
Therapeutic Targeting of the Leukaemia Microenvironment

 , and
, and
Abstract
:1. Introduction
2. The Bone Marrow Niche
3. Leukaemia Niche Regulation
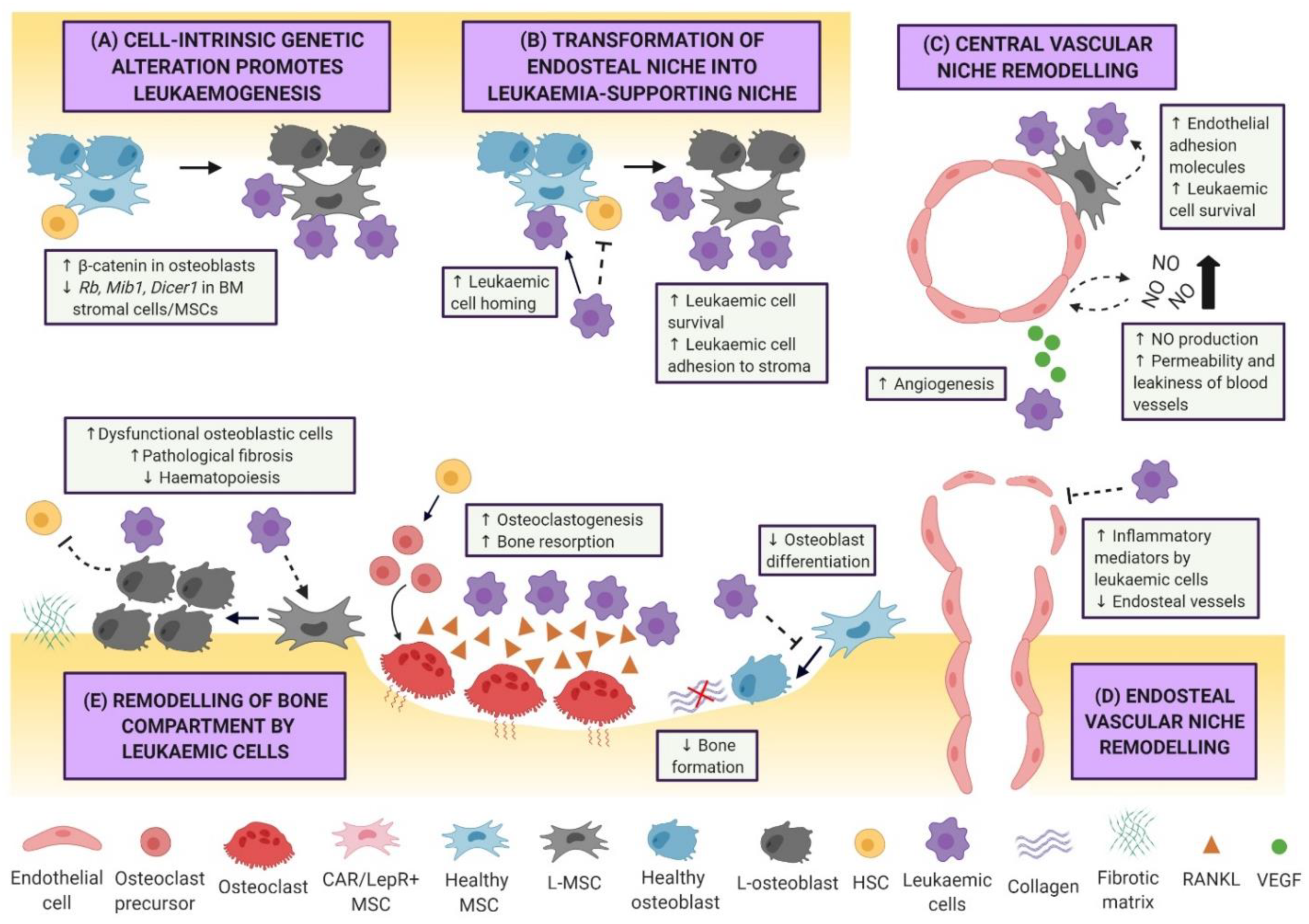
3.1. Niche-Driven Malignant Transformation
3.2. Leukaemic Remodelling of the Vasculature and Endosteal Niche
3.3. Adipocytic Niche in Leukaemia
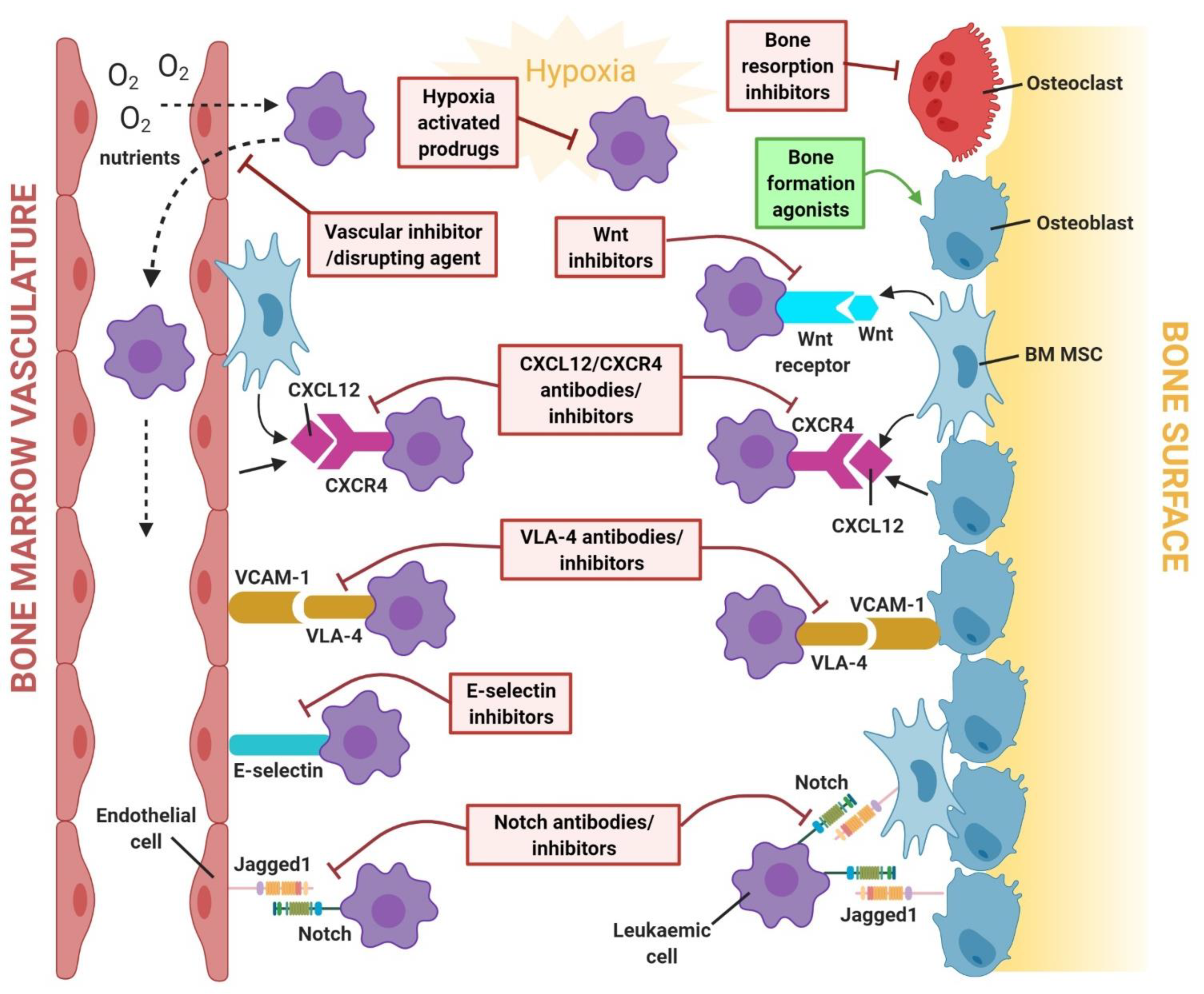
4. Targeting the Leukaemia Microenvironment: Mechanisms of Treatment Resistance and Therapeutic Strategies
4.1. CXCL12/CXCR4 Signalling Pathway
4.2. Notch Signalling Pathway
4.3. Wnt/β-Catenin Signalling Pathway
4.4. Cell–Cell Adhesion
4.5. Bone Remodelling Signalling Pathways
4.6. Hypoxia-Related Signalling Pathways
4.7. The Vasculature
4.8. Emerging Therapies: Immunotherapies and Mitochondrial-Targeted Therapies
5. Open Questions and Future Perspectives
6. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarini, F.; Lonetti, A.; Evangelisti, C.; Buontempo, F.; Orsini, E.; Evangelisti, C.; Cappellini, A.; Neri, L.M.; McCubrey, J.; Martelli, A.M. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Kular, J.; Tickner, J.; Chim, S.M.; Xu, J. An overview of the regulation of bone remodelling at the cellular level. Clin. Biochem. 2012, 45, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Mostoufi-Moab, S.; Halton, J. Bone Morbidity in Childhood Leukemia: Epidemiology, Mechanisms, Diagnosis, and Treatment. Curr. Osteoporos. Rep. 2014, 12, 300–312. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.L.; Ness, K.K. Bone Mineral Density Deficits and Fractures in Survivors of Childhood Cancer. Curr. Osteoporos. Rep. 2013, 11, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Wang, A.; Zhong, H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology 2018, 23, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Arai, F.; Hirao, A.; Ohmura, M.; Sato, H.; Matsuoka, S.; Takubo, K.; Ito, K.; Koh, G.Y.; Suda, T. Tie2/Angiopoietin-1 Signaling Regulates Hematopoietic Stem Cell Quiescence in the Bone Marrow Niche. Cell 2004, 118, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, S.K.; Johnston, H.M.; Whitty, G.A.; Williams, B.; Webb, R.J.; Denhardt, D.T.; Bertoncello, I.; Bendall, L.J.; Simmons, P.J.; Haylock, D.N. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005, 106, 1232–1239. [Google Scholar] [CrossRef]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef]
- Greenbaum, A.; Hsu, Y.-M.S.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Saunders, T.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.C.; Hara, T.; Lim, V.Y.; Herndler-Brandstetter, D.; Nevius, E.; Sugiyama, T.; Tani-Ichi, S.; Schlenner, S.; Richie, E.; Rodewald, H.R.; et al. Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity 2016, 45, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Bejar, R. Clonal Hematopoiesis in Aging. Curr. Stem Cell Rep. 2018, 4, 209–219. [Google Scholar] [CrossRef]
- King, K.Y.; Huang, Y.; Nakada, D.; Goodell, M.A. Environmental influences on clonal hematopoiesis. Exp. Hematol. 2020, 83, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Raaijmakers, M.H.G.P.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Krause, D.S.; Fulzele, K.; Catic, A.; Sun, C.C.; Dombkowski, D.; Hurley, M.P.; Lezeau, S.; Attar, E.; Wu, J.Y.; Lin, H.Y.; et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat. Med. 2013, 19, 1513–1517. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, H.; Rodriguez, S.; Cao, L.; Parish, J.; Mumaw, C.; Zollman, A.; Kamoka, M.M.; Mu, J.; Chen, D.Z.; et al. Notch-dependent repression of miR-155 in the bone marrow niche regulates hematopoiesis in an NF-kappaB-dependent manner. Cell Stem Cell 2014, 15, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.; Yu, W.-M.; Zheng, H.; Loh, M.L.; Bunting, S.T.; Pauly, M.; Huang, G.; Zhou, M.; Broxmeyer, H.E.; Scadden, D.T.; et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature 2016, 539, 304–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, B.J.; Ashton, J.M.; Xing, L.; Becker, M.W.; Jordan, C.T.; Calvi, L.M. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood 2012, 119, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Dührsen, U.; Frenette, P.S. Acute Myelogenous Leukemia-Induced Sympathetic Neuropathy Promotes Malignancy in an Altered Hematopoietic Stem Cell Niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative Neoplasia Remodels the Endosteal Bone Marrow Niche into a Self-Reinforcing Leukemic Niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, E.D.; Duarte, D.; Akinduro, O.; Khorshed, R.A.; Passaro, D.; Nowicka, M.; Straszkowski, L.; Scott, M.K.; Rothery, S.; Ruivo, N.; et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 2016, 538, 518–522. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zimmerman, G.; Huang, X.; Yu, S.; Myers, J.; Wang, Y.; Moreton, S.; Nthale, J.; Awadallah, A.; Beck, R.; et al. Aberrant Notch Signaling in the Bone Marrow Microenvironment of Acute Lymphoid Leukemia Suppresses Osteoblast-Mediated Support of Hematopoietic Niche Function. Cancer Res. 2016, 76, 1641–1652. [Google Scholar] [CrossRef] [Green Version]
- Passaro, D.; di Tullio, A.; Abarrategi, A.; Rouault-Pierre, K.; Foster, K.; Ariza-McNaughton, L.; Montaner, B.; Chakravarty, P.; Bhaw, L.; Diana, G.; et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 324–341.e6. [Google Scholar] [CrossRef] [Green Version]
- Kampen, K.R.; Ter Elst, A.; de Bont, E.S.J.M. Vascular endothelial growth factor signaling in acute myeloid leukemia. Cell. Mol. Life Sci. 2013, 70, 1307–1317. [Google Scholar] [CrossRef] [Green Version]
- Aguayo, A.; Kantarjian, H.M.; Estey, E.H.; Giles, F.J.; Verstovsek, S.; Manshouri, T.; Gidel, C.; O’Brien, S.; Keating, M.J.; Albitar, M. Plasma vascular endothelial growth factor levels have prognostic significance in patients with acute myeloid leukemia but not in patients with myelodysplastic syndromes. Cancer 2002, 95, 1923–1930. [Google Scholar] [CrossRef]
- Poulos, M.G.; Gars, E.J.; Gutkin, M.C.; Kloss, C.C.; Ginsberg, M.; Scandura, J.; Rafii, S.; Butler, J.M. Activation of the vascular niche supports leukemic progression and resistance to chemotherapy. Exp. Hematol. 2014, 42, 976–986.e3. [Google Scholar] [CrossRef] [Green Version]
- Duarte, D.; Hawkins, E.; Akinduro, O.; Ang, H.; de Filippo, K.; Kong, I.Y.; Haltalli, M.; Ruivo, N.; Straszkowski, L.; Vervoort, S.J.; et al. Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Cell Stem Cell 2018, 22, 64–77.e6. [Google Scholar] [CrossRef] [Green Version]
- Pezeshkian, B.; Donnelly, C.; Tamburo, K.; Geddes, T.; Madlambayan, G.J. Leukemia Mediated Endothelial Cell Activation Modulates Leukemia Cell Susceptibility to Chemotherapy through a Positive Feedback Loop Mechanism. PLoS ONE 2013, 8, e60823. [Google Scholar] [CrossRef] [Green Version]
- Barbier, V.; Erbani, J.; Fiveash, C.; Davies, J.M.; Tay, J.; Tallack, M.R.; Lowe, J.; Magnani, J.L.; Pattabiraman, D.R.; Perkins, A.; et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Cheung, L.C.; Tickner, J.; Hughes, A.M.; Skut, P.; Howlett, M.; Foley, B.; Oommen, J.; Wells, J.E.; He, B.; Singh, S.; et al. New therapeutic opportunities from dissecting the pre-B leukemia bone marrow microenvironment. Leukemia 2018, 32, 2326–2338. [Google Scholar] [CrossRef]
