.; Romo, E.; Rios, A.; Escalante, B. The role of nitric oxide in the post-ischemic revascularization process. Pharmacol. Ther. 2006, 112, 553–563. [Google Scholar] [CrossRef]
- Duda, D.G.; Fukumura, D.; Jain, R.K. Role of eNOS in neovascularization: NO for endothelial progenitor cells. Trends Mol. Med. 2004, 10, 143–145. [Google Scholar] [CrossRef]
- Aicher, A.; Heeschen, C.; Mildner-Rihm, C.; Urbich, C.; Ihling, C.; Technau-Ihling, K.; Zeiher, A.M.; Dimmeler, S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat. Med. 2003, 9, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Babaei, S.; Stewart, D.J. Overexpression of endothelial NO synthase induces angiogenesis in a co-culture model. Cardiovasc. Res. 2002, 55, 190–200. [Google Scholar] [CrossRef] [Green Version]
- Aicher, A.; Zeiher, A.M.; Dimmeler, S. Mobilizing endothelial progenitor cells. Hypertension 2005, 45, 321–325. [Google Scholar] [CrossRef]
- Heissig, B.; Werb, Z.; Rafii, S.; Hattori, K. Role of c-kit/Kit ligand signaling in regulating vasculogenesis. Thromb. Haemost. 2003, 90, 570–576. [Google Scholar] [CrossRef]
- Thum, T.; Fraccarollo, D.; Schultheiss, M.; Froese, S.; Galuppo, P.; Widder, J.D.; Tsikas, D.; Ertl, G.; Bauersachs, J. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes 2007, 56, 666–674. [Google Scholar] [CrossRef] [Green Version]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef]
- Vasa, M.; Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ. Res. 2000, 87, 540–542. [Google Scholar] [CrossRef] [Green Version]
- Hoetzer, G.L.; Irmiger, H.M.; Keith, R.S.; Westbrook, K.M.; DeSouza, C.A. Endothelial nitric oxide synthase inhibition does not alter endothelial progenitor cell colony forming capacity or migratory activity. J. Cardiovasc. Pharmacol. 2005, 46, 387–389. [Google Scholar] [CrossRef]
- Li Calzi, S.; Purich, D.L.; Chang, K.H.; Afzal, A.; Nakagawa, T.; Busik, J.V.; Agarwal, A.; Segal, M.S.; Grant, M.B. Carbon monoxide and nitric oxide mediate cytoskeletal reorganization in microvascular cells via vasodilator-stimulated phosphoprotein phosphorylation: Evidence for blunted responsiveness in diabetes. Diabetes 2008, 57, 2488–2494. [Google Scholar] [CrossRef] [Green Version]
- Segal, M.S.; Shah, R.; Afzal, A.; Perrault, C.M.; Chang, K.; Schuler, A.; Beem, E.; Shaw, L.C.; Li Calzi, S.; Harrison, J.K.; et al. Nitric oxide cytoskeletal-induced alterations reverse the endothelial progenitor cell migratory defect associated with diabetes. Diabetes 2006, 55, 102–109. [Google Scholar] [CrossRef]
- Chen, Y.S.; Chen, K.H.; Liu, C.C.; Lee, C.T.; Yang, C.H.; Chuang, K.C.; Lin, C.R. Propofol-induced vascular permeability change is related to the nitric oxide signaling pathway and occludin phosphorylation. J. Biomed. Sci. 2007, 14, 629–636. [Google Scholar] [CrossRef]
- Verma, S.; Szmitko, P.E.; Anderson, T.J. Endothelial function: Ready for prime time? Can. J. Cardiol. 2004, 20, 1335–1339. [Google Scholar]
- Kalka, C.; Masuda, H.; Takahashi, T.; Gordon, R.; Tepper, O.; Gravereaux, E.; Pieczek, A.; Iwaguro, H.; Hayashi, S.I.; Isner, J.M.; et al. Vascular endothelial growth factor(165) gene transfer augments circulating endothelial progenitor cells in human subjects. Circ. Res. 2000, 86, 1198–1202. [Google Scholar] [CrossRef] [Green Version]
- Young, P.P.; Hofling, A.A.; Sands, M.S. VEGF increases engraftment of bone marrow-derived endothelial progenitor cells (EPCs) into vasculature of newborn murine recipients. Proc. Natl. Acad. Sci. USA 2002, 99, 11951–11956. [Google Scholar] [CrossRef] [Green Version]
- Dulak, J.; Jozkowicz, A.; Frick, M.; Alber, H.F.; Dichtl, W.; Schwarzacher, S.P.; Pachinger, O.; Weidinger, F. Vascular endothelial growth factor: Angiogenesis, atherogenesis or both? J. Am. Coll. Cardiol. 2001, 38, 2137–2138. [Google Scholar] [CrossRef] [Green Version]
- Dulak, J.; Jozkowicz, A.; Dembinska-Kiec, A.; Guevara, I.; Zdzienicka, A.; Zmudzinska-Grochot, D.; Florek, I.; Wojtowicz, A.; Szuba, A.; Cooke, J.P. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 659–666. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H.; Esumi, H. Reciprocal regulation between nitric oxide and vascular endothelial growth factor in angiogenesis. Acta Biochim. Pol. 2003, 50, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Di, Y.; Zhang, D.; Hu, T.; Li, D. miR-23 regulate the pathogenesis of patients with coronary artery disease. Int J. Clin. Exp. Med. 2015, 8, 11759–11769. [Google Scholar]
- Qin, W.; Xie, W.; Xia, N.; He, Q.; Sun, T. Silencing of Transient Receptor Potential Channel 4 Alleviates oxLDL-induced Angiogenesis in Human Coronary Artery Endothelial Cells by Inhibition of VEGF and NF-kappaB. Med. Sci. Monit. 2016, 22, 930–936. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, X.; Guan, X.; Cui, X.; Wang, Y.; Chu, H.; Cheng, M. Advanced glycation end products impair the migration, adhesion and secretion potentials of late endothelial progenitor cells. Cardiovasc. Diabetol. 2012, 11, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, N.; Langford-Smith, A.W.W.; Wilkinson, F.L.; Alexander, M.Y. Endothelial Progenitor Cells: New Targets for Therapeutics for Inflammatory Conditions With High Cardiovascular Risk. Front. Med. 2018, 5, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castejon, R.; Jimenez-Ortiz, C.; Rosado, S.; Tutor-Ureta, P.; Mellor-Pita, S.; Yebra-Bango, M. Metabolic syndrome is associated with decreased circulating endothelial progenitor cells and increased arterial stiffness in systemic lupus erythematosus. Lupus 2016, 25, 129–136. [Google Scholar] [CrossRef]
- Mok, C.C. Metabolic syndrome and systemic lupus erythematosus: The connection. Expert Rev. Clin. Immunol. 2019, 15, 765–775. [Google Scholar] [CrossRef]
- Rodriguez-Carrio, J.; Prado, C.; de Paz, B.; Lopez, P.; Gomez, J.; Alperi-Lopez, M.; Ballina-Garcia, F.J.; Suarez, A. Circulating endothelial cells and their progenitors in systemic lupus erythematosus and early rheumatoid arthritis patients. Rheumatology 2012, 51, 1775–1784. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Carrio, J.; de Paz, B.; Lopez, P.; Prado, C.; Alperi-Lopez, M.; Ballina-Garcia, F.J.; Suarez, A. IFNalpha serum levels are associated with endothelial progenitor cells imbalance and disease features in rheumatoid arthritis patients. PLoS ONE 2014, 9, e86069. [Google Scholar] [CrossRef]
