
Ferryl Hemoglobin and Heme Induce A1-Microglobulin in Hemorrhaged Atherosclerotic Lesions with Inhibitory Function against Hemoglobin and Lipid Oxidation

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
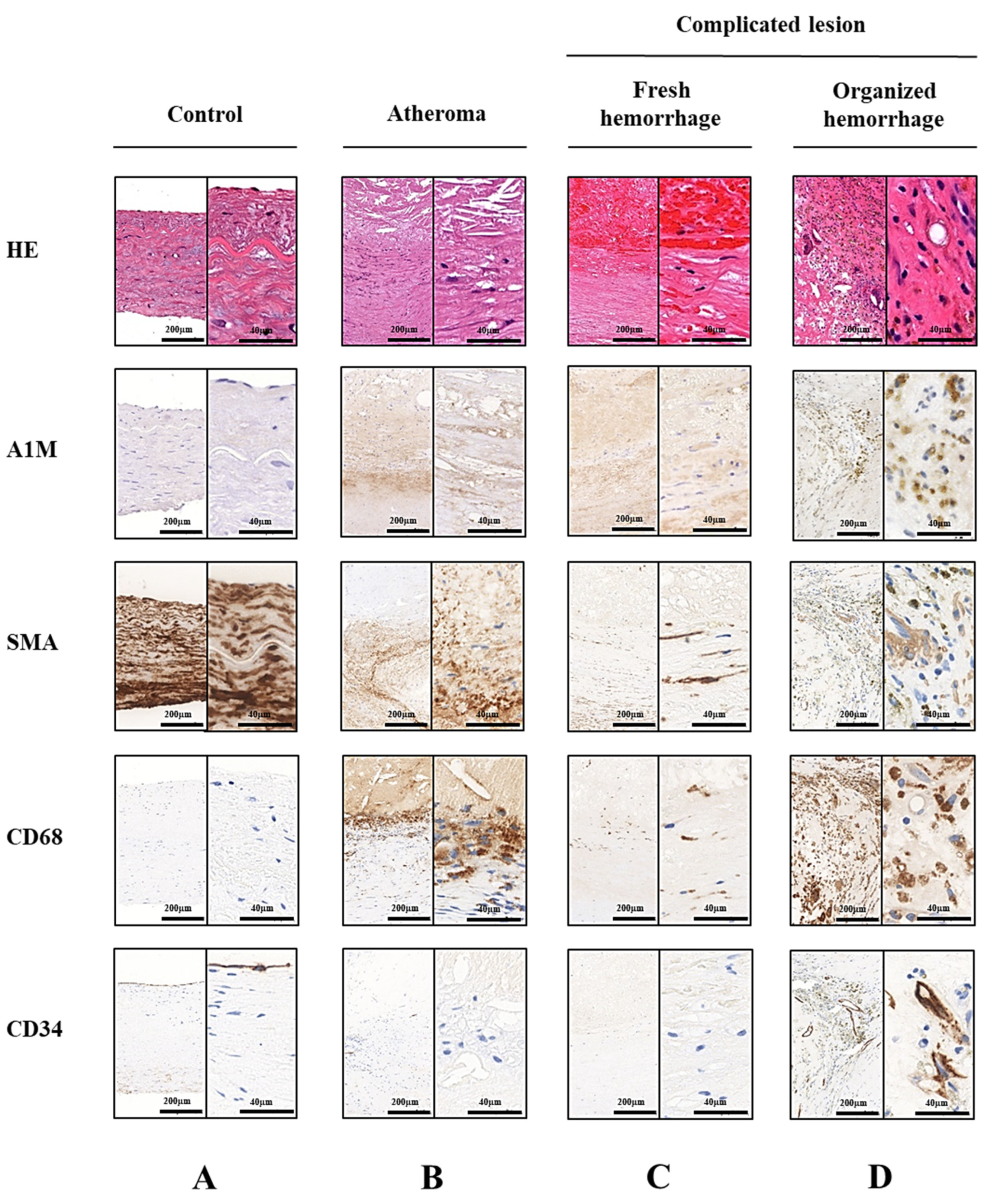
2.1. A1M Protein Is Abundantly Present in Human Carotid Artery Plaques
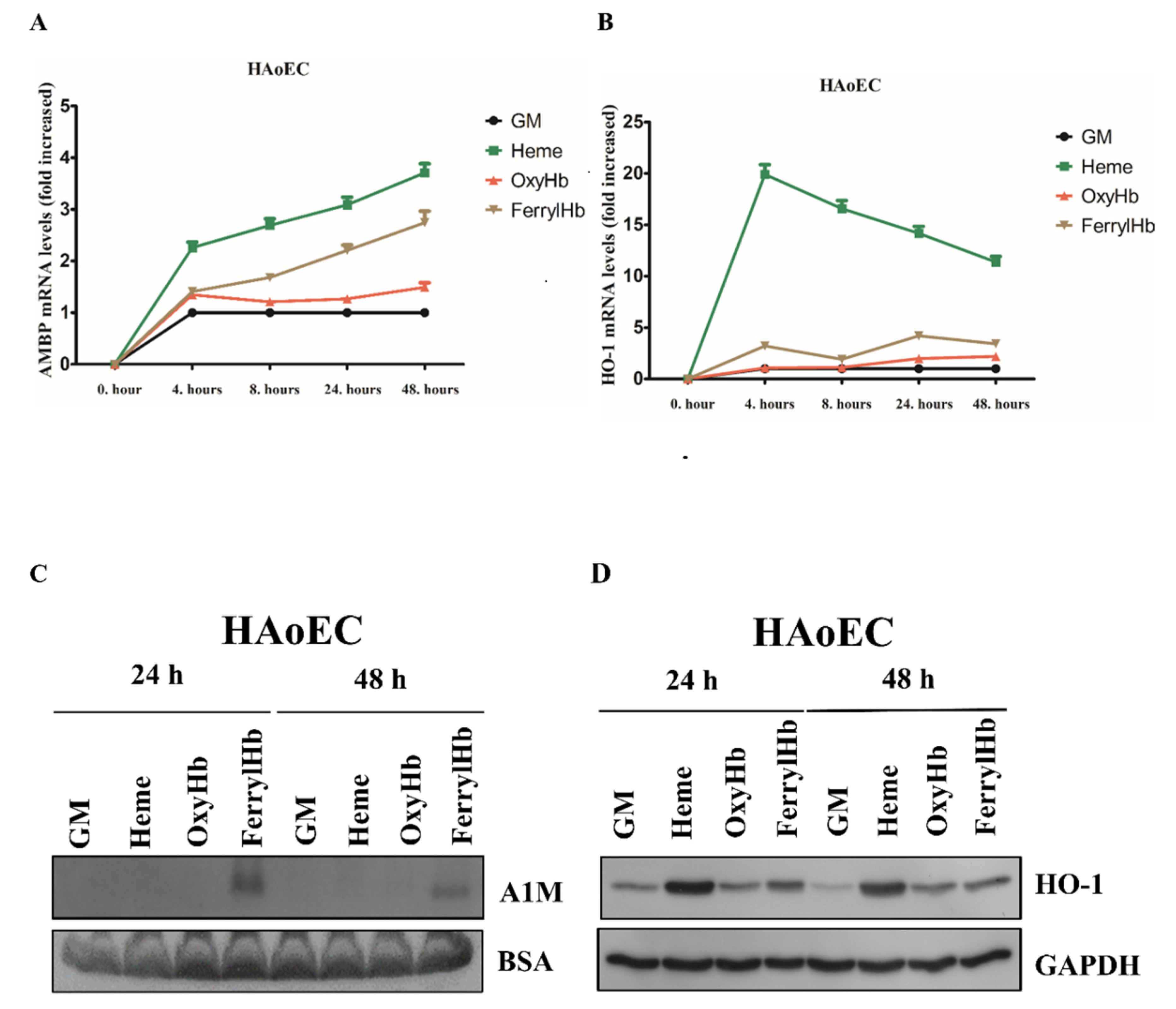
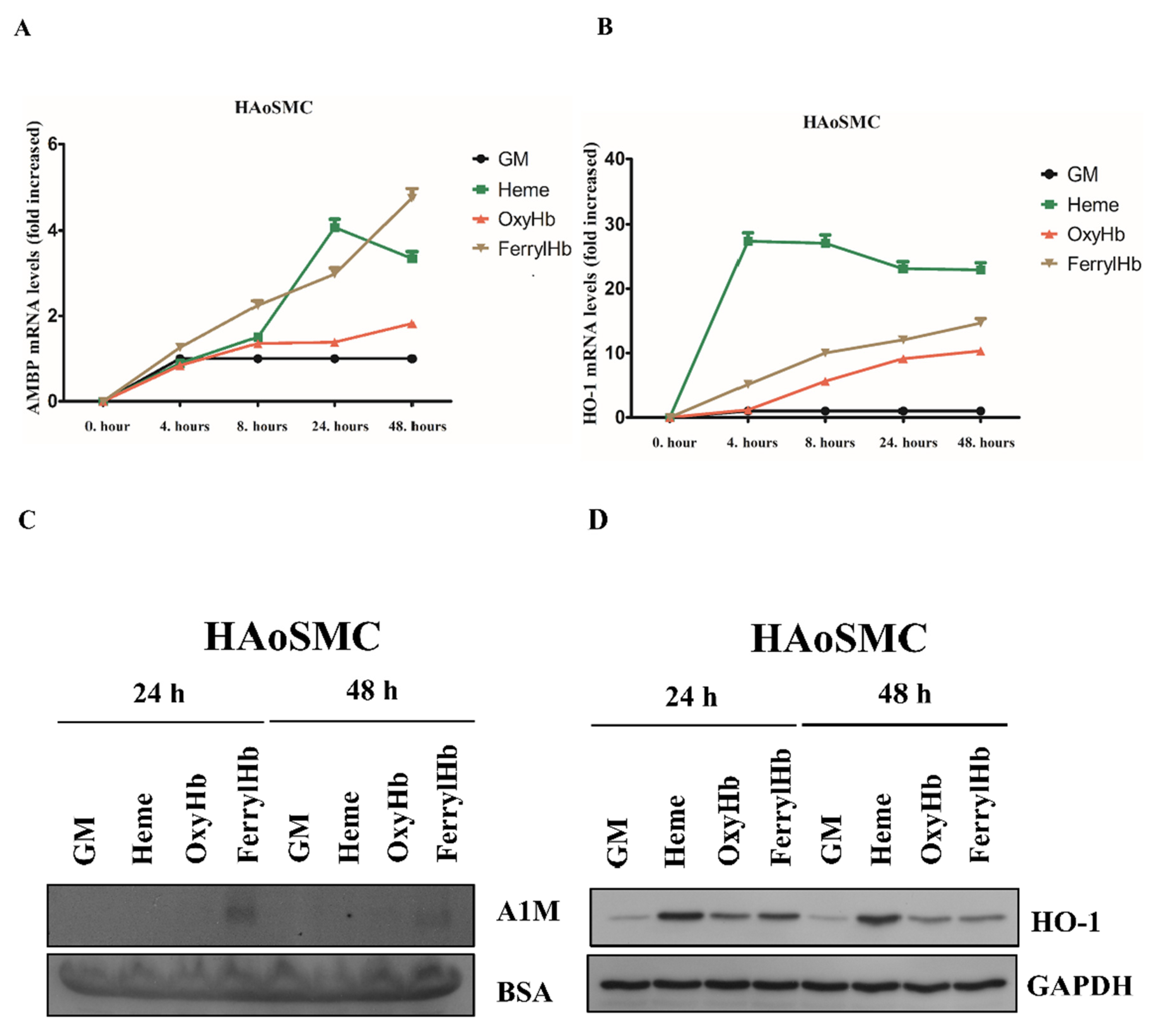
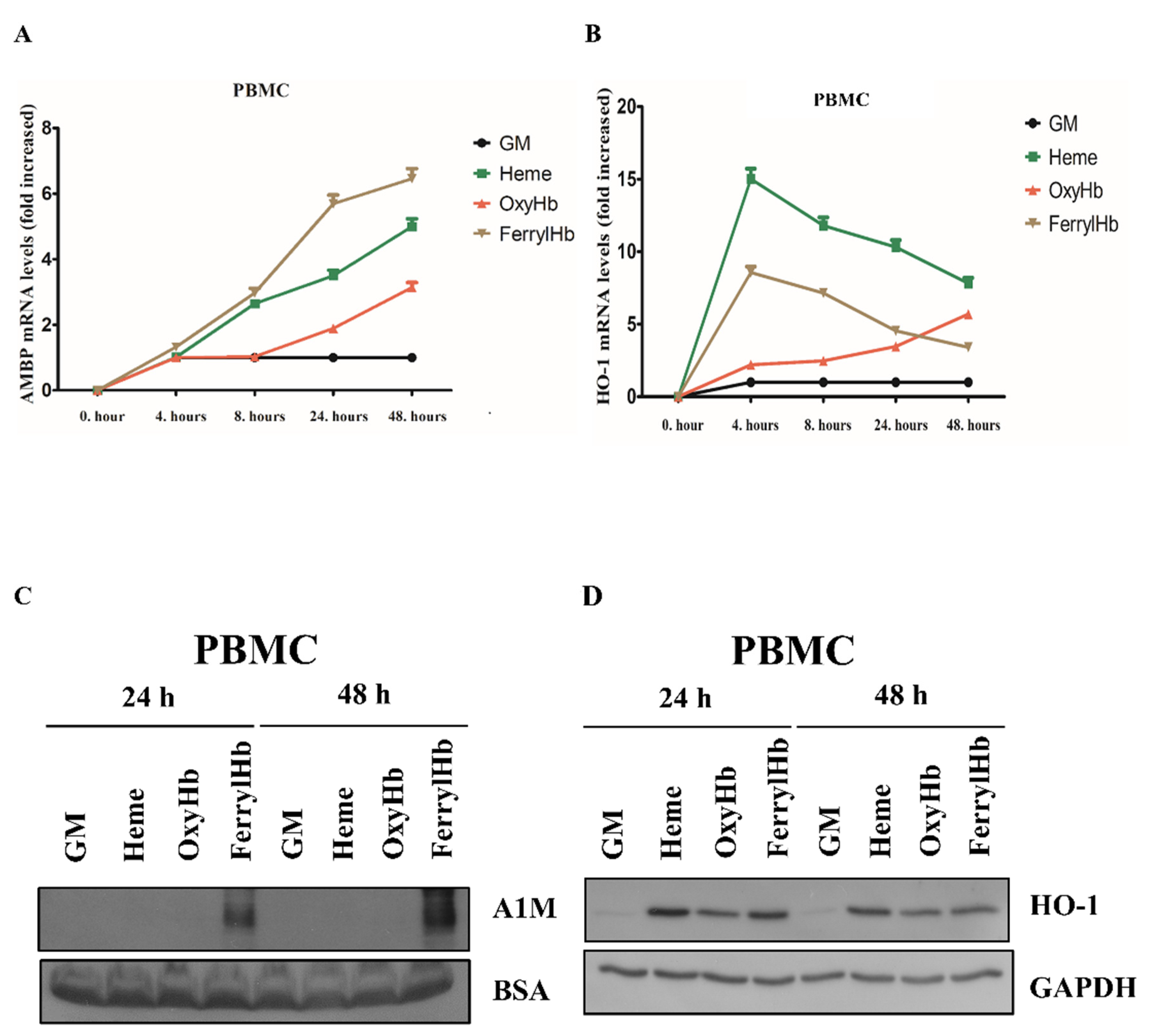
2.2. Heme and FerrylHb Induce A1M Expression in Human Aortic Endothelial Cell-, Human Aortic Smooth Muscle Cell- and Peripheral Blood Mononuclear Cell Cultures
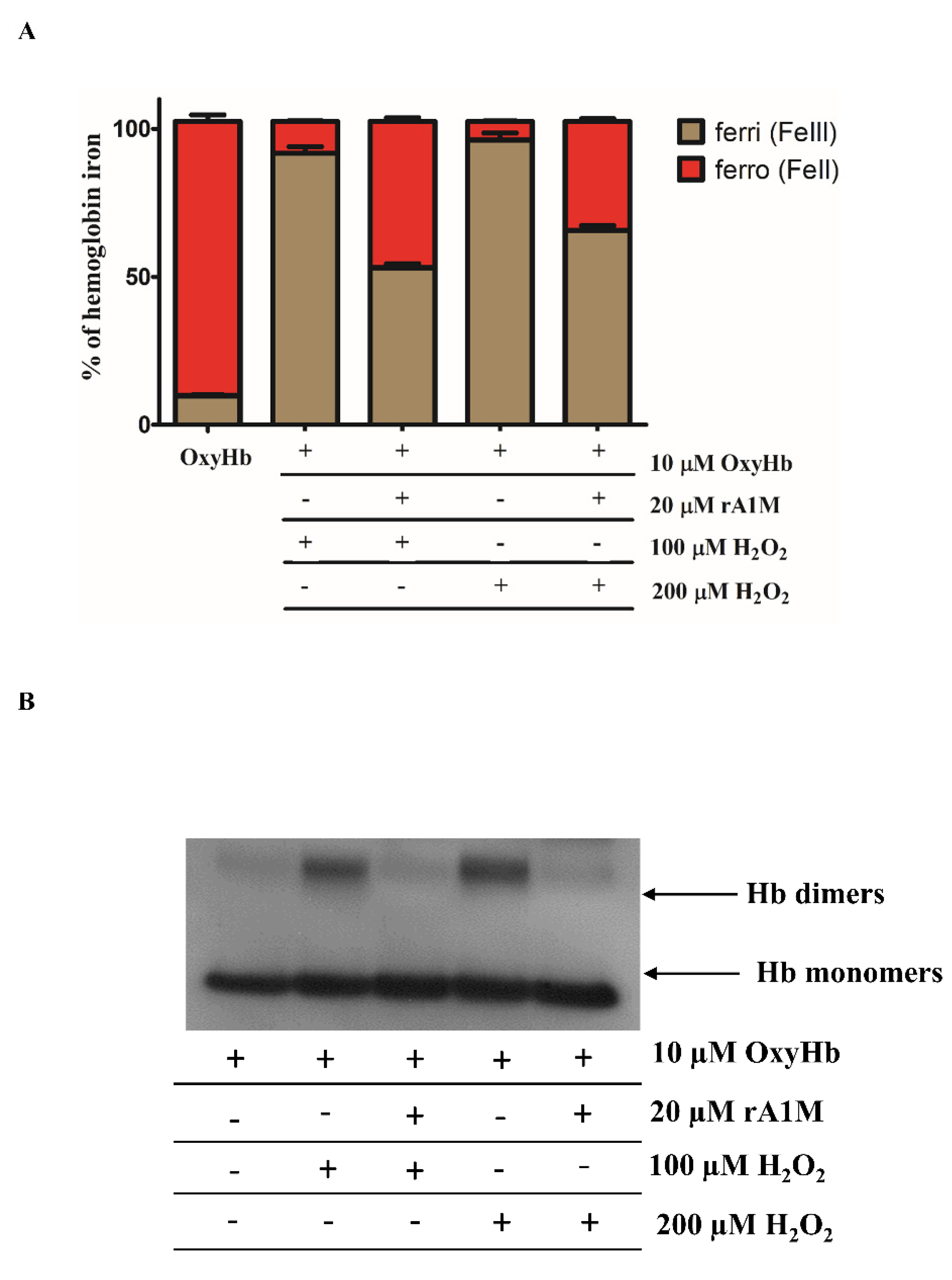
2.3. rA1M Inhibits Hb Oxidation
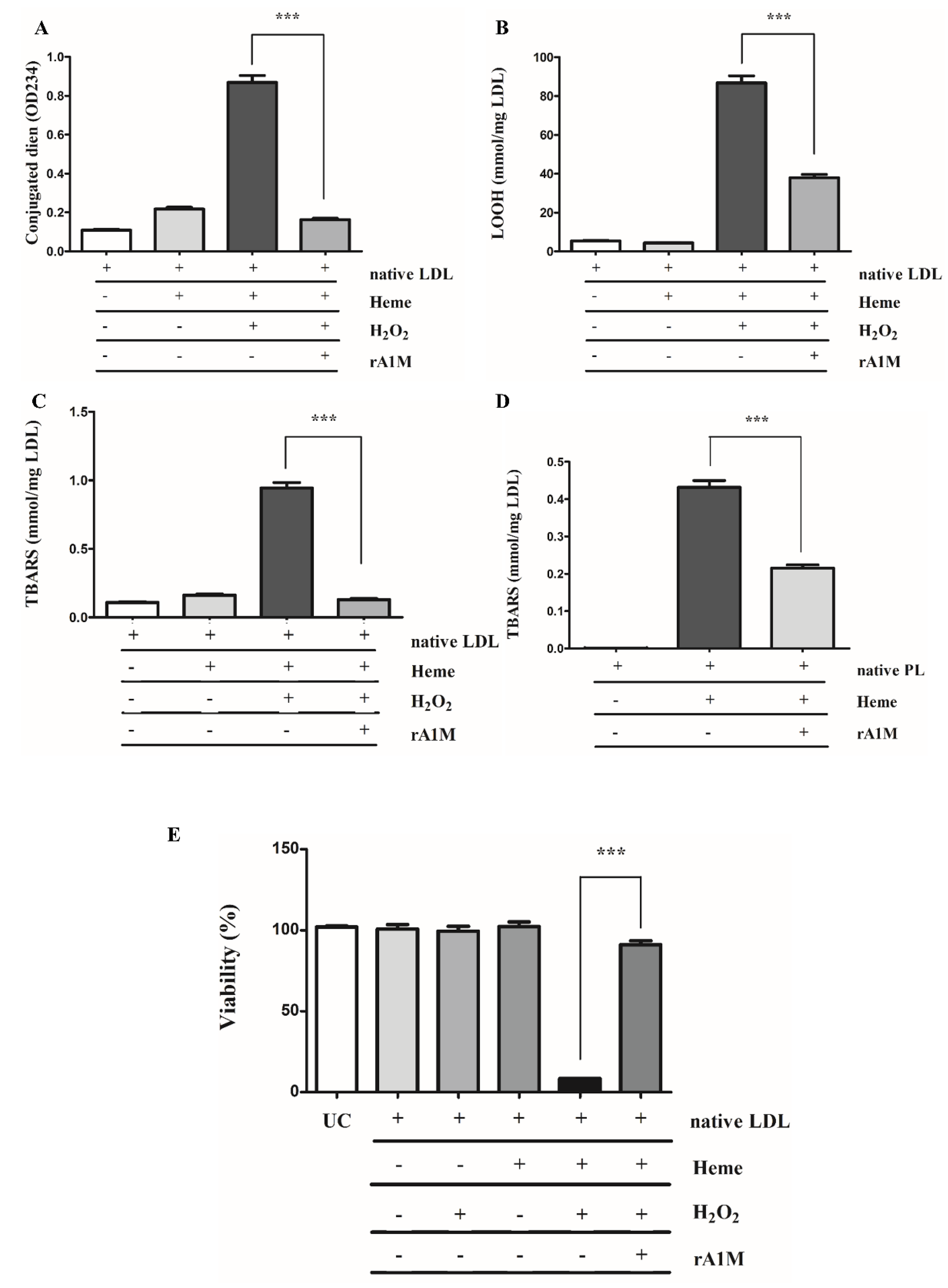
2.4. rA1M Inhibits Heme-Induced of LDL/Plaque Lipid Peroxidation and Subsequent Cell Death
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Hb Preparations
4.3. Analysis of Hb Oxidation by Immunoblot
4.4. Tissue Samples
4.5. Immunohistochemistry
4.6. RNA Isolation and Quantitative Reverse Transcription-Polymerase Chain Reaction
4.7. Immunoblot
4.8. A1M
4.9. Preparation and Oxidation of Low-Density Lipoproteins
4.10. Plaque Lipid Preparation and Oxidation
4.11. Measurement of Lipid Peroxidation Products in LDL and Plaque Lipids
4.12. Effect of Recombinant A1M on Heme-Induced LDL Oxidation
4.13. EC Cytotoxicity Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rifkind, J.M.; Zhang, L.; Levy, A.; Manoharan, P.T. The Hypoxic Stress on Erythrocytes Associated with Superoxide Formation. Free Radic. Res. Commun. 1991, 12–13 Pt 2, 645–652. [Google Scholar] [CrossRef]
- Abugo, O.O.; Rifkind, J.M. Oxidation of Hemoglobin and the Enhancement Produced by Nitroblue Tetrazolium. J. Biol. Chem. 1994, 269, 24845–24853. [Google Scholar] [CrossRef]
- Nagababu, E.; Rifkind, J.M. Formation of Fluorescent Heme Degradation Products during the Oxidation of Hemoglobin by Hydrogen Peroxide. Biochem. Biophys. Res. Commun. 1998, 247, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Balagopalakrishna, C.; Manoharan, P.T.; Abugo, O.O.; Rifkind, J.M. Production of Superoxide from Hemoglobin-Bound Oxygen Under Hypoxic Conditions. Biochemistry 1996, 35, 6393–6398. [Google Scholar] [CrossRef] [PubMed]
- Carrell, R.W.; Winterbourn, C.C.; Rachmilewitz, E.A. Activated Oxygen and Haemolysis. Br. J. Haematol. 1975, 30, 259–264. [Google Scholar] [CrossRef]
- Giulivi, C.; Hochstein, P.; Davies, K.J. Hydrogen Peroxide Production by Red Blood Cells. Free Radic. Biol. Med. 1994, 16, 123–129. [Google Scholar] [CrossRef]
- Rifkind, J.M.; Nagababu, E.; Ramasamy, S.; Ravi, L.B. Hemoglobin Redox Reactions and Oxidative Stress. Redox Rep. 2003, 8, 234–237. [Google Scholar] [CrossRef]
- Chang, J.C.; van der Hoeven, L.H.; Haddox, C.H. Glutathione Reductase in the Red Blood Cells. Ann. Clin. Lab. Sci. 1978, 8, 23–29. [Google Scholar]
- Nagababu, E.; Chrest, F.J.; Rifkind, J.M. Hydrogen-Peroxide-Induced Heme Degradation in Red Blood Cells: The Protective Roles of Catalase and Glutathione Peroxidase. Biochim. Biophys. Acta 2003, 1620, 211–217. [Google Scholar] [CrossRef]