- Anderson, D.; Skut, P.; Hughes, A.M.; Ferrari, E.; Tickner, J.; Xu, J.; Mullin, B.H.; Tang, D.; Malinge, S.; Kees, U.R.; et al. The bone marrow microenvironment of pre-B acute lymphoblastic leukemia at single-cell resolution. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Cahu, X.; Calvo, J.; Poglio, S.; Prade, N.; Colsch, B.; Arcangeli, M.-L.; Leblanc, T.; Petit, A.; Baleydier, F.; Baruchel, A.; et al. Bone marrow sites differently imprint dormancy and chemoresistance to T-cell acute lymphoblastic leukemia. Blood Adv. 2017, 1, 1760–1772. [Google Scholar] [CrossRef]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.; Edwards, D.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef]
- Sheng, X.; Parmentier, J.-H.; Tucci, J.; Pei, H.; Cortez-Toledo, O.; Dieli-Conwright, C.M.; Oberley, M.J.; Neely, M.; Orgel, E.; Louie, S.G.; et al. Adipocytes Sequester and Metabolize the Chemotherapeutic Daunorubicin. Mol. Cancer Res. 2017, 15, 1704–1713. [Google Scholar] [CrossRef] [Green Version]
- Zinngrebe, J.; Debatin, K.-M.; Fischer-Posovszky, P. Adipocytes in hematopoiesis and acute leukemia: Friends, enemies, or innocent bystanders? Leukemia 2020, 34, 2305–2316. [Google Scholar] [CrossRef]
- Samimi, A.; Ghanavat, M.; Shahrabi, S.; Azizidoost, S.; Saki, N. Role of bone marrow adipocytes in leukemia and chemotherapy challenges. Cell. Mol. Life Sci. 2019, 76, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Wasnik, S.; Chen, W.; Ahmed, A.S.I.; Zhang, X.-B.; Tang, X.; Baylink, D.J. Chapter 5—HSC Niche: Regulation of Mobilization and Homing. In Biology and Engineering of Stem Cell Niches; Vishwakarma, A., Karp, J.M., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 63–73. [Google Scholar]
- Welschinger, R.; Liedtke, F.; Basnett, J.; Pena, A.D.; Juarez, J.G.; Bradstock, K.F.; Bendall, L.J. Plerixafor (AMD3100) induces prolonged mobilization of acute lymphoblastic leukemia cells and increases the proportion of cycling cells in the blood in mice. Exp. Hematol. 2013, 41, 293–302.e1. [Google Scholar] [CrossRef] [PubMed]
- Sison, E.A.R.; Rau, R.; McIntyre, E.; Li, L.; Small, D.; Brown, P. MLL-rearranged acute lymphoblastic leukaemia stem cell interactions with bone marrow stroma promote survival and therapeutic resistance that can be overcome with CXCR4 antagonism. Br. J. Haematol. 2013, 160, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juárez, J.; Pena, A.D.; Baraz, R.; Hewson, J.; Khoo, M.; Cisterne, A.; Fricker, S.; Fujii, N.; Bradstock, K.F.; Bendall, L.J. CXCR4 antagonists mobilize childhood acute lymphoblastic leukemia cells into the peripheral blood and inhibit engraftment. Leukemia 2007, 21, 1249–1257. [Google Scholar] [CrossRef] [Green Version]
- Cooper, T.M.; Sison, E.A.R.; Baker, S.; Li, L.; Ahmed, A.; Trippett, T.; Gore, L.; Macy, M.E.; Narendran, A.; August, K.; et al. A phase 1 study of the CXCR4 antagonist plerixafor in combination with high-dose cytarabine and etoposide in children with relapsed or refractory acute leukemias or myelodysplastic syndrome: A Pediatric Oncology Experimental Therapeutics Investigators’ Consortium Study (POE 10-03). Pediatr. Blood Cancer 2017, 64, e26414. [Google Scholar] [CrossRef]
- Michelis, F.V.; Hedley, D.W.; Malhotra, S.; Chow, S.; Loach, D.; Gupta, V.; Kim, D.D.; Kuruvilla, J.; Lipton, J.H.; Viswabandya, A.; et al. Mobilization of Leukemic Cells Using Plerixafor as Part of a Myeloablative Preparative Regimen for Patients with Acute Myelogenous Leukemia Undergoing Allografting: Assessment of Safety and Tolerability. Biol. Blood Marrow Transplant. 2019, 25, 1158–1163. [Google Scholar] [CrossRef]
- Borthakur, G.; Zeng, Z.; Cortes, J.E.; Chen, H.; Huang, X.; Konopleva, M.; Ravandi, F.; Kadia, T.; Patel, K.P.; Daver, N.; et al. Phase 1 study of combinatorial sorafenib, G-CSF, and plerixafor treatment in relapsed/refractory, FLT3-ITD-mutated acute myelogenous leukemia patients. Am. J. Hematol. 2020, 95, 1296–1303. [Google Scholar] [CrossRef]
- Zeng, Z.; Shi, Y.X.; Samudio, I.J.; Wang, R.-Y.; Ling, X.; Frolova, O.; Levis, M.; Rubin, J.B.; Negrin, R.R.; Estey, E.H.; et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 2009, 113, 6215–6224. [Google Scholar] [CrossRef] [Green Version]
- Pitt, L.A.; Tikhonova, A.N.; Hu, H.; Trimarchi, T.; King, B.; Gong, Y.; Sanchez-Martin, M.; Tsirigos, A.; Littman, D.R.; Ferrando, A.A.; et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell 2015, 27, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, R.; Yu, M.; Lim, M.; Groffen, J.; Heisterkamp, N. Combination of drug therapy in acute lymphoblastic leukemia with a CXCR4 antagonist. Leukemia 2011, 25, 1314–1323. [Google Scholar] [CrossRef]
- Abraham, M.; Klein, S.; Bulvik, B.; Wald, H.; Weiss, I.D.; Olam, D.; Weiss, L.; Beider, K.; Eizenberg, O.; Wald, O.; et al. The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by downregulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16-1 expression. Leukemia 2017, 31, 2336–2346. [Google Scholar] [CrossRef]
- Borthakur, G.; Tallman, M.S.; Ofran, Y.; Foran, J.M.; Uy, G.L.; D iPersio, J.F.; Nagler, A.; Rowe, J.M.; Showel, M.M.; Altman, J.; et al. The Selective Anti Leukemic Effect of BL-8040, a Peptidic CXCR4 Antagonist, Is Mediated by Induction of Leukemic Blast Mobilization, Differentiation and Apoptosis: Results of Correlative Studies from a Ph2a Trial in Acute Myeloid Leukemia. Blood 2016, 128, 2745. [Google Scholar] [CrossRef]
- Cho, B.-S.; Zeng, Z.; Mu, H.; Wang, Z.; Konoplev, S.; McQueen, T.; Protopopova, M.; Cortes, J.; Marszalek, J.R.; Peng, S.-B.; et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood 2015, 126, 222–232. [Google Scholar] [CrossRef]
- Boddu, P.; Borthakur, G.; Koneru, M.; Huang, X.; Naqvi, K.; Wierda, W.; Bose, P.; Jabbour, E.; Estrov, Z.; Burger, J.; et al. Initial Report of a Phase I Study of LY2510924, Idarubicin, and Cytarabine in Relapsed/Refractory Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 369. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Guo, H.; Yang, Y.; Meng, J.; Liu, J.; Wang, C.; Xu, H. A designed peptide targeting CXCR4 displays anti-acute myelocytic leukemia activity in vitro and in vivo. Sci. Rep. 2015, 4, 6610. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Guo, H.; Duan, H.; Yang, Y.; Meng, J.; Liu, J.; Wang, C.; Xu, H. Improving chemotherapeutic efficiency in acute myeloid leukemia treatments by chemically synthesized peptide interfering with CXCR4/CXCL12 axis. Sci. Rep. 2015, 5, 16228. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zeng, Z.; Shi, Y.; Jacamo, R.; Ludin, C.; Dembowsky, K.; Frenette, P.S.; Konopleva, M.; Andreeff, M. Targeting CXCR4, SDF1 and Beta-Adrenergic Receptors in the AML Microenvironment by Novel Antagonist POL6326, G-CSF and Isoproterenol. Blood 2010, 116, 2179. [Google Scholar] [CrossRef]