- Lee, P.Y.; Li, Y.; Richards, H.B.; Chan, F.S.; Zhuang, H.; Narain, S.; Butfiloski, E.J.; Sobel, E.S.; Reeves, W.H.; Segal, M.S. Type I interferon as a novel risk factor for endothelial progenitor cell depletion and endothelial dysfunction in systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 3759–3769. [Google Scholar] [CrossRef]
- Thacker, S.G.; Zhao, W.; Smith, C.K.; Luo, W.; Wang, H.; Vivekanandan-Giri, A.; Rabquer, B.J.; Koch, A.E.; Pennathur, S.; Davidson, A.; et al. Type I interferons modulate vascular function, repair, thrombosis, and plaque progression in murine models of lupus and atherosclerosis. Arthritis Rheum. 2012, 64, 2975–2985. [Google Scholar] [CrossRef] [Green Version]
- Kahlenberg, J.M.; Thacker, S.G.; Berthier, C.C.; Cohen, C.D.; Kretzler, M.; Kaplan, M.J. Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J. Immunol. 2011, 187, 6143–6156. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.G.; Zhong, Z.Y.; Sun, G.F.; Zhou, Y.X.; Zhao, Y. Effects of tumour necrosis factor-alpha on activity and nitric oxide synthase of endothelial progenitor cells from peripheral blood. Cell Prolif. 2011, 44, 352–359. [Google Scholar] [CrossRef]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Madeddu, P.; Waltenberger, J.; Fiorina, P. Vascular stem and progenitor cells in diabetic complications. Exp. Diabetes Res. 2012, 2012, 580343. [Google Scholar] [CrossRef]
- Fadini, G.P.; Sartore, S.; Schiavon, M.; Albiero, M.; Baesso, I.; Cabrelle, A.; Agostini, C.; Avogaro, A. Diabetes impairs progenitor cell mobilisation after hindlimb ischaemia-reperfusion injury in rats. Diabetologia 2006, 49, 3075–3084. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Avogaro, A. Diabetes impairs mobilization of stem cells for the treatment of cardiovascular disease: A meta-regression analysis. Int. J. Cardiol. 2013, 168, 892–897. [Google Scholar] [CrossRef]
- Gallagher, K.A.; Liu, Z.J.; Xiao, M.; Chen, H.; Goldstein, L.J.; Buerk, D.G.; Nedeau, A.; Thom, S.R.; Velazquez, O.C. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J. Clin. Investig. 2007, 117, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Barwari, T.; Rienks, M.; Mayr, M. MicroRNA-21 and the Vulnerability of Atherosclerotic Plaques. Mol. Ther. 2018, 26, 938–940. [Google Scholar] [CrossRef] [Green Version]
- Navickas, R.; Gal, D.; Laucevicius, A.; Taparauskaite, A.; Zdanyte, M.; Holvoet, P. Identifying circulating microRNAs as biomarkers of cardiovascular disease: A systematic review. Cardiovasc. Res. 2016, 111, 322–337. [Google Scholar] [CrossRef]
- Qu, K.; Wang, Z.; Lin, X.L.; Zhang, K.; He, X.L.; Zhang, H. MicroRNAs: Key regulators of endothelial progenitor cell functions. Clin. Chim. Acta 2015, 448, 65–73. [Google Scholar] [CrossRef]
- Ito, T.; Yagi, S.; Yamakuchi, M. MicroRNA-34a regulation of endothelial senescence. Biochem. Biophys. Res. Commun. 2010, 398, 735–740. [Google Scholar] [CrossRef]
- Santulli, G. MicroRNAs and Endothelial (Dys) Function. J. Cell. Physiol. 2016, 231, 1638–1644. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Cao, J.T.; Zhang, B.; Zhou, Q.; Shen, C.X.; Wang, C.Q. Downregulation of microRNA-126 in endothelial progenitor cells from diabetes patients, impairs their functional properties, via target gene Spred-1. J. Mol. Cell. Cardiol. 2012, 53, 64–72. [Google Scholar] [CrossRef]
- Meng, S.; Cao, J.; Zhang, X.; Fan, Y.; Fang, L.; Wang, C.; Lv, Z.; Fu, D.; Li, Y. Downregulation of microRNA-130a contributes to endothelial progenitor cell dysfunction in diabetic patients via its target Runx3. PLoS ONE 2013, 8, e68611. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zuo, Q.; Bi, Y.; Zhang, W.; Jin, J.; Zhang, L.; Zhang, Y.N.; Li, B. miR-31 Regulates Spermatogonial Stem Cells Meiosis via Targeting Stra8. J. Cell. Biochem. 2017, 118, 4844–4853. [Google Scholar] [CrossRef]
- Lu, W.C.; Liu, C.J.; Tu, H.F.; Chung, Y.T.; Yang, C.C.; Kao, S.Y.; Chang, K.W.; Lin, S.C. miR-31 targets ARID1A and enhances the oncogenicity and stemness of head and neck squamous cell carcinoma. Oncotarget 2016, 7, 57254–57267. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.W.; Huang, T.S.; Lo, H.H.; Huang, P.H.; Lin, C.C.; Chang, S.J.; Liao, K.H.; Tsai, C.H.; Chan, C.H.; Tsai, C.F.; et al. Deficiency of the microRNA-31-microRNA-720 pathway in the plasma and endothelial progenitor cells from patients with coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 857–869. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Li, J.; Chen, A.F. MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E110–E116. [Google Scholar] [CrossRef] [Green Version]
- Lian, W.; Hu, X.; Shi, R.; Han, S.; Cao, C.; Wang, K.; Li, M. MiR-31 regulates the function of diabetic endothelial progenitor cells by targeting Satb2. Acta Biochim. Biophys. Sin. 2018, 50, 336–344. [Google Scholar] [CrossRef]
- Choi, S.W.; Friso, S. Epigenetics: A New Bridge between Nutrition and Health. Adv. Nutr. 2010, 1, 8–16. [Google Scholar] [CrossRef]
- Wang, C.; Wang, F.; Li, Z.; Cao, Q.; Huang, L.; Chen, S. MeCP2-mediated epigenetic regulation in senescent endothelial progenitor cells. Stem Cell Res. Ther. 2018, 9, 87. [Google Scholar] [CrossRef]
- Fraineau, S.; Palii, C.G.; McNeill, B.; Ritso, M.; Shelley, W.C.; Prasain, N.; Chu, A.; Vion, E.; Rieck, K.; Nilufar, S.; et al. Epigenetic Activation of Pro-angiogenic Signaling Pathways in Human Endothelial Progenitors Increases Vasculogenesis. Stem Cell Rep. 2017, 9, 1573–1587. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, K.; Vlachojannis, G.J.; Koyanagi, M.; Boeckel, J.N.; Urbich, C.; Farcas, R.; Bonig, H.; Marquez, V.E.; Zeiher, A.M.; Dimmeler, S. Epigenetic regulation of endothelial lineage committed genes in pro-angiogenic hematopoietic and endothelial progenitor cells. Circ. Res. 2011, 109, 1219–1229. [Google Scholar] [CrossRef] [Green Version]
- McCully, K.S. Homocysteine and vascular disease. Nat. Med. 1996, 2, 386–389. [Google Scholar] [CrossRef]
- Kang, S.S.; Wong, P.W.; Malinow, M.R. Hyperhomocyst(e)inemia as a risk factor for occlusive vascular disease. Annu. Rev. Nutr. 1992, 12, 279–298. [Google Scholar] [CrossRef]