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklósi, J.; Méhes, G.; Csonka, T.; Smith, A.; et al. Red Cells, Hemoglobin, Heme, Iron, and Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1347–1353. [Google Scholar] [CrossRef]
- Hugelshofer, M.; Buzzi, R.M.; Schaer, C.A.; Richter, H.; Akeret, K.; Anagnostakou, V.; Mahmoudi, L.; Vaccani, R.; Vallelian, F.; Deuel, J.W.; et al. Haptoglobin Administration into the Subarachnoid Space Prevents Hemoglobin-Induced Cerebral Vasospasm. J. Clin. Invest. 2019, 129, 5219–5235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rother, R.P.; Bell, L.; Hillmen, P.; Gladwin, M.T. The Clinical Sequelae of Intravascular Hemolysis and Extracellular Plasma Hemoglobin: A Novel Mechanism of Human Disease. JAMA 2005, 293, 1653–1662. [Google Scholar] [CrossRef]
- Janz, D.R.; Bastarache, J.A.; Peterson, J.F.; Sills, G.; Wickersham, N.; May, A.K.; Roberts, L.J.; Ware, L.B. Association between Cell-Free Hemoglobin, Acetaminophen, and Mortality in Patients with Sepsis: An Observational Study. Crit. Care Med. 2013, 41, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A Definition of Advanced Types of Atherosclerotic Lesions and a Histological Classification of Atherosclerosis. A Report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1995, 92, 1355–1374. [Google Scholar] [CrossRef]
- Michel, J.; Martin-Ventura, J.L.; Nicoletti, A.; Ho-Tin-Noé, B. Pathology of Human Plaque Vulnerability: Mechanisms and Consequences of Intraplaque Haemorrhages. Atherosclerosis 2014, 234, 311–319. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Gold, H.K.; Burke, A.P.; Fowler, D.R.; Kruth, H.S.; Weber, D.K.; Farb, A.; Guerrero, L.J.; Hayase, M.; Kutys, R.; et al. Intraplaque Hemorrhage and Progression of Coronary Atheroma. N. Engl. J. Med. 2003, 349, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.; Virmani, R.; Arbustini, E.; Pasterkamp, G. Intraplaque Haemorrhages as the Trigger of Plaque Vulnerability. Eur. Heart J. 2011, 32, 1977–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeney, V.; Balla, G.; Balla, J. Red Blood Cell, Hemoglobin and Heme in the Progression of Atherosclerosis. Front. Physiol. 2014, 5, 379. [Google Scholar] [CrossRef] [Green Version]
- Winterbourn, C.C. Free-Radical Production and Oxidative Reactions of Hemoglobin. Environ. Health Perspect. 1985, 64, 321–330. [Google Scholar] [CrossRef]
- Alayash, A.I.; Patel, R.P.; Cashon, R.E. Redox Reactions of Hemoglobin and Myoglobin: Biological and Toxicological Implications. Antioxid. Redox Signal 2001, 3, 313–327. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-Free Hemoglobin Limits Nitric Oxide Bioavailability in Sickle-Cell Disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Sadrzadeh, S.M.; Graf, E.; Panter, S.S.; Hallaway, P.E.; Eaton, J.W. Hemoglobin. A Biologic Fenton Reagent. J. Biol. Chem. 1984, 259, 14354–14356. [Google Scholar] [CrossRef]
- Bunn, H.F.; Jandl, J.H. Exchange of Heme among Hemoglobins and between Hemoglobin and Albumin. J. Biol. Chem. 1968, 243, 465–475. [Google Scholar] [CrossRef]
- Wijayanti, N.; Katz, N.; Immenschuh, S. Biology of Heme in Health and Disease. Curr. Med. Chem. 2004, 11, 981–986. [Google Scholar] [CrossRef]