- Kuhne, M.R.; Mulvey, T.; Belanger, B.; Chen, S.; Pan, C.; Chong, C.; Cao, F.; Niekro, W.; Kempe, T.; Henning, K.A.; et al. BMS-936564/MDX-1338: A Fully Human Anti-CXCR4 Antibody Induces Apoptosis In Vitro and Shows Antitumor Activity In Vivo in Hematologic Malignancies. Clin. Cancer Res. 2013, 19, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Chien, S.; Beyerle, L.E.; Wood, B.L.; Estey, E.H.; Appelbaum, F.R.; Cardarelli, P.P.M.; Sabbatini, P.; Shelat, M.-P.S.; Cohen, L.; Becker, P.S. Mobilization of Blasts and Leukemia Stem Cells by Anti-CXCR4 Antibody BMS-936564 (MDX 1338) in Patients with Relapsed/Refractory Acute Myeloid Leukemia. Blood 2013, 122, 3882. [Google Scholar] [CrossRef]
- Becker, P.S.; Foran, J.M.; Altman, J.K.; Yacoub, M.A.; Castro, J.E.; Sabbatini, P.; Dilea, C.; Wade, M.; Xing, G.; Gutierrez, A.; et al. Targeting the CXCR4 Pathway: Safety, Tolerability and Clinical Activity of Ulocuplumab (BMS-936564), an Anti-CXCR4 Antibody, in Relapsed/Refractory Acute Myeloid Leukemia. Blood 2014, 124, 386. [Google Scholar] [CrossRef]
- Peng, S.-B.; Zhang, X.; Paul, D.; Kays, L.M.; Ye, M.; Vaillancourt, P.; Dowless, M.; Stancato, L.F.; Stewart, J.; Uhlik, M.T.; et al. Inhibition of CXCR4 by LY2624587, a Fully Humanized Anti-CXCR4 Antibody Induces Apoptosis of Hematologic Malignancies. PLoS ONE 2016, 11, e0150585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacsovics, T.J.; Mims, A.; Salama, M.E.; Pantin, J.; Rao, N.; Kosak, K.M.; Ahorukomeye, P.; Glenn, M.J.; Deininger, M.W.N.; Boucher, K.M.; et al. Combination of the low anticoagulant heparin CX-01 with chemotherapy for the treatment of acute myeloid leukemia. Blood Adv. 2018, 2, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, P.; Walls, M.; Qiu, M.; Ding, R.; Denlinger, R.H.; Wong, A.; Tsaparikos, K.; Jani, J.P.; Hosea, N.; Sands, M.; et al. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol. Cancer. Ther. 2010, 9, 1618–1628. [Google Scholar] [CrossRef] [Green Version]
- Samon, J.B.; Castillo-Martin, M.; Hadler, M.; Ambesi-Impiobato, A.; Paietta, E.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Jakubczak, J.; Randolph, S.; et al. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol. Cancer. Ther. 2012, 11, 1565–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannidis, C.; De Angelo, D.J.; Stock, W.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; English, P.A.; Ozeck, M.; et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015, 5, e350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavai, A.V.; Quesnelle, C.A.; Norris, D.J.; Han, W.-C.; Gill, P.S.; Shan, W.; Balog, A.; Chen, K.; Tebben, A.J.; Rampulla, R.; et al. Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors. ACS Med. Chem. Lett. 2015, 6, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Zweidler-McKay, M.P.A.; de Angelo, M.D.J.; Douer, D.; Dombret, H.; Ottmann, O.G.; Vey, N.; Thomas, D.A.; Zhu, M.L.; Huang, F.; Bajaj, G.; et al. The Safety and Activity of BMS-906024, a Gamma Secretase Inhibitor (GSI) with Anti-Notch Activity, in Patients with Relapsed T-Cell Acute Lymphoblastic Leukemia (T-ALL): Initial Results of a Phase 1 Trial. Blood 2014, 124, 968. [Google Scholar] [CrossRef]
- Habets, R.A.; de Bock, C.E.; Serneels, L.; Lodewijckx, I.; Verbeke, D.; Nittner, D.; Narlawar, R.; Demeyer, S.; Dooley, J.; Liston, A.; et al. Safe targeting of T cell acute lymphoblastic leukemia by pathology-specific NOTCH inhibition. Sci. Transl. Med. 2019, 11, eaau6246. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Mallampati, S.; Sun, B.; Zhang, J.; Kim, S.-B.; Lee, J.-S.; Gong, Y.; Cai, Z.; Sun, X. Wnt pathway contributes to the protection by bone marrow stromal cells of acute lymphoblastic leukemia cells and is a potential therapeutic target. Cancer Lett. 2013, 333, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Mak, P.Y.; Mu, H.; Mak, D.H.; Zeng, Z.; Cortes, J.; Liu, Q.; Andreeff, M.; Carter, B.Z. Combined inhibition of beta-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia 2017, 31, 2065–2074. [Google Scholar] [CrossRef]
- Jiang, X.; Mak, P.Y.; Mu, H.; Tao, W.; Mak, D.H.; Kornblau, S.; Zhang, Q.; Ruvolo, P.; Burks, J.K.; Zhang, W.; et al. Disruption of Wnt/beta-Catenin Exerts Antileukemia Activity and Synergizes with FLT3 Inhibition in FLT3-Mutant Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 2417–2429. [Google Scholar] [CrossRef] [Green Version]
- Fiskus, W.; Sharma, S.; Saha, S.; Shah, B.; Devaraj, S.G.; Sun, B.; Horrigan, S.; Leveque, C.; Zu, Y.; Iyer, S.; et al. Pre-clinical efficacy of combined therapy with novel beta-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia 2015, 29, 1267–1278. [Google Scholar] [CrossRef]
- Lee, J.-H.; Faderl, S.; Pagel, J.M.; Jung, C.W.; Yoon, S.-S.; Pardanani, A.D.; Becker, P.S.; Lee, H.; Choi, J.; Lee, K.; et al. Phase 1 study of CWP232291 in patients with relapsed or refractory acute myeloid leukemia and myelodysplastic syndrome. Blood Adv. 2020, 4, 2032–2043. [Google Scholar] [CrossRef]
- Ma, S.; Yang, L.-L.; Niu, T.; Cheng, C.; Zhong, L.; Zheng, M.-W.; Xiong, Y.; Li, L.-L.; Xiang, R.; Chen, L.-J.; et al. SKLB-677, an FLT3 and Wnt/β-catenin signaling inhibitor, displays potent activity in models of FLT3-driven AML. Sci. Rep. 2015, 5, 15646. [Google Scholar] [CrossRef]
- Da Cruz, L.; McConkey, F.; Feng, N.; Pereira, D.; Hahn, S.; Rubinstein, D.; Young, D. Anti-CD44 antibody, ARH460-16-2, binds to human AML CD34+CD38− cancer stem cells and demonstrates anti-tumor activity in an AML xenograft model. Cancer Res. 2008, 68 (Suppl. 9), 3976. [Google Scholar]
- Vey, N.; Delaunay, J.; Martinelli, G.; Fiedler, W.; Raffoux, E.; Prebet, T.; Gomez-Roca, C.; Papayannidis, C.; Kebenko, M.; Paschka, P.; et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget 2016, 7, 32532–32542. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, Y.-T.; Jiang, E.; Pham, J.; Kim, H.-N.; Abdel-Azim, H.; Khazal, S.; Bug, G.; Spohn, P.G.; Bonig, H.; Kim, Y.-M. VLA4 Blockade in Acute Myeloid Leukemia. Blood 2013, 122, 3944. [Google Scholar] [CrossRef]
- Hsieh, Y.-T.; Gang, E.J.; Geng, H.; Park, E.; Huantes, S.; Chudziak, R.; Dauber, K.; Schaefer, P.; Scharman, C.; Shimada, H.; et al. Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood 2013, 121, 1814–1818. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, Y.T.; Gang, E.J.; Shishido, S.N.; Kim, H.N.; Pham, J.; Khazal, S.; Osborne, A.; Esguerra, Z.A.; Kwok, E.; Jang, J.; et al. Effects of the small-molecule inhibitor of integrin α4, TBC3486, on pre-B-ALL cells. Leukemia 2014, 28, 2101–2104. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Fukai, F.; Miura, S.; Nakane, Y.; Owaki, T.; Kodama, H.; Tanaka, M.; Nagaya, T.; Takimoto, R.; Takayama, T.; et al. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia 2007, 22, 353–360. [Google Scholar] [CrossRef] [Green Version]
- DeAngelo, D.J.; Jonas, B.A.; Liesveld, J.L.; Bixby, D.L.; Advani, A.S.; Marlton, P.; O’Dwyer, M.E.; Fogler, W.E.; Wolfgang, C.D.; Magnani, J.L.; et al. Uproleselan (GMI-1271), an E-Selectin Antagonist, Improves the Efficacy and Safety of Chemotherapy in Relapsed/Refractory (R/R) and Newly Diagnosed Older Patients with Acute Myeloid Leukemia: Final, Correlative, and Subgroup Analyses. Blood 2018, 132 (Suppl. 1), 331. [Google Scholar] [CrossRef]