- Welch, G.N.; Loscalzo, J. Homocysteine and atherothrombosis. N. Engl. J. Med. 1998, 338, 1042–1050. [Google Scholar] [CrossRef]
- Rasmussen, K.; Moller, J. Total homocysteine measurement in clinical practice. Ann. Clin. Biochem. 2000, 37, 627–648. [Google Scholar] [CrossRef]
- Yang, Q.; He, G.W. Imbalance of Homocysteine and H2S: Significance, Mechanisms, and Therapeutic Promise in Vascular Injury. Oxid. Med. Cell. Longev. 2019, 2019, 7629673. [Google Scholar] [CrossRef] [Green Version]
- Citi, V.; Martelli, A.; Gorica, E.; Brogi, S.; Testai, L.; Calderone, V. Role of hydrogen sulfide in endothelial dysfunction: Pathophysiology and therapeutic approaches. J. Adv. Res. 2021, 27, 99–113. [Google Scholar] [CrossRef]
- Stampfer, M.J.; Malinow, M.R. Can lowering homocysteine levels reduce cardiovascular risk? N. Engl. J. Med. 1995, 332, 328–329. [Google Scholar] [CrossRef]
- Dawson, H.; Collins, G.; Pyle, R.; Deep-Dixit, V.; Taub, D.D. The immunoregulatory effects of homocysteine and its intermediates on T-lymphocyte function. Mech. Ageing Dev. 2004, 125, 107–110. [Google Scholar] [CrossRef]
- Selhub, J.; Jacques, P.F.; Wilson, P.W.; Rush, D.; Rosenberg, I.H. Vitamin status and intake as primary determinants of homocysteinemia in an elderly population. JAMA 1993, 270, 2693–2698. [Google Scholar] [CrossRef]
- Refsum, H.; Smith, A.D.; Ueland, P.M.; Nexo, E.; Clarke, R.; McPartlin, J.; Johnston, C.; Engbaek, F.; Schneede, J.; McPartlin, C.; et al. Facts and recommendations about total homocysteine determinations: An expert opinion. Clin. Chem. 2004, 50, 3–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef] [Green Version]
- Lentz, S.R.; Erger, R.A.; Dayal, S.; Maeda, N.; Malinow, M.R.; Heistad, D.D.; Faraci, F.M. Folate dependence of hyperhomocysteinemia and vascular dysfunction in cystathionine beta-synthase-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H970–H975. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Pacher, P.; Rischak, K.; Szollar, L.; Koller, A. Dysfunction of nitric oxide mediation in isolated rat arterioles with methionine diet-induced hyperhomocysteinemia. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1899–1904. [Google Scholar] [CrossRef] [Green Version]
- Lentz, S.R.; Sobey, C.G.; Piegors, D.J.; Bhopatkar, M.Y.; Faraci, F.M.; Malinow, M.R.; Heistad, D.D. Vascular dysfunction in monkeys with diet-induced hyperhomocyst(e)inemia. J. Clin. Investig. 1996, 98, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Widner, B.; Enzinger, C.; Laich, A.; Wirleitner, B.; Fuchs, D. Hyperhomocysteinemia, pteridines and oxidative stress. Curr. Drug Metab. 2002, 3, 225–232. [Google Scholar] [CrossRef]
- Alvarez, B.; Demicheli, V.; Duran, R.; Trujillo, M.; Cervenansky, C.; Freeman, B.A.; Radi, R. Inactivation of human Cu, Zn superoxide dismutase by peroxynitrite and formation of histidinyl radical. Free Radic. Biol. Med. 2004, 37, 813–822. [Google Scholar] [CrossRef]
- MacMillan-Crow, L.A.; Crow, J.P.; Thompson, J.A. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 1998, 37, 1613–1622. [Google Scholar] [CrossRef]
- Tyagi, N.; Sedoris, K.C.; Steed, M.; Ovechkin, A.V.; Moshal, K.S.; Tyagi, S.C. Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2649–H2656. [Google Scholar] [CrossRef] [Green Version]
- Mayo, J.N.; Beard, R.S., Jr.; Price, T.O.; Chen, C.H.; Erickson, M.A.; Ercal, N.; Banks, W.A.; Bearden, S.E. Nitrative stress in cerebral endothelium is mediated by mGluR5 in hyperhomocysteinemia. J. Cereb. Blood Flow Metab. 2012, 32, 825–834. [Google Scholar] [CrossRef]
- Calcerrada, P.; Peluffo, G.; Radi, R. Nitric oxide-derived oxidants with a focus on peroxynitrite: Molecular targets, cellular responses and therapeutic implications. Curr. Pharm. Des. 2011, 17, 3905–3932. [Google Scholar] [CrossRef]
- Szabo, C.; Mabley, J.G.; Moeller, S.M.; Shimanovich, R.; Pacher, P.; Virag, L.; Soriano, F.G.; Van Duzer, J.H.; Williams, W.; Salzman, A.L.; et al. Part I: Pathogenetic role of peroxynitrite in the development of diabetes and diabetic vascular complications: Studies with FP15, a novel potent peroxynitrite decomposition catalyst. Mol. Med. 2002, 8, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Z.; Zhu, J.H.; Wang, X.X.; Zhu, J.H.; Xie, X.D.; Sun, J.; Shang, Y.P.; Guo, X.G.; Dai, H.M.; Hu, S.J. Effects of homocysteine on number and activity of endothelial progenitor cells from peripheral blood. J. Mol. Cell. Cardiol. 2004, 36, 233–239. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, X.; Chen, J.; Sun, J.; Zhang, F. Reduced number and activity of circulating endothelial progenitor cells from patients with hyperhomocysteinemia. Arch. Med. Res. 2006, 37, 484–489. [Google Scholar]
- Dong, Y.; Sun, Q.; Liu, T.; Wang, H.; Jiao, K.; Xu, J.; Liu, X.; Liu, H.; Wang, W. Nitrative Stress Participates in Endothelial Progenitor Cell Injury in Hyperhomocysteinemia. PLoS ONE 2016, 11, e0158672. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.M.; Mohammad, A.A.; Shuaib, U.; Wang, C.; Ghani, U.; Schwindt, B.; Todd, K.G.; Shuaib, A. Homocysteine reduces endothelial progenitor cells in stroke patients through apoptosis. J. Cereb. Blood Flow Metab. 2009, 29, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Noor, R.; Shuaib, U.; Wang, C.X.; Todd, K.; Ghani, U.; Schwindt, B.; Shuaib, A. High-density lipoprotein cholesterol regulates endothelial progenitor cells by increasing eNOS and preventing apoptosis. Atherosclerosis 2007, 192, 92–99. [Google Scholar] [CrossRef]
- Behera, J.; Tyagi, S.C.; Tyagi, N. Hyperhomocysteinemia induced endothelial progenitor cells dysfunction through hyper-methylation of CBS promoter. Biochem. Biophys. Res. Commun. 2019, 510, 135–141. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Lefer, D.J. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ. Res. 2014, 114, 730–737. [Google Scholar] [CrossRef]
- Ciccone, V.; Genah, S.; Morbidelli, L. Endothelium as a Source and Target of H2S to Improve Its Trophism and Function. Antioxidants 2021, 10, 486. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. H2S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef] [Green Version]