- Silva, G.; Jeney, V.; Chora, A.; Larsen, R.; Balla, J.; Soares, M.P. Oxidized Hemoglobin is an Endogenous Proinflammatory Agonist that Targets Vascular Endothelial Cells. J. Biol. Chem. 2009, 284, 29582–29595. [Google Scholar] [CrossRef] [Green Version]
- Posta, N.; Csősz, É.; Oros, M.; Pethő, D.; Potor, L.; Kalló, G.; Hendrik, Z.; Sikura, K.É.; Méhes, G.; Tóth, C.; et al. Hemoglobin Oxidation Generates Globin-Derived Peptides in Atherosclerotic Lesions and Intraventricular Hemorrhage of the Brain, Provoking Endothelial Dysfunction. Lab. Investig. 2020, 100, 986–1002. [Google Scholar] [CrossRef] [PubMed]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercellotti, G.M. Hemin: A Possible Physiological Mediator of Low Density Lipoprotein Oxidation and Endothelial Injury. Arterioscler. Thromb. 1991, 11, 1700–1711. [Google Scholar] [CrossRef] [Green Version]
- Miller, Y.I.; Altamentova, S.M.; Shaklai, N. Oxidation of Low-Density Lipoprotein by Hemoglobin Stems from a Heme-Initiated Globin Radical: Antioxidant Role of Haptoglobin. Biochemistry 1997, 36, 12189–12198. [Google Scholar] [CrossRef]
- Paganga, G.; Rice-Evans, C.; Rule, R.; Leake, D. The Interaction between Ruptured Erythrocytes and Low-Density Lipoproteins. FEBS Lett. 1992, 303, 154–158. [Google Scholar]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-Oxidant and Cytotoxic Effects of Circulating Heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Schaer, C.A.; Deuel, J.W.; Bittermann, A.G.; Rubio, I.G.; Schoedon, G.; Spahn, D.R.; Wepf, R.A.; Vallelian, F.; Schaer, D.J. Mechanisms of Haptoglobin Protection Against Hemoglobin Peroxidation Triggered Endothelial Damage. Cell Death Differ. 2013, 20, 1569–1579. [Google Scholar] [CrossRef] [Green Version]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.J.; Law, S.K.; Moestrup, S.K. Identification of the Haemoglobin Scavenger Receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Thomsen, J.H.; Etzerodt, A.; Svendsen, P.; Moestrup, S.K. The Haptoglobin-CD163-Heme Oxygenase-1 Pathway for Hemoglobin Scavenging. Oxid. Med. Cell Longev. 2013, 2013, 523652. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.; McCulloh, R.J. Hemopexin and Haptoglobin: Allies Against Heme Toxicity from Hemoglobin Not Contenders. Front. Physiol. 2015, 6, 187. [Google Scholar] [CrossRef]
- Tenhunen, R.; Marver, H.S.; Schmid, R. Microsomal Heme Oxygenase. Characterization of the Enzyme. J. Biol. Chem. 1969, 244, 6388–6394. [Google Scholar] [PubMed]
- Akerstrom, B.; Flower, D.R.; Salier, J.P. Lipocalins: Unity in Diversity. Biochim. Biophys. Acta 2000, 1482, 1–8. [Google Scholar] [CrossRef]
- Berggård, T.; Thelin, N.; Falkenberg, C.; Enghild, J.J.; Akerström, B. Prothrombin, Albumin and Immunoglobulin A Form Covalent Complexes with Alpha1-Microglobulin in Human Plasma. Eur. J. Biochem. 1997, 245, 676–683. [Google Scholar] [CrossRef]
- Berggård, T.; Enghild, J.J.; Badve, S.; Salafia, C.M.; Lögdberg, L.; Akerström, B. Histologic Distribution and Biochemical Properties of Alpha 1-Microglobulin in Human Placenta. Am. J. Reprod. Immunol. 1999, 41, 52–60. [Google Scholar] [CrossRef]
- Larsson, J.; Wingårdh, K.; Berggård, T.; Davies, J.R.; Lögdberg, L.; Strand, S.E.; Akerström, B. Distribution of Iodine 125-Labeled Alpha1-Microglobulin in Rats After Intravenous Injection. J. Lab. Clin. Med. 2001, 137, 165–175. [Google Scholar] [CrossRef]