- Zhang, W.; Ly, C.; Zhang, Q.; Mu, H.; Battula, V.L.; Patel, N.; Schober, W.; Han, X.; Fogler, W.E.; Magnani, J.L.; et al. Dual E-Selectin/CXCR4 Antagonist GMI-1359 Exerts Efficient Anti-Leukemia Effects in a FLT3 ITD Mutated Acute Myeloid Leukemia Patient-Derived Xenograft Murine Model. Blood 2016, 128, 3519. [Google Scholar] [CrossRef]
- Daver, N.; Boumber, Y.; Kantarjian, H.; Ravandi, F.; Cortes, J.; Rytting, M.E.; Kawedia, J.D.; Basnett, J.; Culotta, K.S.; Zeng, Z.; et al. A Phase I/II Study of the mTOR Inhibitor Everolimus in Combination with HyperCVAD Chemotherapy in Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2015, 21, 2704–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Place, A.E.; Pikman, Y.; Stevenson, K.E.; Harris, M.H.; Pauly, M.; Sulis, M.-L.; Hijiya, N.; Gore, L.; Cooper, T.M.; Loh, M.L.; et al. Phase I trial of the mTOR inhibitor everolimus in combination with multi-agent chemotherapy in relapsed childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2018, 65, e27062. [Google Scholar] [CrossRef]
- Lu, J.-W.; Wang, A.-N.; Liao, H.-A.; Chen, C.-Y.; Hou, H.-A.; Hu, C.-Y.; Tien, H.-F.; Ou, D.-L.; Lin, L.-I. Cabozantinib is selectively cytotoxic in acute myeloid leukemia cells with FLT3-internal tandem duplication (FLT3-ITD). Cancer Lett. 2016, 376, 218–225. [Google Scholar] [CrossRef]
- Fathi, A.T.; Blonquist, T.M.; Hernandez, D.; Amrein, P.C.; Ballen, K.K.; McMasters, M.; Avigan, D.E.; Joyce, R.; Logan, E.K.; Hobbs, G.; et al. Cabozantinib is well tolerated in acute myeloid leukemia and effectively inhibits the resistance-conferring FLT3/tyrosine kinase domain/F691 mutation. Cancer 2018, 124, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Hu, X.; Wang, L.; Lü, S.; Cheng, H.; Song, X.; Wang, J. Bortezomib suppresses the growth of leukemia cells with Notch1 overexpression in vivo and in vitro. Cancer Chemother. Pharmacol. 2012, 70, 801–809. [Google Scholar] [CrossRef]
- Schnerch, D.; Schüler, J.; Follo, M.; Felthaus, J.; Wider, D.; Klingner, K.; Greil, C.; Duyster, J.; Engelhardt, M.; Wäsch, R. Proteasome inhibition enhances the efficacy of volasertib-induced mitotic arrest in AML in vitro and prolongs survival in vivo. Oncotarget 2017, 8, 21153–21166. [Google Scholar] [CrossRef] [Green Version]
- Messinger, Y.H.; Bostrom, B.C. Bortezomib-based four-drug induction does induce a response in advanced relapsed ALL but cure remains elusive. Pediatr. Blood Cancer 2019, 67, e28115. [Google Scholar] [CrossRef]
- Aplenc, R.; Meshinchi, S.; Sung, L.; Alonzo, T.; Choi, J.; Fisher, B.; Gerbing, R.; Hirsch, B.; Horton, T.; Kahwash, S.; et al. Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: A report from the Children’s Oncology Group. Haematologica 2020, 105, 1879–1886. [Google Scholar] [CrossRef]
- Liu, C.Y.; Hsieh, F.S.; Chu, P.Y.; Tsai, W.C.; Huang, C.T.; Yu, Y.B.; Huang, T.T.; Ko, P.S.; Hung, M.H.; Wang, W.L.; et al. Carfilzomib induces leukaemia cell apoptosis via inhibiting ELK1/KIAA1524 (Elk-1/CIP2A) and activating PP2A not related to proteasome inhibition. Br. J. Haematol. 2017, 177, 726–740. [Google Scholar] [CrossRef] [Green Version]
- Cheung, L.C.; de Kraa, R.; Oommen, J.; Chua, G.-A.; Singh, S.; Hughes, A.M.; Ferrari, E.; Ford, J.; Chiu, S.K.; Stam, R.W.; et al. Preclinical Evaluation of Carfilzomib for Infant KMT2A-Rearranged Acute Lymphoblastic Leukemia. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Wartman, L.D.; Fiala, M.A.; Fletcher, T.; Hawkins, E.R.; Cashen, A.; Di Persio, J.F.; Jacoby, M.A.; Stockerl-Goldstein, K.E.; Pusic, I.; Uy, G.L.; et al. A phase I study of carfilzomib for relapsed or refractory acute myeloid and acute lymphoblastic leukemia. Leuk. Lymphoma 2015, 57, 728–730. [Google Scholar] [CrossRef] [Green Version]
- Burke, M.J.; Ziegler, D.S.; Sirvent, F.J.B.; Attarbaschi, A.; Gore, L.; Locatelli, F.; O’Brien, M.M.; Pauly, M.; Obreja, M.; Morris, C.L.; et al. Phase 1b Study of Carfilzomib in Combination with Induction Chemotherapy in Children with Relapsed or Refractory Acute Lymphoblastic Leukemia (ALL). Blood 2019, 134 (Suppl. 1), 3873. [Google Scholar] [CrossRef]
- Advani, A.S.; Cooper, B.; Visconte, V.; Elson, P.; Chan, R.; Carew, J.; Wei, W.; Mukherjee, S.; Gerds, A.; Carraway, H.; et al. A Phase I/II Trial of MEC (Mitoxantrone, Etoposide, Cytarabine) in Combination with Ixazomib for Relapsed Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 4231–4237. [Google Scholar] [CrossRef] [Green Version]
- Benito, J.; Shi, Y.; Szymanska, B.; Carol, H.; Boehm, I.; Lu, H.; Konoplev, S.; Fang, W.; Zweidler-McKay, P.A.; Campana, D.; et al. Pronounced Hypoxia in Models of Murine and Human Leukemia: High Efficacy of Hypoxia-Activated Prodrug PR-104. PLoS ONE 2011, 6, e23108. [Google Scholar] [CrossRef]
- Moradi Manesh, D.; El-Hoss, J.; Evans, K.; Richmond, J.; Toscan, C.E.; Bracken, L.S.; Hedrick, A.; Sutton, R.; Marshall, G.M.; Wilson, W.R.; et al. AKR1C3 is a biomarker of sensitivity to PR-104 in preclinical models of T-cell acute lymphoblastic leukemia. Blood 2015, 126, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Thall, P.F.; Yi, C.A.; Borthakur, G.; Coveler, A.; Bueso-Ramos, C.; Benito, J.; Konoplev, S.; Gu, Y.; Ravandi, F.; et al. Phase I/II study of the hypoxia-activated prodrug PR104 in refractory/relapsed acute myeloid leukemia and acute lymphoblastic leukemia. Haematologica 2015, 100, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Portwood, S.; Lal, D.; Hsu, Y.-C.; Vargas, R.; Johnson, M.K.; Wetzler, M.; Hart, C.; Wang, E.S. Activity of the Hypoxia-Activated Prodrug, TH-302, in Preclinical Human Acute Myeloid Leukemia Models. Clin. Cancer Res. 2013, 19, 6506–6519. [Google Scholar] [CrossRef] [Green Version]
- Benito, J.; Ramirez, M.S.; Millward, N.Z.; Velez, J.; Harutyunyan, K.G.; Lu, H.; Shi, Y.-X.; Matre, P.; Jacamo, R.; Ma, H.; et al. Hypoxia-Activated Prodrug TH-302 Targets Hypoxic Bone Marrow Niches in Preclinical Leukemia Models. Clin. Cancer Res. 2016, 22, 1687–1698. [Google Scholar] [CrossRef] [Green Version]
- Badar, T.; Handisides, D.R.; Benito, J.M.; Richie, M.A.; Borthakur, G.; Jabbour, E.; Harutyunyan, K.; Konoplev, S.; Faderl, S.; Kroll, S.; et al. Phase I study of evofosfamide, an investigational hypoxia-activated prodrug, in patients with advanced leukemia. Am. J. Hematol. 2016, 91, 800–805. [Google Scholar] [CrossRef] [Green Version]
- Benezra, M.; Phillips, E.L.; Tilki, D.; Ding, B.-S.; Butler, J.M.; Dobrenkov, K.; Siim, B.G.; Chaplin, D.D.; Rafii, S.; Rabbany, S.Y.; et al. Serial monitoring of human systemic and xenograft models of leukemia using a novel vascular disrupting agent. Leukemia 2012, 26, 1771–1778. [Google Scholar] [CrossRef] [Green Version]
- Uckun, F.M.; Cogle, C.R.; Lin, T.L.; Qazi, S.; Trieu, V.N.; Schiller, G.; Watts, J.M. A Phase 1B Clinical Study of Combretastatin A1 Diphosphate (OXi4503) and Cytarabine (ARA-C) in Combination (OXA) for Patients with Relapsed or Refractory Acute Myeloid Leukemia. Cancers 2019, 12, 74. [Google Scholar] [CrossRef] [Green Version]