- Beltowski, J. Synthesis, Metabolism, and Signaling Mechanisms of Hydrogen Sulfide: An Overview. Methods Mol. Biol. 2019, 2007, 1–8. [Google Scholar]
- Cheng, Z.; Garikipati, V.N.; Nickoloff, E.; Wang, C.; Polhemus, D.J.; Zhou, J.; Benedict, C.; Khan, M.; Verma, S.K.; Rabinowitz, J.E.; et al. Restoration of Hydrogen Sulfide Production in Diabetic Mice Improves Reparative Function of Bone Marrow Cells. Circulation 2016, 134, 1467–1483. [Google Scholar] [CrossRef]
- Cai, W.J.; Wang, M.J.; Moore, P.K.; Jin, H.M.; Yao, T.; Zhu, Y.C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 2007, 76, 29–40. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [Green Version]
- Szabo, C.; Papapetropoulos, A. Hydrogen sulphide and angiogenesis: Mechanisms and applications. Br. J. Pharmacol. 2011, 164, 853–865. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [Green Version]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol. Cell. 2012, 45, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Modis, K.; Panopoulos, P.; Asimakopoulou, A.; Gero, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef] [Green Version]
- Altaany, Z.; Ju, Y.; Yang, G.; Wang, R. The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci. Signal. 2014, 7, ra87. [Google Scholar] [CrossRef]
- Altaany, Z.; Yang, G.; Wang, R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J. Cell. Mol. Med. 2013, 17, 879–888. [Google Scholar] [CrossRef]
- Liu, F.; Chen, D.D.; Sun, X.; Xie, H.H.; Yuan, H.; Jia, W.; Chen, A.F. Hydrogen sulfide improves wound healing via restoration of endothelial progenitor cell functions and activation of angiopoietin-1 in type 2 diabetes. Diabetes 2014, 63, 1763–1778. [Google Scholar] [CrossRef] [Green Version]
- Obi, S.; Yamamoto, K.; Ando, J. Effects of shear stress on endothelial progenitor cells. J. Biomed. Nanotechnol. 2014, 10, 2586–2597. [Google Scholar] [CrossRef]
- Obi, S.; Masuda, H.; Shizuno, T.; Sato, A.; Yamamoto, K.; Ando, J.; Abe, Y.; Asahara, T. Fluid shear stress induces differentiation of circulating phenotype endothelial progenitor cells. Am. J. Physiol. Cell. Physiol. 2012, 303, C595–C606. [Google Scholar] [CrossRef]
- Xia, W.H.; Yang, Z.; Xu, S.Y.; Chen, L.; Zhang, X.Y.; Li, J.; Liu, X.; Qiu, Y.X.; Shuai, X.T.; Tao, J. Age-related decline in reendothelialization capacity of human endothelial progenitor cells is restored by shear stress. Hypertension 2012, 59, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Chen, C.T.; Chen, C.S.; Wang, Y.M.; Hsieh, H.J.; Wang, D.L. Laminar shear flow increases hydrogen sulfide and activates a nitric oxide producing signaling cascade in endothelial cells. Biochem. Biophys. Res. Commun. 2015, 464, 1254–1259. [Google Scholar] [CrossRef]
- Hu, Q.; Zhang, B.; Liu, Y.; Guo, Y.; Zhang, T.; Nie, R.; Ke, X.; Dong, X. The effect of fluid shear stress in hydrogen sulphide production and cystathionine gamma-lyase expression in human early endothelial progenitor cells. Ann. Transl. Med. 2020, 8, 1318. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C.; Golding, J.; Kuh, D.; Wadsworth, M.E. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 1989, 298, 564–567. [Google Scholar] [CrossRef] [Green Version]
- Roseboom, T.; de Rooij, S.; Painter, R. The Dutch famine and its long-term consequences for adult health. Early Hum. Dev. 2006, 82, 485–491. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Kong, Y.; Zhang, J.H.; Zeng, Q. The Great Chinese Famine leads to shorter and overweight females in Chongqing Chinese population after 50 years. Obesity 2010, 18, 588–592. [Google Scholar] [CrossRef]
- Cohen, E.; Wong, F.Y.; Horne, R.S.; Yiallourou, S.R. Intrauterine growth restriction: Impact on cardiovascular development and function throughout infancy. Pediatr. Res. 2016, 79, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Fattal-Valevski, A.; Bernheim, J.; Leitner, Y.; Redianu, B.; Bassan, H.; Harel, S. Blood pressure values in children with intrauterine growth retardation. Isr. Med. Assoc. J. 2001, 3, 805–808. [Google Scholar]
- Rossi, P.; Tauzin, L.; Marchand, E.; Boussuges, A.; Gaudart, J.; Frances, Y. Respective roles of preterm birth and fetal growth restriction in blood pressure and arterial stiffness in adolescence. J. Adolesc. Health 2011, 48, 520–522. [Google Scholar] [CrossRef]
- Nilsson, P.M.; Ostergren, P.O.; Nyberg, P.; Soderstrom, M.; Allebeck, P. Low birth weight is associated with elevated systolic blood pressure in adolescence: A prospective study of a birth cohort of 149378 Swedish boys. J. Hypertens. 1997, 15, 1627–1631. [Google Scholar] [CrossRef]
- Leon, D.A.; Johansson, M.; Rasmussen, F. Gestational age and growth rate of fetal mass are inversely associated with systolic blood pressure in young adults: An epidemiologic study of 165,136 Swedish men aged 18 years. Am. J. Epidemiol. 2000, 152, 8. [Google Scholar] [CrossRef]
- Curhan, G.C.; Chertow, G.M.; Willett, W.C.; Spiegelman, D.; Colditz, G.A.; Manson, J.E.; Speizer, F.E.; Stampfer, M.J. Birth weight and adult hypertension and obesity in women. Circulation 1996, 94, 1310–1315. [Google Scholar] [CrossRef]
- Law, C.M.; Shiell, A.W. Is blood pressure inversely related to birth weight? The strength of evidence from a systematic review of the literature. J. Hypertens. 1996, 14, 935–941. [Google Scholar] [CrossRef]
- Martyn, C.N.; Barker, D.J.; Jespersen, S.; Greenwald, S.; Osmond, C.; Berry, C. Growth in utero, adult blood pressure, and arterial compliance. Br. Heart J. 1995, 73, 6. [Google Scholar] [CrossRef] [Green Version]
- Yzydorczyk, C.; Armengaud, J.B.; Peyter, A.C.; Chehade, H.; Cachat, F.; Juvet, C.; Siddeek, B.; Simoncini, S.; Sabatier, F.; Dignat-George, F.; et al. Endothelial dysfunction in individuals born after fetal growth restriction: Cardiovascular and renal consequences and preventive approaches. J. Dev. Orig. Health Dis. 2017, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Leeson, C.; Whincup, P.; Cook, D.; Donald, A.; Papacosta, O.; Lucas, A.; Deanfield, J. Flow-mediated dilation in 9- to 11-year-old children: The influence of intrauterine and childhood factors. Circulation 1997, 96, 2233–2238. [Google Scholar] [CrossRef]