- Akerström, B.; Maghzal, G.J.; Winterbourn, C.C.; Kettle, A.J. The Lipocalin Alpha1-Microglobulin has Radical Scavenging Activity. J. Biol. Chem. 2007, 282, 31493–31503. [Google Scholar] [CrossRef] [Green Version]
- Rutardottir, S.; Karnaukhova, E.; Nantasenamat, C.; Songtawee, N.; Prachayasittikul, V.; Rajabi, M.; Rosenlöf, L.W.; Alayash, A.I.; Åkerström, B. Structural and Biochemical Characterization of Two Heme Binding Sites on A1-Microglobulin using Site Directed Mutagenesis and Molecular Simulation. Biochim. Biophys. Acta 2016, 1864, 29–41. [Google Scholar] [CrossRef]
- Åkerström, B.; Gram, M. A1M, an Extravascular Tissue Cleaning and Housekeeping Protein. Free Radic. Biol. Med. 2014, 74, 274–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Huang, A.L.; Kyaw, T.S.; Bobik, A.; Peter, K. Atherosclerotic Plaque Rupture: Identifying the Straw that Breaks the Camel’s Back. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 63. [Google Scholar] [CrossRef] [Green Version]
- Alayash, A.I. βCysteine 93 in Human Hemoglobin: A Gateway to Oxidative Stability in Health and Disease. Lab. Invest. 2021, 101, 4–11. [Google Scholar] [CrossRef]
- Heinecke, J.W. Mechanisms of Oxidative Damage of Low Density Lipoprotein in Human Atherosclerosis. Curr. Opin. Lipidol. 1997, 8, 268–274. [Google Scholar] [CrossRef]
- Kita, T.; Kume, N.; Minami, M.; Hayashida, K.; Murayama, T.; Sano, H.; Moriwaki, H.; Kataoka, H.; Nishi, E.; Horiuchi, H.; et al. Role of Oxidized LDL in Atherosclerosis. Ann. N. Y. Acad. Sci. 2001, 947, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Kristiansson, A.; Gram, M.; Flygare, J.; Hansson, S.R.; Åkerström, B.; Storry, J.R. The Role of A1-Microglobulin (A1M) in Erythropoiesis and Erythrocyte Homeostasis-Therapeutic Opportunities in Hemolytic Conditions. Int. J. Mol. Sci. 2020, 21, 7234. [Google Scholar] [CrossRef] [PubMed]
- Cipollone, F.; Fazia, M.L.; Mezzetti, A. Oxidative Stress, Inflammation and Atherosclerotic Plaque Development. Int. Congr. Ser. 2007, 1303, 35–40. [Google Scholar] [CrossRef]
- Singh, U.; Jialal, I. Oxidative Stress and Atherosclerosis. Pathophysiology (Amsterdam) 2006, 13, 129–142. [Google Scholar] [CrossRef]
- Bergwik, J.; Kristiansson, A.; Larsson, J.; Ekström, S.; Åkerström, B.; Allhorn, M. Binding of the Human Antioxidation Protein A1-Microglobulin (A1M) to Heparin and Heparan Sulfate. Mapping of Binding Site, Molecular and Functional Characterization, and Co-Localization in Vivo and in Vitro. Redox Biol. 2021, 41, 101892. [Google Scholar] [CrossRef]
- Tejler, L.; Eriksson, S.; Grubb, A.; Astedt, B. Production of Protein HC by Human Fetal Liver Explants. Biochim. Biophys. Acta 1978, 542, 506–514. [Google Scholar] [CrossRef]
- Wester, L.; Fast, J.; Labuda, T.; Cedervall, T.; Wingårdh, K.; Olofsson, T.; Akerström, B. Carbohydrate Groups of Alpha1-Microglobulin are Important for Secretion and Tissue Localization but Not for Immunological Properties. Glycobiology 2000, 10, 891–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristiansson, A.; Davidsson, S.; Johansson, M.E.; Piel, S.; Elmér, E.; Hansson, M.J.; Åkerström, B.; Gram, M. A1-Microglobulin (A1M) Protects Human Proximal Tubule Epithelial Cells from Heme-Induced Damage in Vitro. Int. J. Mol. Sci. 2020, 21, 5825. [Google Scholar] [CrossRef]