- Sison, E.A.R.; McIntyre, E.; Magoon, D.; Brown, P. Dynamic Chemotherapy-Induced Upregulation of CXCR4 Expression: A Mechanism of Therapeutic Resistance in Pediatric AML. Mol. Cancer Res. 2013, 11, 1004–1016. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, A.; Kollet, O.; Peled, A.; Abel, L.; Nagler, A.; Bielorai, B.; Rechavi, G.; Vormoor, J.; Lapidot, T. Unique SDF-1–induced activation of human precursor-B ALL cells as a result of altered CXCR4 expression and signaling. Blood 2004, 103, 2900–2907. [Google Scholar] [CrossRef]
- Armstrong, F.; de la Grange, P.B.; Gerby, B.; Rouyez, M.-C.; Calvo, J.; Fontenay, M.; Boissel, N.; Dombret, H.; Baruchel, A.; Landman-Parker, J.; et al. NOTCH is a key regulator of human T-cell acute leukemia initiating cell activity. Blood 2009, 113, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- García-Peydró, M.; Fuentes, P.; Mosquera, M.; García-León, M.J.; Alcain, J.; Rodríguez, A.; de Miguel, P.G.; Menéndez, P.; Weijer, K.; Spits, H.; et al. The NOTCH1/CD44 axis drives pathogenesis in a T cell acute lymphoblastic leukemia model. J. Clin. Investig. 2018, 128, 2802–2818. [Google Scholar] [CrossRef] [PubMed]
- Nwabo Kamdje, A.H.; Mosna, F.; Bifari, F.; Lisi, V.; Bassi, G.; Malpeli, G.; Ricciardi, M.; Perbellini, O.; Scupoli, M.T.; Pizzolo, G.; et al. Notch-3 and Notch-4 signaling rescue from apoptosis human B-ALL cells in contact with human bone marrow-derived mesenchymal stromal cells. Blood 2011, 118, 380–389. [Google Scholar] [CrossRef]
- Kamga, P.T.; dal Collo, G.; Midolo, M.; Adamo, A.; Delfino, P.; Mercuri, A.; Cesaro, S.; Mimiola, E.; Bonifacio, M.; Andreini, A.; et al. Inhibition of Notch Signaling Enhances Chemosensitivity in B-cell Precursor Acute Lymphoblastic Leukemia. Cancer Res. 2019, 79, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamga, P.T.; Bassi, G.; Cassaro, A.; Midolo, M.; di Trapani, M.; Gatti, A.; Carusone, R.; Resci, F.; Perbellini, O.; Gottardi, M.; et al. Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget 2016, 7, 21713–21727. [Google Scholar] [CrossRef] [Green Version]
- Indraccolo, S.; Minuzzo, S.; Masiero, M.; Pusceddu, I.; Persano, L.; Moserle, L.; Reboldi, A.; Favaro, E.; Mecarozzi, M.; di Mario, G.; et al. Cross-talk between Tumor and Endothelial Cells Involving the Notch3-Dll4 Interaction Marks Escape from Tumor Dormancy. Cancer Res. 2009, 69, 1314–1323. [Google Scholar] [CrossRef] [Green Version]
- Minuzzo, S.; Agnusdei, V.; Pusceddu, I.; Pinazza, M.; Moserle, L.; Masiero, M.; Rossi, E.; Crescenzi, M.; Hoey, T.; Ponzoni, M.; et al. DLL4 regulates NOTCH signaling and growth of T acute lymphoblastic leukemia cells in NOD/SCID mice. Carcinogenesis 2015, 36, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.C.; Eisenberg, P.D.; Manikhas, G.; Chugh, R.; Gubens, M.A.; Stagg, R.J.; Kapoun, A.M.; Xu, L.; Dupont, J.; Sikic, B. A Phase I Dose Escalation and Expansion Study of the Anticancer Stem Cell Agent Demcizumab (Anti-DLL4) in Patients with Previously Treated Solid Tumors. Clin. Cancer Res. 2014, 20, 6295–6303. [Google Scholar] [CrossRef] [Green Version]
- Chiorean, E.G.; Lorusso, P.M.; Strother, R.M.; Diamond, J.R.; Younger, A.; Messersmith, W.A.; Adriaens, L.; Liu, L.; Kao, R.J.; DiCioccio, A.T.; et al. A Phase I First-in-Human Study of Enoticumab (REGN421), a Fully Human Delta-like Ligand 4 (Dll4) Monoclonal Antibody in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 2695–2703. [Google Scholar] [CrossRef] [Green Version]
- Tejada, F.N.H.; Silva, J.R.G.; Zweidler-McKay, P.A. The challenge of targeting notch in hematologic malignancies. Front. Pediatr. 2014, 2, 54. [Google Scholar]
- DeAngelo, D.J.; Stone, R.M.; Silverman, L.B.; Stock, W.; Attar, E.C.; Fearen, I.; Dallob, A.; Matthews, C.; Stone, J.; Freedman, S.J.; et al. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J. Clin. Oncol. 2006, 24, 6585. [Google Scholar] [CrossRef]
- Cullion, K.; Draheim, K.M.; Hermance, N.; Tammam, J.; Sharma, V.M.; Ware, C.; Nikov, G.; Krishnamoorthy, V.; Majumder, P.K.; Kelliher, M.A. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood 2009, 113, 6172–6181. [Google Scholar] [CrossRef] [Green Version]
- Real, P.J.; Tosello, V.; Palomero, T.; Castillo, M.; Hernando, E.; de Stanchina, E.; Sulis, M.L.; Barnes, K.; Sawai, C.; Homminga, I.; et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat. Med. 2009, 15, 50–58. [Google Scholar] [CrossRef] [Green Version]
- López-Guerra, M.; Xargay-Torrent, S.; Rosich, L.; Montraveta, A.; Roldán, J.; Matas-Céspedes, A.; Villamor, N.; Aymerich, M.; López-Otín, C.; Pérez-Galán, P.; et al. The gamma-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells. Leukemia 2015, 29, 96–106. [Google Scholar] [CrossRef]
- Hogan, L.E.; Meyer, J.A.; Yang, J.; Wang, J.; Wong, N.; Yang, W.; Condos, G.; Hunger, S.P.; Raetz, E.; Saffery, R.; et al. Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood 2011, 118, 5218–5226. [Google Scholar] [CrossRef] [Green Version]
- Gruszka, A.M.; Valli, D.; Alcalay, M. Wnt Signalling in Acute Myeloid Leukaemia. Cells 2019, 8, 1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kode, A.; Mosialou, I.; Manavalan, S.J.; Rathinam, C.V.; Friedman, R.A.; Teruya-Feldstein, J.; Bhagat, G.; Berman, E.; Kousteni, S. FoxO1-dependent induction of acute myeloid leukemia by osteoblasts in mice. Leukemia 2016, 30, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, S.; Haq, S.U.; Pawlus, M.; Moon, R.T.; Estey, E.H.; Appelbaum, F.R.; Othus, M.; Magnani, J.L.; Becker, P.S. Adhesion of Acute Myeloid Leukemia Blasts To E-Selectin In The Vascular Niche Enhances Their Survival By Mechanisms Such As Wnt Activation. Blood 2013, 122, 61. [Google Scholar] [CrossRef]
- Yang, S.; Gu, Y.; Wang, G.; Hu, Q.; Chen, S.; Wang, Y.; Zhao, M. HMGA2 regulates acute myeloid leukemia progression and sensitivity to daunorubicin via Wnt/beta-catenin signaling. Int. J. Mol. Med. 2019, 44, 427–436. [Google Scholar] [PubMed] [Green Version]
- Pak, S.; Park, S.; Kim, Y.; Park, J.H.; Park, C.H.; Lee, K.J.; Kim, C.S.; Ahn, H. The small molecule WNT/beta-catenin inhibitor CWP232291 blocks the growth of castration-resistant prostate cancer by activating the endoplasmic reticulum stress pathway. J. Exp. Clin. Cancer Res. 2019, 38, 342. [Google Scholar]
- Cha, J.Y.; Jung, J.-E.; Lee, K.-H.; Briaud, I.; Tenzin, F.; Jung, H.K.; Pyon, Y.; Lee, N.; Chung, J.U.; Lee, J.H.; et al. Anti-Tumor Activity of Novel Small Molecule Wnt Signaling Inhibitor, CWP232291, In Multiple Myeloma. Blood 2010, 116, 3038. [Google Scholar] [CrossRef]
- Griffiths, E.A.; Golding, M.C.; Srivastava, P.; Povinelli, B.J.; James, S.R.; Ford, L.A.; Wetzler, M.; Wang, E.S.; Nemeth, M.J. Pharmacological targeting of beta-catenin in normal karyotype acute myeloid leukemia blasts. Haematologica 2015, 100, e49–e52. [Google Scholar] [CrossRef] [Green Version]
- Winter, S.S.; Sweatman, J.J.; Lawrence, M.B.; Rhoades, T.H.; Hart, A.L.; Larson, R.S. Enhanced T-lineage acute lymphoblastic leukaemia cell survival on bone marrow stroma requires involvement of LFA-1 and ICAM-1. Br. J. Haematol. 2001, 115, 862–871. [Google Scholar] [CrossRef]
- Shalapour, S.; Hof, J.; Kirschner-Schwabe, R.; Bastian, L.; Eckert, C.; Prada, J.; Henze, G.; von Stackelberg, A.; Seeger, K. High VLA-4 expression is associated with adverse outcome and distinct gene expression changes in childhood B-cell precursor acute lymphoblastic leukemia at first relapse. Haematologica 2011, 96, 1627–1635. [Google Scholar] [CrossRef]