- Martin, H.; Gazelius, B.; Norman, M. Impaired acetylcholine-induced vascular relaxation in low birth weight infants: Implications for adult hypertension? Pediatr. Res. 2000, 47, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Martyn, C.N.; Greenwald, S.E. Impaired synthesis of elastin in walls of aorta and large conduit arteries during early development as an initiating event in pathogenesis of systemic hypertension. Lancet 1997, 350, 953–955. [Google Scholar] [CrossRef]
- Joo Turoni, C.; Chaila, Z.; Chahla, R.; Bazan de Casella, M.C.; Peral de Bruno, M. Vascular Function in Children with Low Birthweight and Its Relationship with Early Markers of Cardiovascular Risk. Horm. Res. Paediatr. 2016, 85, 396–405. [Google Scholar] [CrossRef]
- Krause, B.J.; Costello, P.M.; Munoz-Urrutia, E.; Lillycrop, K.A.; Hanson, M.A.; Casanello, P. Role of DNA methyltransferase 1 on the altered eNOS expression in human umbilical endothelium from intrauterine growth restricted fetuses. Epigenetics 2013, 8, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Caniuguir, A.; Krause, B.J.; Hernandez, C.; Uauy, R.; Casanello, P. Markers of early endothelial dysfunction in intrauterine growth restriction-derived human umbilical vein endothelial cells revealed by 2D-DIGE and mass spectrometry analyses. Placenta 2016, 41, 14–26. [Google Scholar] [CrossRef]
- Grandvuillemin, I.; Buffat, C.; Boubred, F.; Lamy, E.; Fromonot, J.; Charpiot, P.; Simoncini, S.; Sabatier, F.; Dignat-George, F.; Peyter, A.C.; et al. Arginase up-regulation and eNOS uncoupling contribute to impaired endothelium-dependent vasodilation in a rat model of intrauterine growth restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R509–R520. [Google Scholar] [CrossRef]
- Cambonie, G.; Comte, B.; Yzydorczyk, C.; Ntimbane, T.; Germain, N.; Le, N.L.; Pladys, P.; Gauthier, C.; Lahaie, I.; Abran, D.; et al. Antenatal antioxidant prevents adult hypertension, vascular dysfunction, and microvascular rarefaction associated with in utero exposure to a low-protein diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1236–R1245. [Google Scholar] [CrossRef]
- Yzydorczyk, C.; Comte, B.; Cambonie, G.; Lavoie, J.C.; Germain, N.; Ting Shun, Y.; Wolff, J.; Deschepper, C.; Touyz, R.M.; Lelievre-Pegorier, M.; et al. Neonatal oxygen exposure in rats leads to cardiovascular and renal alterations in adulthood. Hypertension 2008, 52, 889–895. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J.; Clark, P.M.; Cox, L.J.; Fall, C.; Osmond, C.; Winter, P.D. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 1991, 303, 1019–1022. [Google Scholar] [CrossRef] [Green Version]
- Zarrati, M.; Shidfar, F.; Razmpoosh, E.; Nezhad, F.N.; Keivani, H.; Hemami, M.R.; Asemi, Z. Does low birth weight predict hypertension and obesity in schoolchildren? Ann. Nutr. Metab. 2013, 63, 69–76. [Google Scholar] [CrossRef]
- Ramadhani, M.K.; Grobbee, D.E.; Bots, M.L.; Castro Cabezas, M.; Vos, L.E.; Oren, A.; Uiterwaal, C.S. Lower birth weight predicts metabolic syndrome in young adults: The Atherosclerosis Risk in Young Adults (ARYA)-study. Atherosclerosis 2006, 184, 21–27. [Google Scholar] [CrossRef]
- Nobili, V.; Marcellini, M.; Marchesini, G.; Vanni, E.; Manco, M.; Villani, A.; Bugianesi, E. Intrauterine growth retardation, insulin resistance, and nonalcoholic fatty liver disease in children. Diabetes Care 2007, 30, 2638–2640. [Google Scholar] [CrossRef] [Green Version]
- Fraser, A.; Ebrahim, S.; Davey Smith, G.; Lawlor, D.A. The associations between height components (leg and trunk length) and adult levels of liver enzymes. J. Epidemiol. Community Health 2008, 62, 48–53. [Google Scholar] [CrossRef]
- Meister, B.; Totsch, M.; Mayr, A.; Widschwendter, M.; Huter, O.; Sperl, W. Identification of CD34+ cord blood cells and their subpopulations in preterm and term neonates using three-color flow cytometry. Biol. Neonate 1994, 66, 272–279. [Google Scholar] [CrossRef]
- Borghesi, A.; Massa, M.; Campanelli, R.; Bollani, L.; Tzialla, C.; Figar, T.A.; Ferrari, G.; Bonetti, E.; Chiesa, G.; de Silvestri, A.; et al. Circulating endothelial progenitor cells in preterm infants with bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2009, 180, 540–546. [Google Scholar] [CrossRef]
- Calcaterra, F.; Taddeo, A.; Colombo, E.; Cappelletti, M.; Martinelli, A.; Calabrese, S.; Mavilio, D.; Cetin, I.; Della Bella, S. Reduction of maternal circulating endothelial progenitor cells in human pregnancies with intrauterine growth restriction. Placenta 2014, 35, 431–436. [Google Scholar] [CrossRef]
- Monga, R.; Buck, S.; Sharma, P.; Thomas, R.; Chouthai, N.S. Effect of preeclampsia and intrauterine growth restriction on endothelial progenitor cells in human umbilical cord blood. J. Matern. Fetal Neonatal. Med. 2012, 25, 2385–2389. [Google Scholar] [CrossRef]
- Ligi, I.; Simoncini, S.; Tellier, E.; Vassallo, P.F.; Sabatier, F.; Guillet, B.; Lamy, E.; Sarlon, G.; Quemener, C.; Bikfalvi, A.; et al. A switch toward angiostatic gene expression impairs the angiogenic properties of endothelial progenitor cells in low birth weight preterm infants. Blood 2011, 118, 1699–1709. [Google Scholar] [CrossRef]
- Simoncini, S.; Chateau, A.L.; Robert, S.; Todorova, D.; Yzydorczyk, C.; Lacroix, R.; Ligi, I.; Louis, L.; Bachelier, R.; Simeoni, U.; et al. Biogenesis of Pro-senescent Microparticles by Endothelial Colony Forming Cells from Premature Neonates is driven by SIRT1-Dependent Epigenetic Regulation of MKK6. Sci. Rep. 2017, 7, 8277. [Google Scholar] [CrossRef] [Green Version]
- Darby, J.R.T.; Varcoe, T.J.; Orgeig, S.; Morrison, J.L. Cardiorespiratory consequences of intrauterine growth restriction: Influence of timing, severity and duration of hypoxaemia. Theriogenology 2020, 150, 84–95. [Google Scholar] [CrossRef]
- Pike, K.; Jane Pillow, J.; Lucas, J.S. Long term respiratory consequences of intrauterine growth restriction. Semin. Fetal Neonatal. Med. 2012, 17, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Briana, D.D.; Malamitsi-Puchner, A. Small for gestational age birth weight: Impact on lung structure and function. Paediatr. Respir. Rev. 2013, 14, 256–262. [Google Scholar] [CrossRef]
- Zana-Taieb, E.; Pham, H.; Franco-Montoya, M.L.; Jacques, S.; Letourneur, F.; Baud, O.; Jarreau, P.H.; Vaiman, D. Impaired alveolarization and intra-uterine growth restriction in rats: A postnatal genome-wide analysis. J. Pathol. 2015, 235, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.F.; Lv, Y.; Gu, W.Z.; Tang, L.L.; Wei, J.K.; Zhang, L.Y.; Du, L.Z. Epigenetics of hypoxic pulmonary arterial hypertension following intrauterine growth retardation rat: Epigenetics in PAH following IUGR. Respir. Res. 2013, 14, 20. [Google Scholar] [CrossRef] [Green Version]
- Junhui, Z.; Xingxiang, W.; Guosheng, F.; Yunpeng, S.; Furong, Z.; Junzhu, C. Reduced number and activity of circulating endothelial progenitor cells in patients with idiopathic pulmonary arterial hypertension. Respir. Med. 2008, 102, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Diller, G.P.; van Eijl, S.; Okonko, D.O.; Howard, L.S.; Ali, O.; Thum, T.; Wort, S.J.; Bedard, E.; Gibbs, J.S.; Bauersachs, J.; et al. Circulating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertension. Circulation 2008, 117, 3020–3030. [Google Scholar] [CrossRef] [Green Version]
- Assad, T.R.; Hemnes, A.R. Metabolic Dysfunction in Pulmonary Arterial Hypertension. Curr. Hypertens. Rep. 2015, 17, 20. [Google Scholar] [CrossRef] [Green Version]
- Renaud, S.; de Lorgeril, M. Wine, alcohol, platelets, and the French paradox for coronary heart disease. Lancet 1992, 339, 1523–1526. [Google Scholar] [CrossRef]
- Renaud, S.C.; Gueguen, R.; Schenker, J.; d’Houtaud, A. Alcohol and mortality in middle-aged men from eastern France. Epidemiology 1998, 9, 184–188. [Google Scholar] [CrossRef]
- Weiskirchen, S.; Weiskirchen, R. Resveratrol: How Much Wine Do You Have to Drink to Stay Healthy? Adv. Nutr. 2016, 7, 706–718. [Google Scholar] [CrossRef] [Green Version]
- Piotrowska, H.; Kucinska, M.; Murias, M. Biological activity of piceatannol: Leaving the shadow of resveratrol. Mutat. Res. 2012, 750, 60–82. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.Z.; Liu, R.X.; Wang, D.P.; Wang, X.; Dai, C.C. Biocatalysis and biotransformation of resveratrol in microorganisms. Biotechnol. Lett. 2015, 37, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Manganiello, V.; Dyck, J.R. Resveratrol as a calorie restriction mimetic: Therapeutic implications. Trends Cell. Biol. 2012, 22, 546–554. [Google Scholar] [CrossRef] [Green Version]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Heiss, E.H.; Dirsch, V.M. Effect of resveratrol on endothelial cell function: Molecular mechanisms. Biofactors 2010, 36, 342–349. [Google Scholar] [CrossRef]
- Wang, H.; Yang, Y.J.; Qian, H.Y.; Zhang, Q.; Xu, H.; Li, J.J. Resveratrol in cardiovascular disease: What is known from current research? Heart Fail. Rev. 2012, 17, 437–448. [Google Scholar] [CrossRef]
- Dyck, G.J.B.; Raj, P.; Zieroth, S.; Dyck, J.R.B.; Ezekowitz, J.A. The Effects of Resveratrol in Patients with Cardiovascular Disease and Heart Failure: A Narrative Review. Int. J. Mol. Sci. 2019, 20, 904. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.B.; Huang, J.; Zou, J.G.; Su, E.B.; Shan, Q.J.; Yang, Z.J.; Cao, K.J. Effects of resveratrol on number and activity of endothelial progenitor cells from human peripheral blood. Clin. Exp. Pharmacol. Physiol. 2007, 34, 1109–1115. [Google Scholar] [PubMed]
- Wallerath, T.; Deckert, G.; Ternes, T.; Anderson, H.; Li, H.; Witte, K.; Forstermann, U. Resveratrol, a polyphenolic phytoalexin present in red wine, enhances expression and activity of endothelial nitric oxide synthase. Circulation 2002, 106, 1652–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gracia-Sancho, J.; Villarreal, G., Jr.; Zhang, Y.; Garcia-Cardena, G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc. Res. 2010, 85, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Chen, Z.; Chen, J.Z.; Xie, J.; Xu, B. Resveratrol Improves Tube Formation in AGE-Induced Late Endothelial Progenitor Cells by Suppressing Syndecan-4 Shedding. Oxid. Med. Cell. Longev. 2018, 2018, 9045976. [Google Scholar] [CrossRef] [Green Version]
- Xia, N.; Daiber, A.; Forstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, L.; Wang, X.X.; Hu, X.S.; Guo, X.G.; Shang, Y.P.; Chen, H.J.; Zeng, C.L.; Zhang, F.R.; Chen, J.Z. Resveratrol reduces endothelial progenitor cells senescence through augmentation of telomerase activity by Akt-dependent mechanisms. Br. J. Pharmacol. 2008, 155, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.B.; Zhu, L.; Huang, J.; Yin, Y.G.; Kong, X.Q.; Rong, Q.F.; Shi, A.W.; Cao, K.J. Resveratrol-induced augmentation of telomerase activity delays senescence of endothelial progenitor cells. Chin. Med. J. 2011, 124, 4310–4315. [Google Scholar]
- Wu, H.; Li, G.N.; Xie, J.; Li, R.; Chen, Q.H.; Chen, J.Z.; Wei, Z.H.; Kang, L.N.; Xu, B. Resveratrol ameliorates myocardial fibrosis by inhibiting ROS/ERK/TGF-beta/periostin pathway in STZ-induced diabetic mice. BMC Cardiovasc. Disord. 2016, 16, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spanier, G.; Xu, H.; Xia, N.; Tobias, S.; Deng, S.; Wojnowski, L.; Forstermann, U.; Li, H. Resveratrol reduces endothelial oxidative stress by modulating the gene expression of superoxide dismutase 1 (SOD1), glutathione peroxidase 1 (GPx1) and NADPH oxidase subunit (Nox4). J. Physiol. Pharmacol. 2009, 60, 111–116. [Google Scholar]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.F.; Chen, L.L.; Zeng, T.S.; Li, Y.M.; Fan, Y.; Hu, L.J.; Ling, Y. Number of circulating endothelial progenitor cells as a marker of vascular endothelial function for type 2 diabetes. Vasc. Med. 2010, 15, 279–285. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Musi, N.; Hirshman, M.F.; Nygren, J.; Svanfeldt, M.; Bavenholm, P.; Rooyackers, O.; Zhou, G.; Williamson, J.M.; Ljunqvist, O.; Efendic, S.; et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 2002, 51, 2074–2081. [Google Scholar] [CrossRef] [Green Version]
- Kao, J.; Tobis, J.; McClelland, R.L.; Heaton, M.R.; Davis, B.R.; Holmes, D.R., Jr.; Currier, J.W. Investigators in the Prevention of Restenosis With Tranilast and its Outcomes Trial. Relation of metformin treatment to clinical events in diabetic patients undergoing percutaneous intervention. Am. J. Cardiol. 2004, 93, 1347–1350. [Google Scholar] [CrossRef]
- Zheng, Z.; Chen, H.; Li, J.; Li, T.; Zheng, B.; Zheng, Y.; Jin, H.; He, Y.; Gu, Q.; Xu, X. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes 2012, 61, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Arunachalam, G.; Samuel, S.M.; Marei, I.; Ding, H.; Triggle, C.R. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br. J. Pharmacol. 2014, 171, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Bakhashab, S.; Ahmed, F.W.; Schulten, H.J.; Bashir, A.; Karim, S.; Al-Malki, A.L.; Gari, M.A.; Abuzenadah, A.M.; Chaudhary, A.G.; Alqahtani, M.H.; et al. Metformin improves the angiogenic potential of human CD34(+) cells co-incident with downregulating CXCL10 and TIMP1 gene expression and increasing VEGFA under hyperglycemia and hypoxia within a therapeutic window for myocardial infarction. Cardiovasc. Diabetol. 2016, 15, 27. [Google Scholar] [CrossRef] [Green Version]
- Haendeler, J.; Dimmeler, S. Inseparably tied: Functional and antioxidative capacity of endothelial progenitor cells. Circ. Res. 2006, 98, 157–158. [Google Scholar] [CrossRef] [Green Version]
- Formoso, G.; De Filippis, E.A.; Michetti, N.; Di Fulvio, P.; Pandolfi, A.; Bucciarelli, T.; Ciabattoni, G.; Nicolucci, A.; Davi, G.; Consoli, A. Decreased in vivo oxidative stress and decreased platelet activation following metformin treatment in newly diagnosed type 2 diabetic subjects. Diabetes Metab. Res. Rev. 2008, 24, 231–237. [Google Scholar] [CrossRef]
- Sena, C.M.; Louro, T.; Matafome, P.; Nunes, E.; Monteiro, P.; Seica, R. Antioxidant and vascular effects of gliclazide in type 2 diabetic rats fed high-fat diet. Physiol. Res. 2009, 58, 203–209. [Google Scholar] [CrossRef]
- Chen, L.L.; Liao, Y.F.; Zeng, T.S.; Yu, F.; Li, H.Q.; Feng, Y. Effects of metformin plus gliclazide compared with metformin alone on circulating endothelial progenitor cell in type 2 diabetic patients. Endocrine 2010, 38, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Xiao-Yun, X.; Zhao-Hui, M.; Ke, C.; Hong-Hui, H.; Yan-Hong, X. Glucagon-like peptide-1 improves proliferation and differentiation of endothelial progenitor cells via upregulating VEGF generation. Med. Sci. Monit. 2011, 17, BR35–BR41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves, A.; Leal, E.; Paiva, A.; Teixeira Lemos, E.; Teixeira, F.; Ribeiro, C.F.; Reis, F.; Ambrosio, A.F.; Fernandes, R. Protective effects of the dipeptidyl peptidase IV inhibitor sitagliptin in the blood-retinal barrier in a type 2 diabetes animal model. Diabetes Obes. Metab. 2012, 14, 454–463. [Google Scholar] [CrossRef]
- Won, S.; Sayeed, I.; Peterson, B.L.; Wali, B.; Kahn, J.S.; Stein, D.G. Vitamin D prevents hypoxia/reoxygenation-induced blood-brain barrier disruption via vitamin D receptor-mediated NF-kB signaling pathways. PLoS ONE 2015, 10, e0122821. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Meza, C.A.; Clarke, H.; Kim, J.S.; Hickner, R.C. Vitamin D and Endothelial Function. Nutrients 2020, 12, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cianciolo, G.; La Manna, G.; Della Bella, E.; Cappuccilli, M.L.; Angelini, M.L.; Dormi, A.; Capelli, I.; Laterza, C.; Costa, R.; Alviano, F.; et al. Effect of vitamin D receptor activator therapy on vitamin D receptor and osteocalcin expression in circulating endothelial progenitor cells of hemodialysis patients. Blood Purif. 2013, 35, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Hammer, Y.; Soudry, A.; Levi, A.; Talmor-Barkan, Y.; Leshem-Lev, D.; Singer, J.; Kornowski, R.; Lev, E.I. Effect of vitamin D on endothelial progenitor cells function. PLoS ONE 2017, 12, e0178057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, M.; Pabst, A.M.; Houdek, J.P.; Ziebart, T.; Konerding, M.A. Priming with proangiogenic growth factors and endothelial progenitor cells improves revascularization in linear diabetic wounds. Int. J. Mol. Med. 2014, 33, 833–839. [Google Scholar] [CrossRef] [Green Version]
- Mees, B.; Recalde, A.; Loinard, C.; Tempel, D.; Godinho, M.; Vilar, J.; van Haperen, R.; Levy, B.; de Crom, R.; Silvestre, J.S. Endothelial nitric oxide synthase overexpression restores the efficiency of bone marrow mononuclear cell-based therapy. Am. J. Pathol. 2011, 178, 55–60. [Google Scholar] [CrossRef]
- Essaadi, A.; Nollet, M.; Moyon, A.; Stalin, J.; Simoncini, S.; Balasse, L.; Bertaud, A.; Bachelier, R.; Leroyer, A.S.; Sarlon, G.; et al. Stem cell properties of peripheral blood endothelial progenitors are stimulated by soluble CD146 via miR-21: Potential use in autologous cell therapy. Sci. Rep. 2018, 8, 9387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masaki, T. Possible role of endothelin in endothelial regulation of vascular tone. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 235–255. [Google Scholar] [CrossRef]
- Johnston, C.I. Franz Volhard Lecture. Renin-angiotensin system: A dual tissue and hormonal system for cardiovascular control. J. Hypertens. Suppl. 1992, 10, S13–S26. [Google Scholar] [CrossRef]
- Rossi, G.P.; Sacchetto, A.; Cesari, M.; Pessina, A.C. Interactions between endothelin-1 and the renin-angiotensin-aldosterone system. Cardiovasc. Res. 1999, 43, 300–307. [Google Scholar] [CrossRef]
- Bahlmann, F.H.; de Groot, K.; Mueller, O.; Hertel, B.; Haller, H.; Fliser, D. Stimulation of endothelial progenitor cells: A new putative therapeutic effect of angiotensin II receptor antagonists. Hypertension 2005, 45, 526–529. [Google Scholar] [CrossRef] [Green Version]
- Steinmetz, M.; Brouwers, C.; Nickenig, G.; Wassmann, S. Synergistic effects of telmisartan and simvastatin on endothelial progenitor cells. J. Cell. Mol. Med. 2010, 14, 1645–1656. [Google Scholar] [CrossRef] [Green Version]
- Reinhard, H.; Jacobsen, P.K.; Lajer, M.; Pedersen, N.; Billestrup, N.; Mandrup-Poulsen, T.; Parving, H.H.; Rossing, P. Multifactorial treatment increases endothelial progenitor cells in patients with type 2 diabetes. Diabetologia 2010, 53, 2129–2133. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.; Rafnsson, A.; Brismar, K.; Pernow, J. Endothelial progenitor cells in relation to endothelin-1 and endothelin receptor blockade: A randomized, controlled trial. Int. J. Cardiol. 2013, 168, 1017–1022. [Google Scholar] [CrossRef] [Green Version]
