
Perturbations of the Proteome and of Secreted Metabolites in Primary Astrocytes from the hSOD1(G93A) ALS Mouse Model

 , , , and
, , , and
Abstract
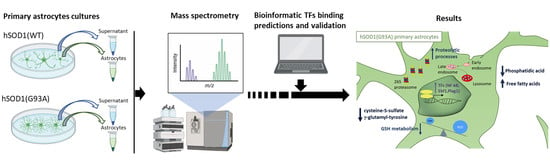
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Conditioned Medium Analysis by Ultra-High Performance Liquid Chromatography-High Resolution Mass Spectrometry (UHPLC-HRMS)
2.2. Proteomics Analysis of Primary Cultured Spinal Astrocytes
2.3. Validation of TMT-Based Proteomic Results by Western Blot (WB) Analysis
2.4. Computational Identification of Altered TFs in hSOD1(G93A) Astrocytes
2.5. Evaluation of TF Alterations in hSOD1(G93A) Primary Astrocytes
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Primary Cultures of Murine Spinal Cord Astrocytes
4.3. Untargeted Metabolomic Analysis of the Secretome of Primary Cultures of Astrocytes
4.4. Metabolomics Data Analysis
4.5. Astrocytes Lysis and Protein Extraction
4.6. Proteomics Profiling of Primary Astrocytes
4.7. UHPLC-HRMS/MS Analysis
4.8. Untargeted MS Data Analysis
4.9. Subcellular Fractionation
4.10. WB Analysis and Antibodies
4.11. Promoters Analysis
4.12. Total RNA Extraction and Retrotranscription
4.13. Primer Design and Quantitative Real-Time PCR (qRT-PCR) Analysis
4.14. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cleveland, D.W.; Rothstein, J.D. From charcot to lou gehrig: Deciphering selective motor neuron death in als. Nat. Rev. Neurosci. 2001, 2, 806–819. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17085. [Google Scholar] [CrossRef] [PubMed]
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Berg, L.H.V.D. Therapy of amyotrophic lateral sclerosis remains a challenge. Lancet Neurol. 2014, 13, 1062–1063. [Google Scholar] [CrossRef]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laferriere, F.; Polymenidou, M. Advances and challenges in understanding the multifaceted pathogenesis of amyotrophic lateral sclerosis. Swiss Med. Wkly. 2015, 145, w14054. [Google Scholar] [CrossRef]
- Taylor, J.P., Jr.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.C.; Hentati, A.; Donaldson, D.H.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Pfohl, S.R.; Halicek, M.T.; Mitchell, C.S. Characterization of the Contribution of Genetic Background and Gender to Disease Progression in the SOD1 G93A Mouse Model of Amyotrophic Lateral Sclerosis: A Meta-Analysis. J. Neuromuscul. Dis. 2015, 2, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Hyeon, S.J.; Im, H.; Ryu, H.; Kim, Y.; Ryu, H. Astrocytes and Microglia as Non-cell Autonomous Players in the Pathogenesis of ALS. Exp. Neurobiol. 2016, 25, 233–240. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Huang, H.; Peterson, A.; Hu, B.; Maragakis, N.J.; Ming, G.-L.; Chen, H.; Zhang, S.-C. Sporadic ALS Astrocytes Induce Neuronal Degeneration In Vivo. Stem Cell Rep. 2017, 8, 843–855. [Google Scholar] [CrossRef]
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of Disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; McManus, R.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Valbuena, G.N.; Tortarolo, M.; Bendotti, C.; Cantoni, L.; Keun, H.C. Altered Metabolic Profiles Associate with Toxicity in SOD1G93A Astrocyte-Neuron Co-Cultures. Sci. Rep. 2017, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Veyrat-Durebex, C.; Corcia, P.; Piver, E.; Devos, D.; Dangoumau, A.; Gouel, F.; Vourc, H.P.; Emond, P.; Laumonnier, F.; Nadal-Desbarats, L.; et al. Disruption of TCA Cycle and Glutamate Metabolism Identified by Metabolomics in an In Vitro Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2015, 53, 6910–6924. [Google Scholar] [CrossRef]
- Hounoum, B.M.; Mavel, S.; Coque, E.; Patin, F.; Vourc, H.P.; Marouillat, S.; Nadal-Desbarats, L.; Emond, P.; Corcia, P.; Andres, C.; et al. Wildtype motoneurons, ALS-Linked SOD1 mutation and glutamate profoundly modify astrocyte metabolism and lactate shuttling. Glia 2017, 65, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; De León, A.; Robinson, K.M.; Mason, R.P.; Beckman, J.S.; Barbeito, L.; et al. Mitochondrial Dysfunction in SOD1G93A-Bearing Astrocytes Promotes Motor Neuron Degeneration: Prevention by Mitochondrial-Targeted Antioxidants. J. Neurosci. 2008, 28, 4115–4122. [Google Scholar] [CrossRef] [Green Version]