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-kappaB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef]
- Ho, P.-R.; Koendgen, H.; Campbell, N.; Haddock, B.; Richman, S.; Chang, I. Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: A retrospective analysis of data from four clinical studies. Lancet Neurol. 2017, 16, 925–933. [Google Scholar] [CrossRef]
- Gadhoum, S.; Madhoun, N.; AbuElela, A.; Merzaban, J.S. Anti-CD44 antibodies inhibit both mTORC1 and mTORC2: A new rationale supporting CD44-induced AML differentiation therapy. Leukemia 2016, 30, 2397–2401. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.G.G.; Watts, N.B.; Ebetino, F.H.; Rogers, M. Mechanisms of action of bisphosphonates: Similarities and differences and their potential influence on clinical efficacy. Osteoporos. Int. 2008, 19, 733–759. [Google Scholar] [CrossRef]
- Padhye, B.; dalla-Pozza, L.; Little, D.; Munns, C. Incidence and outcome of osteonecrosis in children and adolescents after intensive therapy for acute lymphoblastic leukemia (ALL). Cancer Med. 2016, 5, 960–967. [Google Scholar] [CrossRef] [Green Version]
- Kneissel, M.; Luong-Nguyen, N.-H.; Baptist, M.; Cortesi, R.; Zumstein-Mecker, S.; Kossida, S.; O’Reilly, T.; Lane, H.; Susa, M. Everolimus suppresses cancellous bone loss, bone resorption, and cathepsin K expression by osteoclasts. Bone 2004, 35, 1144–1156. [Google Scholar] [CrossRef]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [Green Version]
- Fioramonti, M.; Santini, D.; Iuliani, M.; Ribelli, G.; Manca, P.; Papapietro, N.; Spiezia, F.; Vincenzi, B.; Denaro, V.; Russo, A.; et al. Cabozantinib targets bone microenvironment modulating human osteoclast and osteoblast functions. Oncotarget 2017, 8, 20113–20121. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Sezer, O.; Croucher, P.; Dimopoulos, M. Myeloma bone disease and proteasome inhibition therapies. Blood 2007, 110, 1098–1104. [Google Scholar] [CrossRef]
- Mohty, M.; Malard, F.; Mohty, B.; Savani, B.; Moreau, P.; Terpos, E. The effects of bortezomib on bone disease in patients with multiple myeloma. Cancer 2014, 120, 618–623. [Google Scholar] [CrossRef] [Green Version]
- Accardi, F.; Toscani, D.; Bolzoni, M.; Palma, A.B.D.; Aversa, F.; Giuliani, N. Mechanism of Action of Bortezomib and the New Proteasome Inhibitors on Myeloma Cells and the Bone Microenvironment: Impact on Myeloma-Induced Alterations of Bone Remodeling. BioMed Res. Int. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hurchla, M.A.; Garcia, A.; Hornick, M.C.; Ocio, E.M.; Li, A.; Blanco, J.F.; Collins, L.; Kirk, C.J.; Piwnica-Worms, D.; Vij, R.; et al. The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects. Leukemia 2012, 27, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.; Quwaider, D.; Canavese, M.; Ocio, E.M.; Tian, Z.; Blanco, J.F.; Berger, A.J.; de Solórzano, C.O.; Hernández-Iglesias, T.; Martens, A.C.; et al. Preclinical Activity of the Oral Proteasome Inhibitor MLN9708 in Myeloma Bone Disease. Clin. Cancer Res. 2014, 20, 1542–1554. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.O.; Mortensen, B.T.; Hodgkiss, R.J.; Iversen, P.O.; Christensen, I.J.; Helledie, N.; Larsen, J.K. Increased cellular hypoxia and reduced proliferation of both normal and leukaemic cells during progression of acute myeloid leukaemia in rats. Cell Prolif. 2000, 33, 381–395. [Google Scholar] [CrossRef]
- Frolova, O.; Samudio, I.; Benito, J.M.; Jacamo, R.; Kornblau, S.M.; Markovic, A.; Schober, W.; Lu, H.; Qiu, Y.H.; Buglio, D.; et al. Regulation of HIF-1α signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol. Ther. 2012, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, M.; Garcia-Ruiz, J.C.; Berra, E. The hypoxia signalling pathway in haematological malignancies. Oncotarget 2017, 8, 36832–36844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco-Hernandez, T.; Hyrenius-Wittsten, A.; Rehn, M.; Bryder, D.; Cammenga, J. HIF-1α can act as a tumor suppressor gene in murine acute myeloid leukemia. Blood 2014, 124, 3597–3607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco-Hernandez, T.; Tornero, D.; Cammenga, J. Loss of HIF-1α accelerates murine FLT-3ITD-induced myeloproliferative neoplasia. Leukemia 2015, 29, 2366–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, A.; Ferry, D.M.; Edmunds, S.; Gu, Y.; Singleton, R.S.; Patel, K.; Pullen, S.M.; Hicks, K.O.; Syddall, S.P.; Atwell, G.J.; et al. Mechanism of Action and Preclinical Antitumor Activity of the Novel Hypoxia-Activated DNA Cross-Linking Agent PR-104. Clin. Cancer Res. 2007, 13, 3922–3932. [Google Scholar] [CrossRef] [Green Version]
- Guise, C.P.; Abbattista, M.R.; Singleton, R.S.; Holford, S.D.; Connolly, J.; Dachs, G.U.; Fox, S.B.; Pollock, R.; Harvey, J.; Guilford, P.; et al. The Bioreductive Prodrug PR-104A Is Activated under Aerobic Conditions by Human Aldo-Keto Reductase 1C3. Cancer Res. 2010, 70, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.D.; Liu, Q.; Wang, J.; Ahluwalia, D.; Ferraro, D.; Wang, Y.; Duan, J.-X.; Ammons, W.S.; Curd, J.G.; Matteucci, M.D.; et al. Selective Tumor Hypoxia Targeting by Hypoxia-Activated Prodrug TH-302 Inhibits Tumor Growth in Preclinical Models of Cancer. Clin. Cancer Res. 2012, 18, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Mulkey, F.; Hasserjian, R.P.; Sanford, B.L.; Vij, R.; Hurd, D.D.; Odenike, O.M.; Bloomfield, C.D.; Owzar, K.; Stone, R.M.; et al. A phase II study of the oral VEGF receptor tyrosine kinase inhibitor vatalanib (PTK787/ZK222584) in myelodysplastic syndrome: Cancer and Leukemia Group B study 10105 (Alliance). Investig. New Drugs 2013, 31, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Serve, H.; Krug, U.; Wagner, R.; Sauerland, M.C.; Heinecke, A.; Brunnberg, U.; Schaich, M.; Ottmann, O.; Duyster, J.; Wandt, H.; et al. Sorafenib in Combination with Intensive Chemotherapy in Elderly Patients with Acute Myeloid Leukemia: Results from a Randomized, Placebo-Controlled Trial. J. Clin. Oncol. 2013, 31, 3110–3118. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Brown, P.A.; Gerbing, R.B.; Fox, E.; Choi, J.K.; Fisher, D.B.T.; Hirsch, B.A.; Kahwash, S.; Levine, M.J.E.; et al. Sorafenib in Combination with Standard Chemotherapy for Children with High Allelic Ratio FLT3/ITD+ AML Improves Event-Free Survival and Reduces Relapse Risk: A Report from the Children’s Oncology Group Protocol AAML1031. Blood 2019, 134 (Suppl. 1), 292. [Google Scholar] [CrossRef]
- Madlambayan, G.J.; Meacham, A.M.; Hosaka, K.; Mir, S.; Jorgensen, M.; Scott, E.W.; Siemann, D.W.; Cogle, C.R. Leukemia regression by vascular disruption and antiangiogenic therapy. Blood 2010, 116, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Petty, A.J.; Yang, Y. Tumor-Associated Macrophages in Hematologic Malignancies: New Insights and Targeted Therapies. Cells 2019, 8, 1526. [Google Scholar] [CrossRef] [Green Version]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 1–21. [Google Scholar] [CrossRef]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.-X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Pehlivan, K.C.; Duncan, B.B.; Lee, D.W. CAR-T Cell Therapy for Acute Lymphoblastic Leukemia: Transforming the Treatment of Relapsed and Refractory Disease. Curr. Hematol. Malig. Rep. 2018, 13, 396–406. [Google Scholar] [CrossRef]
- Griessinger, E.; Moschoi, R.; Biondani, G.; Peyron, J.-F. Mitochondrial Transfer in the Leukemia Microenvironment. Trends Cancer 2017, 3, 828–839. [Google Scholar] [CrossRef]
- Sahinbegovic, H.; Jelinek, T.; Hrdinka, M.; Bago, J.R.; Turi, M.; Sevcikova, T.; Kurtovic-Kozaric, A.; Hajek, R.; Simicek, M. Intercellular Mitochondrial Transfer in the Tumor Microenvironment. Cancers 2020, 12, 1787. [Google Scholar] [CrossRef]
- Panuzzo, C.; Jovanovski, A.; Pergolizzi, B.; Pironi, L.; Stanga, S.; Fava, C.; Cilloni, D. Mitochondria: A Galaxy in the Hematopoietic and Leukemic Stem Cell Universe. Int. J. Mol. Sci. 2020, 21, 3928. [Google Scholar] [CrossRef] [PubMed]
- Polson, A.G.; Fuji, R.N. The successes and limitations of preclinical studies in predicting the pharmacodynamics and safety of cell-surface-targeted biological agents in patients. Br. J. Pharmacol. 2012, 166, 1600–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.M.B.; Chao, N.; Chhabra, S.; Corbet, K.; Gasparetto, C.; Horwitz, A.; Li, Z.; Venkata, J.K.; Long, G.; Mims, A.; et al. Plerixafor (a CXCR4 antagonist) following myeloablative allogeneic hematopoietic stem cell transplantation enhances hematopoietic recovery. J. Hematol. Oncol. 2016, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopeck, A.T.; Lipton, A.; Body, J.-J.; Steger, G.G.; Tonkin, K.; de Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.A.; Viniegra, M.; et al. Denosumab Compared with Zoledronic Acid for the Treatment of Bone Metastases in Patients with Advanced Breast Cancer: A Randomized, Double-Blind Study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef] [Green Version]
- Kotecha, R.S.; Gottardo, N.G.; Kees, U.R.; Cole, C.H. The evolution of clinical trials for infant acute lymphoblastic leukemia. Blood Cancer J. 2014, 4, e200. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Target Pathways | Target Strategy | Agents | Mechanism of Action in the BMM | Leukaemia Type Investigated | Animal Studies | Clinical Studies |
|---|---|---|---|---|---|---|
| CXCL12/ CXCR4 signalling pathway | CXCR4 Inhibitor (bicyclams) | Plerixafor (AMD3100) | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. | AML, pre-B ALL | [43,44,45] | [46,47,48] FDA-A |
| AMD3465 | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. | AML, Pre-B ALL, T-ALL | [45,49,50] | N/R | ||
| AMD11070 | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. | Pre-B ALL | [51] | N/R | ||
| CXCR4 inhibitor (synthetic peptides) | BL-8040 (BKT140) | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. Induces apoptosis in leukaemic cells. | AML | [52] | [53] | |
| LY2510924 | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. Inhibits proliferation of leukaemic cells. | AML | [54] | [55] | ||
| E5 Peptide | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. Induces apoptosis in leukaemic cells. | AML | [56,57] | N/R | ||
| POL6326 | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. | AML | [58] | NCT01413568Phase I/II # | ||
| Anti-CXCR4 monoclonal antibody | Ulocuplumab | Induces apoptosis and blocks CXCL12-induced leukaemic cell migration. Induces mobilisation of leukaemic cells. | AML | [59] | [60,61] | |
| LY2624587 | Induces apoptosis and blocks CXCL12-induced leukaemic cell migration. | T-ALL | [62] | N/R | ||
| CXCL12 inhibitor | CX-01 | Inhibits binding of CXCL12 to immobilised heparin and enhances treatment efficacy. | AML | N/R | [63] | |
| Notch signalling pathway | Gamma-secretase inhibitor | PF-03084014 | Inhibits stromal-mediated chemoresistance and potentiates sensitivity of leukaemic cells to chemotherapy. Inhibits proliferation and induces apoptosis in leukaemic cells. | T-ALL | [64,65] | [66] |
| BMS-906024 | Inhibits growth and survival of leukaemic cells by targeting Notch signalling. | T-ALL | [67] | [68] | ||
| MRK-560 | Promote leukaemic cell cycle arrest by targeting PSEN1, a subclass of gamma-secretase complexes highly involved in activation of mutant Notch1. | T-ALL | [69] | N/R | ||
| Wnt/β-catenin signalling pathway | Wnt/β-catenin inhibitor | XAV939 | Attenuates BMM-induced protection of leukaemic cells by inhibiting Wnt signalling. Inhibits proliferation of leukaemic cells. | Pre-B ALL | [70] | N/R |
| PRI-724 | Attenuates BMM-induced protection of leukaemic cells by inhibiting Wnt signalling. Induces apoptosis in leukaemic cells. | AML, CML | [71,72] | NCT01606579Phase I/II # | ||
| BC2059 | Induces apoptosis of leukaemic cells by synergistically enhancing the effect of drug treatment. | AML | [73] | N/R | ||
| CWP232291 | Promotes endoplasmic reticulum stress activation, leading to degradation of β-catenin and apoptosis induction in leukaemic cells. | AML | N/R | [74] | ||
| Wnt/β-catenin/FLT3 inhibitor | SKLB-677 | Induces apoptosis in leukaemic cells. | AML | [75] | N/R | |
| Adhesion molecules signalling pathway | Anti-CD44 monoclonal antibody | RG7356/ ARH460-16-2 | Blocks leukaemia–stroma interaction by targeting CD44. | AML | [76] | [77] |
| Anti-α4β1/ VLA-4 monoclonal antibody | Natalizumab | Blocks leukaemia–stroma interaction by targeting VLA-4/VCAM-1, sensitising leukaemic cells to chemotherapy. | AML, Pre-B ALL | [78,79] | N/R FDA-A | |
| α4 inhibitor | TBC3486 | Blocks leukaemia–stroma interaction by targeting integrin α4, sensitising leukaemic cells to chemotherapy. | Pre-B ALL | [80] | N/R | |
| VLA-4 peptide antagonist | FNIII14 | Blocks cell adhesion by targeting VLA-4 to fibronectin interaction, sensitising leukaemic cells to chemotherapy. | AML | [81] | N/R | |
| E-selectin inhibitor | Uproleselan (GMI-1271) | Attenuates cell surface adhesion, regeneration and survival of leukaemic cells by antagonising E-selectin. Sensitises leukaemic cells to chemotherapy. | AML | [34] | [82] NCT03701308 NCT03616470 Phase II/III # | |
| Dual CXCR4/ E-selectin inhibitor | GMI-1359 | Promotes leukaemic cell mobilisation and restores normal haematopoiesis. | AML | [83] | N/R | |
| Bone remodelling signalling pathway | Bone resorption inhibitor | Zoledronic acid | Inhibits osteoclast resorption. | Pre-B ALL | [35] | N/R FDA-A |
| mTOR inhibitor | Everolimus | Inhibits osteoclast resorption. * | ALL | N/R | [84,85] FDA-A | |
| Receptor tyrosine kinase inhibitor | Cabozantinib | Inhibits osteoclast differentiation and resorption, modulates RANKL/osteoprotegerin in osteoblasts. * Induces apoptosis in leukaemic cells. | AML | [86] | [87] FDA-A | |
| Proteasome inhibitor | Bortezomib | Promotes osteoblast differentiation and suppress osteoclast activity. * Induces apoptosis in leukaemic cells. | AML, Pre-B ALL, T-ALL | [88,89] | [90,91] FDA-A | |
| Carfilzomib | Promotes osteoblast differentiation and suppress osteoclast activity. * Induces apoptosis in leukaemic cells. | AML, ALL | [92,93] | [94,95] NCT02303821 NCT02512926 Phase I FDA-A | ||
| Ixazomib | Promotes osteoblast differentiation and suppress osteoclast activity. * | AML | N/R | [96] FDA-A | ||
| Hypoxia-related signalling pathway | Hypoxia activated prodrug | PR-104 | Induces cytotoxicity in hypoxic leukaemic cells. | AML, Pre-B ALL, T-ALL | [97,98] | [99] |
| Evofosfamide (TH-302) | Induces cytotoxicity in hypoxic leukaemic cells. | AML, ALL | [100,101] | [102] | ||
| Vasculature-associated pathway | Vascular disrupting agent | CA1P (OXi4503) | Induces breakdown of vascular architecture. | AML | [103] | [104] |
| Agent | Studies/ Clinical Trial ID (Phases) | Treatment Design | Age Range (Median) | Disease Type (No. Patients Enrolled) | Ref | Outcomes |
|---|---|---|---|---|---|---|
| Plerixafor | NCT01319864 (Phase 1) | Dose escalation at four different dose levels (6, 9, 12, and 15 mg/m2/dose) as part of cytarabine and etoposide therapy. | AML 3–17 (13) ALL 12–21 (14) AML/ MDS 20 (20) | R/R AML (13) R/R ALL (5) R/R AML/MDS (1) | [46] |
|
| Plerixafor | NCT01141543 (Phase 1) | Dose escalation (240 μg/kg) as part of myeloablative conditioning regimen for patients undergoing allogeneic haematopoietic stem cell transplant. | 38–58 (49) | De novo AML (10) Secondary AML (2) | [47] |
|
| Plerixafor | NCT00943943 (Phase 1) | Plerixafor (240 μg/kg adjusted body weight, subcutaneously) combined with G-CSF and sorafenib dose-escalation. | 18–84 (58) | R/R AML with FLT3-ITD mutation (28) | [48] |
|
| BL-8040 | NCT01838395 (Phase 2a) | Once daily dose of BL-8040 (0.5–2 mg/kg) administered as monotherapy for days 1–2, followed by combination of BL-8040 with cytarabine. | (61) | R/R AML (45) | [53] |
|
| LY2510924 | NCT02652871 (Phase 1) | Dose escalation at two doses (10, 20 mg/day), administered as monotherapy daily for 7 days, followed by idarubicin and cytarabine combined with LY2510924. | 19–70 (55) | R/R AML (11) | [55] |
|
| Ulocuplumab | N/R (Phase 1) | Dose escalation at 0.3, 1, 3, and 10 mg/kg. Cohorts at 0.3 mg/kg received three weekly doses as monotherapy, followed by the same doses with MEC chemotherapy. Cohorts at 1, 3, and 10 mg/kg received 1 weekly dose as monotherapy, followed by the same combination regimen. | N/R | R/R AML (24) | [60] |
|
| Ulocuplumab | N/R (Phase 1) | Dose escalation (0.3, 1, 3, and 10 mg/kg) was given as a single infusion a week prior to MEC treatment, followed by 3 additional weekly doses per MEC cycle thereafter. For expansion, 10 mg/kg ulocuplumab + MEC regimen was used. | 21–79 (58) | R/R AML (73) | [61] |
|
| CX-01 | NCT02056782 (Phase 1) | For cytarabine/idarubicin induction, CX-01 (4 mg/kg) was given over 30 min after the first dose of idarubicin, followed by continuous infusion of 0.25 mg/kg per hour thereafter. For consolidation, CX-01 (4 mg/kg) was given over 30 min after the first dose of cytarabine, followed by continuous infusion of 0.25 mg/kg per hour thereafter. | 22–74 (54) | De novo AML (11) CMML-2 (1) | [63] |
|
| PF-03084014 | A8641014 (Phase 1) | PF-03084014 administered twice weekly (150 mg) in continuous cycles. | 18–43 (31) | R/R T-ALL (3) R/R T-LBL (5) | [66] |
|
| BMS-906024 | CA216002 (Phase 1) | BMS-906024 was given intravenously weekly at doses of 0.6, 4, and 6 mg. | 18–74 | R/R T-ALL/T-LBL (25) | [68] |
|
| CWP232291 | NCT01398462 (Phase 1) | Dose escalation at 4–334 mg/m2. Agent was administered intravenously daily for 7 days every 21 days. MTD was defined at 257 mg/m2. | 25–81 (64) | R/R AML (64) R/R MDS (5) | [74] |
|
| RG7356/ ARH460-16-2 | NCT01641250 (Phase 1) | RG7356 was administered intravenously at dosages ≤2400 mg every other week, or ≤1200 mg weekly or twice weekly. | 20–82 (69) | R/R AML (37) TN AML (7) | [77] |
|
| Uproleselan (GMI-1271) | N/R (Phase 1/2) | Dose escalation at 5–20 mg/kg in combination with MEC in patients with R/R AML. In phase 2, patients were given uproleselan with chemotherapy. RP2D was 10 mg/kg. | R/R patients 26–84 (59) TN patients 60–79 (67) | R/R AML (66) TN AML (25) | [82] |
|
| Everolimus | NCT00968253 (Phase 1/2) | Everolimus was given continuously at 5 or 10 mg/day with HyperCVAD treatment. MTD was defined at 5 mg/day. | 11–64 (25) | R/R Pre-B ALL (13) R/R T-ALL (10) MPAL (1) | [84] |
|
| Everolimus | NCT01523977 (Phase 1b) | Dose escalation at 2, 3, and 5 mg/m2/day for 32 days, co-administered with multi-agent reinduction chemotherapy. | 2.4–22.8 (11) | Relapsed B-ALL (21) Relapsed T-ALL (1) | [85] |
|
| Cabozantinib | NCT01961765 (Phase 1) | Dose escalation at 40, 60, and 80 mg daily in 28-day cycles. MTD was defined at 40 mg daily. | 27–85 (68) | R/R AML (16) Newly diagnosed AML (2) | [87] |
|
| Bortezomib | NCT01371981 (Phase 3) | Bortezomib (1.3 mg/m2) was incorporated into chemotherapy. Patients were randomly assigned to either standard AML therapy or standard therapy with bortezomib. | 0–29.5 (9.2) | De novo AML (1231) | [91] |
|
| Carfilzomib | NCT01137747 (Phase 1) | Dose escalation at 36, 45, and 56 mg/m2, administered as a 30 min infusion on a 28-day cycle. | 32–78 (70) | R/R AML (17) R/R ALL (1) | [94] |
|
| Carfilzomib | NCT02303821 (Phase 1b) | Dose escalation at 27–56 mg/m2 for patients treated with VXLD. Patients received one 4-week cycle of induction chemotherapy with VXLD, plus carfilzomib administered intravenously. | 1–19 (11) | R/R B-ALL (7) R/R T-ALL (8) | [95] |
|
| Ixazomib | NCT02070458 (Phase 1/2) | Dose escalation at 1, 2, and 3 mg in combination with MEC treatment. MTD was defined at 1 mg. | 31–70 (58) | R/R AML (30) | [96] |
|
| PR-104 | NCT01037556 (Phase 1/2) | Patients received PR-104 at doses ranging from 1.1–4 g/m2. For dose expansion, patients were treated at 3 or 4 g/m2. | 20–79 (62) | R/R AML (40) R/R ALL (10) | [99] |
|
| Evofosfamide (TH-302) | NCT01149915 (Phase 1) | Evifosfamide administered daily as a 30–60 min infusion (120–550 mg/m2; MTD 460 mg/m2), or as a continuous intravenous infusion over 120 h (MTD 330 mg/m2), on days 1–5 of 21-day cycles. | 23–76 (58) | R/R AML (39) R/R ALL (9) CML (1) | [102] |
|
| CA1P (OXi4503) | NCT02576301 (Phase 1B) | CA1P administered at doses ranging from 3.75 to 12.2 mg/m2 in combination with cytarabine. MTD defined at 9.76 mg/m2. | 26–78 (61) | R/R AML (27) R/R MDS (2) | [104] |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuek, V.; Hughes, A.M.; Kotecha, R.S.; Cheung, L.C. Therapeutic Targeting of the Leukaemia Microenvironment. Int. J. Mol. Sci. 2021, 22, 6888. https://doi.org/10.3390/ijms22136888
Kuek V, Hughes AM, Kotecha RS, Cheung LC. Therapeutic Targeting of the Leukaemia Microenvironment. International Journal of Molecular Sciences. 2021; 22(13):6888. https://doi.org/10.3390/ijms22136888
Chicago/Turabian StyleKuek, Vincent, Anastasia M. Hughes, Rishi S. Kotecha, and Laurence C. Cheung. 2021. "Therapeutic Targeting of the Leukaemia Microenvironment" International Journal of Molecular Sciences 22, no. 13: 6888. https://doi.org/10.3390/ijms22136888
APA StyleKuek, V., Hughes, A. M., Kotecha, R. S., & Cheung, L. C. (2021). Therapeutic Targeting of the Leukaemia Microenvironment. International Journal of Molecular Sciences, 22(13), 6888. https://doi.org/10.3390/ijms22136888