- Ociepa, T.; Bartnik, M.; Zielezinska, K.; Prokowska, M.; Urasinska, E.; Urasinski, T. Abnormal correlation of circulating endothelial progenitor cells and endothelin-1 concentration may contribute to the development of arterial hypertension in childhood acute lymphoblastic leukemia survivors. Hypertens. Res. 2016, 39, 530–535. [Google Scholar] [CrossRef]
- Zhai, L.; Liu, Y.; Zhao, W.; Chen, Q.; Guo, T.; Wei, W.; Luo, Z.; Huang, Y.; Ma, C.; Huang, F.; et al. Aerobic and resistance training enhances endothelial progenitor cell function via upregulation of caveolin-1 in mice with type 2 diabetes. Stem Cell Res. Ther. 2020, 11, 10. [Google Scholar] [CrossRef]
- Sonnenschein, K.; Horvath, T.; Mueller, M.; Markowski, A.; Siegmund, T.; Jacob, C.; Drexler, H.; Landmesser, U. Exercise training improves in vivo endothelial repair capacity of early endothelial progenitor cells in subjects with metabolic syndrome. Eur. J. Cardiovasc. Prev. Rehabil. 2011, 18, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, J.M.; Rosado-Alvarez, D.; Da Silva Grigoletto, M.E.; Rangel-Zuniga, O.A.; Landaeta-Diaz, L.L.; Caballero-Villarraso, J.; Lopez-Miranda, J.; Perez-Jimenez, F.; Fuentes-Jimenez, F. Moderate-to-high-intensity training and a hypocaloric Mediterranean diet enhance endothelial progenitor cells and fitness in subjects with the metabolic syndrome. Clin. Sci. 2012, 123, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Whiteman, M.; Guan, Y.Y.; Neo, K.L.; Cheng, Y.; Lee, S.W.; Zhao, Y.; Baskar, R.; Tan, C.H.; Moore, P.K. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): New insights into the biology of hydrogen sulfide. Circulation 2008, 117, 2351–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Han, Y.; Li, L.; Lu, H.; Meng, G.; Li, X.; Shirhan, M.; Peh, M.T.; Xie, L.; Zhou, S.; et al. The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E(-/-) mice. Br. J. Pharmacol. 2013, 169, 1795–1809. [Google Scholar] [CrossRef] [Green Version]
- Tomasova, L.; Pavlovicova, M.; Malekova, L.; Misak, A.; Kristek, F.; Grman, M.; Cacanyiova, S.; Tomasek, M.; Tomaskova, Z.; Perry, A.; et al. Effects of AP39, a novel triphenylphosphonium derivatised anethole dithiolethione hydrogen sulfide donor, on rat haemodynamic parameters and chloride and calcium Cav3 and RyR2 channels. Nitric Oxide 2015, 46, 131–144. [Google Scholar] [CrossRef]
- Xiong, Y.; Chang, L.L.; Tran, B.; Dai, T.; Zhong, R.; Mao, Y.C.; Zhu, Y.Z. ZYZ-803, a novel hydrogen sulfide-nitric oxide conjugated donor, promotes angiogenesis via cross-talk between STAT3 and CaMKII. Acta Pharmacol. Sin. 2020, 41, 218–228. [Google Scholar] [CrossRef]
- Hu, Q.; Ke, X.; Zhang, T.; Chen, Y.; Huang, Q.; Deng, B.; Xie, S.; Wang, J.; Nie, R. Hydrogen sulfide improves vascular repair by promoting endothelial nitric oxide synthase-dependent mobilization of endothelial progenitor cells. J. Hypertens. 2019, 37, 972–984. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; JH, O.K.; McCarty, M.F. Boosting endogenous production of vasoprotective hydrogen sulfide via supplementation with taurine and N-acetylcysteine: A novel way to promote cardiovascular health. Open Heart 2017, 4, e000600. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, B.; Li, Y.; Sun, F.; Li, P.; Xia, W.; Zhou, X.; Li, Q.; Wang, X.; Chen, J.; et al. Taurine Supplementation Lowers Blood Pressure and Improves Vascular Function in Prehypertension: Randomized, Double-Blind, Placebo-Controlled Study. Hypertension 2016, 67, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Benavides, G.A.; Squadrito, G.L.; Mills, R.W.; Patel, H.D.; Isbell, T.S.; Patel, R.P.; Darley-Usmar, V.M.; Doeller, J.E.; Kraus, D.W. Hydrogen sulfide mediates the vasoactivity of garlic. Proc. Natl. Acad. Sci. USA 2007, 104, 17977–17982. [Google Scholar] [CrossRef] [Green Version]
- Polhemus, D.; Kondo, K.; Bhushan, S.; Bir, S.C.; Kevil, C.G.; Murohara, T.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ. Heart Fail. 2013, 6, 1077–1086. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.K.; Manral, A.; Saini, V.; Singh, A.; Srinivasan, B.P.; Tiwari, M. Novel diallyldisulfide analogs ameliorate cardiovascular remodeling in rats with L-NAME-induced hypertension. Eur. J. Pharmacol. 2012, 691, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Yoder, M.C. Circulating and tissue resident endothelial progenitor cells. J. Cell. Physiol. 2014, 229, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Yoder, M.C. Endothelial progenitor cell: A blood cell by many other names may serve similar functions. J. Mol. Med. 2013, 91, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.S.; Ng, W.K.; Chan, J.K. Concise Review: Endothelial Progenitor Cells in Regenerative Medicine: Applications and Challenges. Stem Cells Transl. Med. 2016, 5, 530–538. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.X.; Zhang, F.R.; Shang, Y.P.; Zhu, J.H.; Xie, X.D.; Tao, Q.M.; Zhu, J.H.; Chen, J.Z. Transplantation of autologous endothelial progenitor cells may be beneficial in patients with idiopathic pulmonary arterial hypertension: A pilot randomized controlled trial. J. Am. Coll. Cardiol. 2007, 49, 1566–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peyter, A.-C.; Armengaud, J.-B.; Guillot, E.; Yzydorczyk, C. Endothelial Progenitor Cells Dysfunctions and Cardiometabolic Disorders: From Mechanisms to Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 6667. https://doi.org/10.3390/ijms22136667
Peyter A-C, Armengaud J-B, Guillot E, Yzydorczyk C. Endothelial Progenitor Cells Dysfunctions and Cardiometabolic Disorders: From Mechanisms to Therapeutic Approaches. International Journal of Molecular Sciences. 2021; 22(13):6667. https://doi.org/10.3390/ijms22136667
Chicago/Turabian StylePeyter, Anne-Christine, Jean-Baptiste Armengaud, Estelle Guillot, and Catherine Yzydorczyk. 2021. "Endothelial Progenitor Cells Dysfunctions and Cardiometabolic Disorders: From Mechanisms to Therapeutic Approaches" International Journal of Molecular Sciences 22, no. 13: 6667. https://doi.org/10.3390/ijms22136667
APA StylePeyter, A. -C., Armengaud, J. -B., Guillot, E., & Yzydorczyk, C. (2021). Endothelial Progenitor Cells Dysfunctions and Cardiometabolic Disorders: From Mechanisms to Therapeutic Approaches. International Journal of Molecular Sciences, 22(13), 6667. https://doi.org/10.3390/ijms22136667