- Martorana, F.; Brambilla, L.; Valori, C.F.; Bergamaschi, C.; Roncoroni, C.; Aronica, E.; Volterra, A.; Bezzi, P.; Rossi, D. The BH4 domain of Bcl-X(L) rescues astrocyte degeneration in amyotrophic lateral sclerosis by modulating intracellular calcium signals. Hum. Mol. Genet. 2012, 21, 826–840. [Google Scholar] [CrossRef] [Green Version]
- Kawamata, H.; Ng, S.K.; Diaz, N.; Burstein, S.; Morel, L.; Osgood, A.; Sider, B.; Higashimori, H.; Haydon, P.G.; Manfredi, G.; et al. Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 2331–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norante, R.P.; Peggion, C.; Rossi, D.; Martorana, F.; De Mario, A.; Lia, A.; Massimino, M.L.; Bertoli, A.; Mario, D.; Lia, A.L.S. Associated SOD1(G93A) Decreases SERCA Pump Levels and Increases Store-Operated Ca2 + Entry in Primary Spinal Cord Astrocytes from a Transgenic Mouse Model. Int. J. Mol. Sci. 2019, 20, 5151. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, F.P.; Carrasco, M.; Siao, M.C.; Maniatis, T.; Eggan, K. Non–cell autonomous effect of glia on motor neurons in an embryonic stem cell–based ALS model. Nat. Neurosci. 2007, 10, 608–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, P.; Rodriguez-Muela, N.; Klim, J.R.; de Boer, A.S.; Agrawal, S.; Sandoe, J.; Lopes, C.S.; Ogliari, K.S.; Williams, L.A.; Shear, M.; et al. Reactive Astrocytes Promote ALS-like Degeneration and Intracellular Protein Aggregation in Human Motor Neurons by Disrupting Autophagy through TGF-beta. Stem Cell Rep. 2017, 9, 667–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Côté, R.G.; Reisinger, F.; Barsnes, H.; Foster, J.M.; Rameseder, J.; Hermjakob, H.; Martens, L. The Proteomics Identifications database: 2010 update. Nucleic Acids Res. 2009, 38, D736–D742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Cha, S.J.; Choi, H.-J.; Kim, K. Omega Class Glutathione S-Transferase: Antioxidant Enzyme in Pathogenesis of Neurodegenerative Diseases. Oxidative Med. Cell. Longev. 2017, 2017, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Van De Giessen, E.; Fogh, I.; Gopinath, S.; Smith, B.; Hu, X.; Powell, J.; Andersen, P.; Nicholson, G.; Al Chalabi, A.; Shaw, C.E. Association study on glutathione S?transferase omega 1 and 2 and familial ALS. Amyotroph. Lateral Scler. 2008, 9, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.; Ke, Y.; Luo, C.; Gozal, D.; Liu, R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience 2007, 144, 991–1003. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Bai, Z.; Qin, X.; Cheng, Y. Aberrations in Oxidative Stress Markers in Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Oxidative Med. Cell. Longev. 2019, 2019, 1–9. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, G.; Calcagno, E.; Tartari, S.; Rizzardini, M.; Invernizzi, R.W.; Cantoni, L. Glutamate and glutathione interplay in a motor neuronal model of amyotrophic lateral sclerosis reveals altered energy metabolism. Neurobiol. Dis. 2011, 43, 346–355. [Google Scholar] [CrossRef]
- Silacci, P.; Mazzolai, L.; Gauci, C.; Stergiopulos, N.; Yin, H.L.; Hayoz, D. Gelsolin superfamily proteins: Key regulators of cellular functions. Cell. Mol. Life Sci. 2004, 61, 2614–2623. [Google Scholar] [CrossRef] [Green Version]
- Merienne, N.; Meunier, C.; Schneider, A.; Seguin, J.; Nair, S.S.; Rocher, A.B.; Le Gras, S.; Keime, C.; Faull, R.; Pellerin, L.; et al. Cell-Type-Specific Gene Expression Profiling in Adult Mouse Brain Reveals Normal and Disease-State Signatures. Cell Rep. 2019, 26, 2477–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, H.; Fujii, Y.; Kuwayama, N.; Kawaji, K.; Gotoh, Y.; Kishi, Y. Plag1 regulates neuronal gene expression and neuronal differentiation of neocortical neural progenitor cells. Genes Cells 2019, 24, 650–666. [Google Scholar] [CrossRef]
- Chung, Y.H.; Joo, K.M.; Lee, Y.J.; Kim, M.J.; Cha, C.I. Reactive astrocytes express cAMP-response-element-binding protein (CREB) binding protein (CBP) in the central nervous system of transgenic mice expressing a human Cu/Zn superoxide dismutase mutation. Neurosci. Lett. 2003, 343, 159–162. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L. The non-cell-autonomous component of ALS: New in vitro models and future challenges. Biochem. Soc. Trans. 2014, 42, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Rostalski, H.; Leskelä, S.; Huber, N.; Katisko, K.; Cajanus, A.; Solje, E.; Marttinen, M.; Natunen, T.; Remes, A.M.; Hiltunen, M.; et al. Astrocytes and Microglia as Potential Contributors to the Pathogenesis of C9orf72 Repeat Expansion-Associated FTLD and ALS. Front. Neurosci. 2019, 13, 486. [Google Scholar] [CrossRef]
- Gil, R.S.; Clarke, B.; Ecroyd, H.; Kalmar, B.; Greensmith, L. Regional Differences in Heat Shock Protein 25 Expression in Brain and Spinal Cord Astrocytes of Wild-Type and SOD1 G93A Mice. Cells 2021, 10, 1257. [Google Scholar] [CrossRef]
- Baker, D.J.; Blackburn, D.J.; Keatinge, M.; Sokhi, D.; Viskaitis, P.; Heath, P.R.; Ferraiuolo, L.; Kirby, J.; Shaw, P. Lysosomal and phagocytic activity is increased in astrocytes during disease progression in the SOD1 G93 A mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2015, 9, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetto, M.C.; Muotri, A.R.; Mu, Y.; Smith, A.M.; Cezar, G.G.; Gage, F.H. Non-Cell-Autonomous Effect of Human SOD1G37R Astrocytes on Motor Neurons Derived from Human Embryonic Stem Cells. Cell Stem Cell 2008, 3, 649–657. [Google Scholar] [CrossRef] [Green Version]
- Tortarolo, M.; Vallarola, A.; Lidonnici, D.; Battaglia, E.; Gensano, F.; Spaltro, G.; Fiordaliso, F.; Corbelli, A.; Garetto, S.; Martini, E.; et al. Lack of TNF-alpha receptor type 2 protects motor neurons in a cellular model of amyotrophic lateral sclerosis and in mutant SOD1 mice but does not affect disease progression. J. Neurochem. 2015, 135, 109–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendonça, D.; Chimelli, L.; Martinez, A. Expression of ubiquitin and proteasome in motorneurons and astrocytes of spinal cords from patients with amyotrophic lateral sclerosis. Neurosci. Lett. 2006, 404, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Seilhean, D.; Takahashi, J.; El Hachimi, K.H.; Fujigasaki, H.; Lebre, A.-S.; Biancalana, V.; Durr, A.; Salachas, F.; Hogenhuis, J.; De Thé, H.; et al. Amyotrophic lateral sclerosis with neuronal intranuclear protein inclusions. Acta Neuropathol. 2004, 108, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Dykes-Hoberg, M.; Culotta, V.C.; Price, D.L.; Wong, P.C.; Rothstein, J.D. Histological Evidence of Protein Aggregation in Mutant SOD1 Transgenic Mice and in Amyotrophic Lateral Sclerosis Neural Tissues. Neurobiol. Dis. 2001, 8, 933–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pišlar, A.; Kos, J. Cysteine Cathepsins in Neurological Disorders. Mol. Neurobiol. 2014, 49, 1017–1030. [Google Scholar] [CrossRef]
- Nassif, M.; Valenzuela, V.; Rojas-Rivera, D.; Vidal, R.; Matus, S.; Castillo, K.; Fuentealba, Y.; Kroemer, G.; Levine, B.; Hetz, C. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy 2014, 10, 1256–1271. [Google Scholar] [CrossRef] [Green Version]
- Conus, S.; Simon, H.-U. Cathepsins: Key modulators of cell death and inflammatory responses. Biochem. Pharmacol. 2008, 76, 1374–1382. [Google Scholar] [CrossRef]
- Fukada, Y.; Yasui, K.; Kitayama, M.; Doi, K.; Nakano, T.; Watanabe, Y.; Nakashima, K. Gene expression analysis of the murine model of amyotrophic lateral sclerosis: Studies of the Leu126delTT mutation in SOD. Brain Res. 2007, 1160, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Aguilar, J.-L.G.; Niederhauser-Wiederkehr, C.; Halter, B.; de Tapia, M.; Di Scala, F.; Demougin, P.; Dupuis, L.; Primig, M.; Meininger, V.; Loeffler, J.-P.; et al. Gene profiling of skeletal muscle in an amyotrophic lateral sclerosis mouse model. Physiol. Genom. 2008, 32, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Wendt, W.; Lufcbbert, H.; Stichel, C.C. Upregulation of cathepsin S in the aging and pathological nervous system of mice. Brain Res. 2008, 1232, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Wootz, H.; Weber, E.; Korhonen, L.; Lindholm, D. Altered distribution and levels of cathepsinD and cystatins in amyotrophic lateral sclerosis transgenic mice: Possible roles in motor neuron survival. Neuroscience 2006, 143, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Yamada, T.; Furuya, H.; Doh-Ura, K.; Ohyagi, Y.; Iwaki, T.; Kira, J.-I. Involvement of cathepsin B in the motor neuron degeneration of amyotrophic lateral sclerosis. Acta Neuropathol. 2003, 105, 462–468. [Google Scholar] [CrossRef]
- Tropak, M.B.; Mahuran, D. Lending a helping hand, screening chemical libraries for compounds that enhance β-hexosaminidase A activity in GM2 gangliosidosis cells. FEBS J. 2007, 274, 4951–4961. [Google Scholar] [CrossRef] [Green Version]
- