
Oral Pathogenic Bacteria-Inducing Neurodegenerative Microgliosis in Human Neural Cell Platform

 , and
, and
Abstract
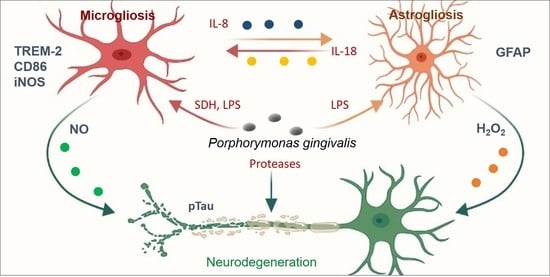
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Neurodegeneration Derived from Microgliosis and Astrogliosis-Induced Bacteria
2.2. Bacterial Conditioned Media Induced Microglial Inflammatory Responses
2.3. Neurodegeneration Induced by Both Microglial and Bacterial Condition Media
2.4. Investigating the Interaction of Astrogliosis and Microgliosis
2.5. Microglial Migration and Investigation of New Bacterial Metabolites
2.6. Inhibition of SDH and LPS Reduces Microglial Inflammatory Responses
3. Discussion
4. Materials and Methods
4.1. Preparation of Bacterial Conditioned Medium
4.2. Liquid Chromatography-Mass Spectrometry (LC-MS) Analysis
4.3. Preparation of Membrane Stained-Microglia
4.4. Microglial Cells Culture Platform
4.5. Proliferation of Neurons/Astrocytes
4.6. Differentiation of Neurons/Astrocytes
4.7. Microfluidic Chip Fabrication
4.8. Microglia Loading into the Microfluidic Chip
4.9. Fluorescent Immunostaining
4.10. Viability Assay
4.11. Nitric Oxide Measurement
4.12. Detection of Multiple Cytokines
4.13. Inhibition Assay
4.14. Fluorescence Imaging
4.15. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for alzheimer’s disease. Signal. Transduct. Target. Ther. 2019, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Diep, Y.N.; Tran, M.; Cho, H. Therapeutic targeting strategies for early-to late-staged alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 9591. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.Y.; Frost, G.R.; Wu, X.; Crump, C.; Pan, S.J.; Wong, E.; Barros, M.; Li, T.; Nie, P.; Zhai, Y.; et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature 2020, 586, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Ennerfelt, H.E.; Lukens, J.R. The role of innate immunity in Alzheimer’s disease. Immunol. Rev. 2020, 297, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Deuschl, G. Neuroinflammation—A common thread in neurological disorders. Nat. Rev. Neurol. 2019, 15, 429–430. [Google Scholar] [CrossRef]
- Shen, H.; Guan, Q.; Zhang, X.; Yuan, C.; Tan, Z.; Zhai, L.; Hao, Y.; Gu, Y.; Han, C. New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 100, 109884. [Google Scholar] [CrossRef]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; de Oliveira, A.C.P. Lipopolysaccharide-induced neuroinflammation as a bridge to understand neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293. [Google Scholar] [CrossRef] [Green Version]
- Angelucci, F.; Cechova, K.; Amlerova, J.; Hort, J. Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 1–10. [Google Scholar] [CrossRef]
- How, K.Y.; Song, K.P.; Chan, K.G. Porphyromonas gingivalis: An overview of periodontopathic pathogen below the gum line. Front. Microbiol. 2016, 7, 53. [Google Scholar] [CrossRef]
- Choi, S.; Kim, K.; Chang, J.; Kim, S.M.; Kim, S.J.; Cho, H.; Park, S.M. Association of Chronic Periodontitis on Alzheimer’s Disease or Vascular Dementia. J. Am. Geriatr. Soc. 2019, 67, 1234–1239. [Google Scholar] [CrossRef] [Green Version]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, S.; Singhrao, S.K.; Kesavalu, L.; Curtis, M.A.; Crean, S. Determining the Presence of Periodontopathic Virulence Factors in Short-Term Postmortem Alzheimer’s Disease Brain Tissue. Adv. Alzheimers Dis. 2017, 5, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Wing, M.L.; Rettori, V.; Al-Shekhlee, A.; Bongiorno, P.B.; Canteros, G.; Mccann, S.M.; Gold, P.W.; Licinio, J. Inducible nitric oxide synthase gene expression in the brain during systemic inflammation. Nat. Med. 1996, 2, 581–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, J.W.; Iturria-Medina, Y.; Strandberg, O.T.; Smith, R.; Levitis, E.; Evans, A.C.; Hansson, O. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 2020, 11, 2612. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Dervartanian, D.V.; Veeger, C. Studies on succinate dehydrogenase. I. Spectral properties of the purified enzyme and formation of enzyme-competitive inhibitor complexes. Biochim. Biophys. Acta (BBA) Enzymol. Subj. 1964, 92, 233–247. [Google Scholar] [CrossRef]
- Lehnardt, S. Innate immunity and neuroinflammation in the CNS: The role of microglia in toll-like receptor-mediated neuronal injury. Glia 2010, 58, 253–263. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, Z.; Nakanishi, Y.; Ni, J.; Hayashi, Y.; Takayama, F.; Zhou, Y.; Kadawaki, T.; Nakanishi, H. Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Klein, R.S.; Garber, C.; Funk, K.E.; Salimi, H.; Soung, A.; Kanmogne, M.; Manivasagam, S.; Agner, S.; Cain, M. Neuroinflammation During RNA Viral Infections. Annu. Rev. Immunol. 2019, 37, 73–95. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Huang, Z.; Sun, X.; Zhu, X.; Zhou, L.; Li, M.; Cheng, B.; Liu, X.; He, C. Microglia polarization with M1/M2 phenotype changes in rd1 mouse model of retinal degeneration. Front. Neuroanat. 2017, 11, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, X.; Han, X.; Li, Q.; Yang, Q.W.; Wang, J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 2017, 13, 420–433. [Google Scholar] [CrossRef] [Green Version]
- Alsadany, M.A.; Shehata, H.H.; Mohamad, M.I.; Mahfouz, R.G. Histone deacetylases enzyme, copper, and IL-8 levels in patients with Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demen. 2013, 28, 54–61. [Google Scholar] [CrossRef]
- Lue, L.F.; Rydel, R.; Brigham, E.F.; Yang, L.B.; Hampel, H.; Murphy, G.M.; Brachova, L.; Yan, S.D.; Walker, D.G.; Shen, Y.; et al. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 2001, 35, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Buckley, P.F. Intrathecal Chemokine Synthesis in Mild Cognitive Impairment and Alzheimer Disease. Yearb. Psychiatry Appl. Ment. Health 2007, 2007, 303–304. [Google Scholar] [CrossRef]
- Dorothée, G. Neuroinflammation in neurodegeneration: Role in pathophysiology, therapeutic opportunities and clinical perspectives. J. Neural Transm. 2018, 125, 749–750. [Google Scholar] [CrossRef] [Green Version]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Santos, R.; Clark, A.T.; Gkountela, S.; Chen, D.; Liu, W.; Sosa, E.; Sukhwani, M.; Hennebold, J.D.; Orwig, K.E. Differentiation of Inflammation-Responsive Astrocytes from Glial Progenitors Generated from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Thirumangalakudi, L.; Yin, L.; Rao, H.V.; Grammas, P. IL-8 induces expression of matrix metalloproteinases, cell cycle and pro-apoptotic proteins, and cell death in cultured neurons. J. Alzheimers Dis. 2007, 11, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, V.T.A.; Kang, Y.J.; Kim, H.-K.; Kim, H.-R.; Cho, H. Oral Pathogenic Bacteria-Inducing Neurodegenerative Microgliosis in Human Neural Cell Platform. Int. J. Mol. Sci. 2021, 22, 6925. https://doi.org/10.3390/ijms22136925
Tran VTA, Kang YJ, Kim H-K, Kim H-R, Cho H. Oral Pathogenic Bacteria-Inducing Neurodegenerative Microgliosis in Human Neural Cell Platform. International Journal of Molecular Sciences. 2021; 22(13):6925. https://doi.org/10.3390/ijms22136925
Chicago/Turabian StyleTran, Van Thi Ai, You Jung Kang, Hyun-Kyoung Kim, Hyung-Ryong Kim, and Hansang Cho. 2021. "Oral Pathogenic Bacteria-Inducing Neurodegenerative Microgliosis in Human Neural Cell Platform" International Journal of Molecular Sciences 22, no. 13: 6925. https://doi.org/10.3390/ijms22136925
APA StyleTran, V. T. A., Kang, Y. J., Kim, H. -K., Kim, H. -R., & Cho, H. (2021). Oral Pathogenic Bacteria-Inducing Neurodegenerative Microgliosis in Human Neural Cell Platform. International Journal of Molecular Sciences, 22(13), 6925. https://doi.org/10.3390/ijms22136925