- Olsson, M.G.; Allhorn, M.; Larsson, J.; Cederlund, M.; Lundqvist, K.; Schmidtchen, A.; Sørensen, O.E.; Mörgelin, M.; Akerström, B. Up-Regulation of A1M/A1-Microglobulin in Skin by Heme and Reactive Oxygen Species Gives Protection from Oxidative Damage. PLoS ONE 2011, 6, e27505. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.G.; Allhorn, M.; Olofsson, T.; Akerström, B. Up-Regulation of Alpha1-Microglobulin by Hemoglobin and Reactive Oxygen Species in Hepatoma and Blood Cell Lines. Free Radic. Biol. Med. 2007, 42, 842–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaer, D.J.; Schaer, C.A.; Buehler, P.W.; Boykins, R.A.; Schoedon, G.; Alayash, A.I.; Schaffner, A. CD163 is the Macrophage Scavenger Receptor for Native and Chemically Modified Hemoglobins in the Absence of Haptoglobin. Blood 2006, 107, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Kassa, T.; Jana, S.; Meng, F.; Alayash, A.I. Differential Heme Release from various Hemoglobin Redox States and the Upregulation of Cellular Heme Oxygenase-1. FEBS Open Bio 2016, 6, 876–884. [Google Scholar] [CrossRef]
- Zavaczki, E.; Gáll, T.; Zarjou, A.; Hendrik, Z.; Potor, L.; Tóth, C.Z.; Méhes, G.; Gyetvai, Á.; Agarwal, A.; Balla, G.; et al. Ferryl Hemoglobin Inhibits Osteoclastic Differentiation of Macrophages in Hemorrhaged Atherosclerotic Plaques. Oxid. Med. Cell Longev. 2020, 2020, 3721383. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.R.; Karaca, M.; Adamski, K.N.; Chorley, B.N.; Wang, X.; Bell, D.A. Novel Hematopoietic Target Genes in the NRF2-Mediated Transcriptional Pathway. Oxid. Med. Cell Longev. 2013, 2013, 120305. [Google Scholar] [CrossRef] [Green Version]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’N’Collar Transcription Factor, Regulates Induction of the Heme Oxygenase-1 Gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.A.; Haggard, J.J.; Croatt, A.J.; Grande, J.P.; Poss, K.D.; Alam, J. The Indispensability of Heme Oxygenase-1 in Protecting Against Acute Heme Protein-Induced Toxicity in Vivo. Am. J. Pathol. 2000, 156, 1527–1535. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Bandyopadhyay, U. Free Heme Toxicity and its Detoxification Systems in Human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Vetr, H.; Gebhard, W. Structure of the Human Alpha 1-Microglobulin-Bikunin Gene. Biol. Chem. Hoppe Seyler 1990, 371, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Rouet, P.; Raguenez, G.; Tronche, F.; Mfou’ou, V.; Salier, J.P. Hierarchy and Positive/Negative Interplays of the Hepatocyte Nuclear Factors HNF-1, -3 and -4 in the Liver-Specific Enhancer for the Human Alpha-1-Microglobulin/Bikunin Precursor. Nucleic Acids Res. 1995, 23, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of Novel NRF2-Regulated Genes by ChIP-Seq: Influence on Retinoid X Receptor Alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [Green Version]
- Harel, S.; Salan, M.A.; Kanner, J. Iron Release from Metmyoglobin, Methaemoglobin and Cytochrome C by a System Generating Hydrogen Peroxide. Free Radic. Res. Commun. 1988, 5, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Harel, S.; Kanner, J. The Generation of Ferryl or Hydroxyl Radicals during Interaction of Haemproteins with Hydrogen Peroxide. Free Radic. Res. Commun. 1988, 5, 21–33. [Google Scholar] [CrossRef]
- Jia, Y.; Buehler, P.W.; Boykins, R.A.; Venable, R.M.; Alayash, A.I. Structural Basis of Peroxide-Mediated Changes in Human Hemoglobin: A Novel Oxidative Pathway. J. Biol. Chem. 2007, 282, 4894–4907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.P.; Svistunenko, D.A.; Darley-Usmar, V.M.; Symons, M.C.; Wilson, M.T. Redox Cycling of Human Methaemoglobin by H2O2 Yields Persistent Ferryl Iron and Protein Based Radicals. Free Radic. Res. 1996, 25, 117–123. [Google Scholar] [CrossRef]
- Reeder, B.J.; Cutruzzola, F.; Bigotti, M.G.; Hider, R.C.; Wilson, M.T. Tyrosine as a Redox-Active Center in Electron Transfer to Ferryl Heme in Globins. Free Radic. Biol. Med. 2008, 44, 274–283. [Google Scholar] [CrossRef]
- Reeder, B.J.; Grey, M.; Silaghi-Dumitrescu, R.; Svistunenko, D.A.; Bülow, L.; Cooper, C.E.; Wilson, M.T. Tyrosine Residues as Redox Cofactors in Human Hemoglobin: Implications for Engineering Nontoxic Blood Substitutes. J. Biol. Chem. 2008, 283, 30780–30787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deterding, L.J.; Ramirez, D.C.; Dubin, J.R.; Mason, R.P.; Tomer, K.B. Identification of Free Radicals on Hemoglobin from its Self-Peroxidation using Mass Spectrometry and Immuno-Spin Trapping: Observation of a Histidinyl Radical. J. Biol. Chem. 2004, 279, 11600–11607. [Google Scholar] [CrossRef] [Green Version]
- Lardinois, O.M.; Detweiler, C.D.; Tomer, K.B.; Mason, R.P.; Deterding, L.J. Identifying the Site of Spin Trapping in Proteins by a Combination of Liquid Chromatography, ELISA, and Off-Line Tandem Mass Spectrometry. Free Radic. Biol. Med. 2008, 44, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, D.C.; Chen, Y.; Mason, R.P. Immunochemical Detection of Hemoglobin-Derived Radicals Formed by Reaction with Hydrogen Peroxide: Involvement of a Protein-Tyrosyl Radical. Free Radic. Biol. Med. 2003, 34, 830–839. [Google Scholar] [CrossRef]
- Jana, S.; Strader, M.B.; Alayash, A.I. The Providence Mutation (βK82D) in Human Hemoglobin Substantially Reduces βCysteine 93 Oxidation and Oxidative Stress in Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 9453. [Google Scholar] [CrossRef]
- Pimenova, T.; Pereira, C.P.; Gehrig, P.; Buehler, P.W.; Schaer, D.J.; Zenobi, R. Quantitative Mass Spectrometry Defines an Oxidative Hotspot in Hemoglobin that is Specifically Protected by Haptoglobin. J. Proteome Res. 2010, 9, 4061–4070. [Google Scholar] [CrossRef]
- Nagababu, E.; Rifkind, J.M. Reaction of Hydrogen Peroxide with Ferrylhemoglobin: Superoxide Production and Heme Degradation. Biochemistry 2000, 39, 12503–12511. [Google Scholar] [CrossRef] [PubMed]
- Romantsik, O.; Agyemang, A.A.; Sveinsdóttir, S.; Rutardóttir, S.; Holmqvist, B.; Cinthio, M.; Mörgelin, M.; Gumus, G.; Karlsson, H.; Hansson, S.R.; et al. The Heme and Radical Scavenger A1-Microglobulin (A1M) Confers Early Protection of the Immature Brain Following Preterm Intraventricular Hemorrhage. J. Neuroinflamm. 2019, 16, 122. [Google Scholar] [CrossRef] [Green Version]
- Alayash, A.I. Mechanisms of Toxicity and Modulation of Hemoglobin-Based Oxygen Carriers. Shock 2019, 52, 41–49. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Goel, A.; Mehta, J.L. LOX-1: Regulation, Signaling and its Role in Atherosclerosis. Antioxidants (Basel) 2019, 8, 218. [Google Scholar] [CrossRef] [Green Version]
- Miller, Y.I.; Smith, A.; Morgan, W.T.; Shaklai, N. Role of Hemopexin in Protection of Low-Density Lipoprotein Against Hemoglobin-Induced Oxidation. Biochemistry 1996, 35, 13112–13117. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, J.P.; Major, A.S. How Oxidized Low-Density Lipoprotein Activates Inflammatory Responses. Crit. Rev. Immunol. 2018, 38, 333–342. [Google Scholar] [CrossRef]
- Cederlund, M.; Deronic, A.; Pallon, J.; Sørensen, O.E.; Åkerström, B. A1M/A1-Microglobulin is Proteolytically Activated by Myeloperoxidase, Binds Its Heme Group and Inhibits Low Density Lipoprotein Oxidation. Front. Physiol. 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutteridge, J.M.; Smith, A. Antioxidant Protection by Haemopexin of Haem-Stimulated Lipid Peroxidation. Biochem. J. 1988, 256, 861–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalamanoglu, A.; Deuel, J.W.; Hunt, R.C.; Baek, J.H.; Hassell, K.; Redinius, K.; Irwin, D.C.; Schaer, D.J.; Buehler, P.W. Depletion of Haptoglobin and Hemopexin Promote Hemoglobin-Mediated Lipoprotein Oxidation in Sickle Cell Disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L765–L774. [Google Scholar] [CrossRef] [Green Version]
- Vinchi, F.; Gastaldi, S.; Silengo, L.; Altruda, F.; Tolosano, E. Hemopexin Prevents Endothelial Damage and Liver Congestion in a Mouse Model of Heme Overload. Am. J. Pathol. 2008, 173, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Vinchi, F.; De Franceschi, L.; Ghigo, A.; Townes, T.; Cimino, J.; Silengo, L.; Hirsch, E.; Altruda, F.; Tolosano, E. Hemopexin Therapy Improves Cardiovascular Function by Preventing Heme-Induced Endothelial Toxicity in Mouse Models of Hemolytic Diseases. Circulation 2013, 127, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A Cytoprotective Antioxidant Strategem of Endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [CrossRef]
- Balla, G.; Vercellotti, G.M.; Muller-Eberhard, U.; Eaton, J.; Jacob, H.S. Exposure of Endothelial Cells to Free Heme Potentiates Damage Mediated by Granulocytes and Toxic Oxygen Species. Lab. Invest. 1991, 64, 648–655. [Google Scholar]
- Kristiansson, A.; Bergwik, J.; Alattar, A.G.; Flygare, J.; Gram, M.; Hansson, S.R.; Olsson, M.L.; Storry, J.R.; Allhorn, M.; Åkerström, B. Human Radical Scavenger A1-Microglobulin Protects Against Hemolysis in Vitro and A1-Microglobulin Knockout Mice Exhibit a Macrocytic Anemia Phenotype. Free Radic. Biol. Med. 2021, 162, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Oxidative Reactions of Hemoglobin. Methods Enzymol. 1990, 186, 265–272. [Google Scholar] [PubMed]
- Vallelian, F.; Pimenova, T.; Pereira, C.P.; Abraham, B.; Mikolajczyk, M.G.; Schoedon, G.; Zenobi, R.; Alayash, A.I.; Buehler, P.W.; Schaer, D.J. The Reaction of Hydrogen Peroxide with Hemoglobin Induces Extensive Alpha-Globin Crosslinking and Impairs the Interaction of Hemoglobin with Endogenous Scavenger Pathways. Free Radic. Biol. Med. 2008, 45, 1150–1158. [Google Scholar] [CrossRef]
- Meng, F.; Alayash, A.I. Determination of Extinction Coefficients of Human Hemoglobin in various Redox States. Anal. Biochem. 2017, 521, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwasek, A.; Osmark, P.; Allhorn, M.; Lindqvist, A.; Akerström, B.; Wasylewski, Z. Production of Recombinant Human Alpha1-Microglobulin and Mutant Forms Involved in Chromophore Formation. Protein Exp. Purif. 2007, 53, 145–152. [Google Scholar] [CrossRef]
- Åkerström, B.; Rosenlöf, L.; Hägerwall, A.; Rutardottir, S.; Ahlstedt, J.; Johansson, M.E.; Erlandsson, L.; Allhorn, M.; Gram, M. rA1M-035, a Physicochemically Improved Human Recombinant A1-Microglobulin, has Therapeutic Effects in Rhabdomyolysis-Induced Acute Kidney Injury. Antioxid. Redox Signal 2019, 30, 489–504. [Google Scholar] [CrossRef] [Green Version]
- Potor, L.; Bányai, E.; Becs, G.; Soares, M.P.; Balla, G.; Balla, J.; Jeney, V. Atherogenesis may Involve the Prooxidant and Proinflammatory Effects of Ferryl Hemoglobin. Oxidative Med. Cell Longev. 2013, 2013, 676425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bligh, E.G.; Dyer, W.J. A Rapid Method of Total Lipid Extraction and Purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.Y.; Hunt, J.V.; Wolff, S.P. Ferrous Ion Oxidation in the Presence of Xylenol Orange for Detection of Lipid Hydroperoxide in Low Density Lipoprotein. Anal. Biochem. 1992, 202, 384–389. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pethő, D.; Gáll, T.; Hendrik, Z.; Nagy, A.; Beke, L.; Gergely, A.P.; Méhes, G.; Tóth, C.; Gram, M.; Åkerström, B.; et al. Ferryl Hemoglobin and Heme Induce A1-Microglobulin in Hemorrhaged Atherosclerotic Lesions with Inhibitory Function against Hemoglobin and Lipid Oxidation. Int. J. Mol. Sci. 2021, 22, 6668. https://doi.org/10.3390/ijms22136668
Pethő D, Gáll T, Hendrik Z, Nagy A, Beke L, Gergely AP, Méhes G, Tóth C, Gram M, Åkerström B, et al. Ferryl Hemoglobin and Heme Induce A1-Microglobulin in Hemorrhaged Atherosclerotic Lesions with Inhibitory Function against Hemoglobin and Lipid Oxidation. International Journal of Molecular Sciences. 2021; 22(13):6668. https://doi.org/10.3390/ijms22136668
Chicago/Turabian StylePethő, Dávid, Tamás Gáll, Zoltán Hendrik, Annamária Nagy, Lívia Beke, Attila Péter Gergely, Gábor Méhes, Csaba Tóth, Magnus Gram, Bo Åkerström, and et al. 2021. "Ferryl Hemoglobin and Heme Induce A1-Microglobulin in Hemorrhaged Atherosclerotic Lesions with Inhibitory Function against Hemoglobin and Lipid Oxidation" International Journal of Molecular Sciences 22, no. 13: 6668. https://doi.org/10.3390/ijms22136668
APA StylePethő, D., Gáll, T., Hendrik, Z., Nagy, A., Beke, L., Gergely, A. P., Méhes, G., Tóth, C., Gram, M., Åkerström, B., Balla, G., & Balla, J. (2021). Ferryl Hemoglobin and Heme Induce A1-Microglobulin in Hemorrhaged Atherosclerotic Lesions with Inhibitory Function against Hemoglobin and Lipid Oxidation. International Journal of Molecular Sciences, 22(13), 6668. https://doi.org/10.3390/ijms22136668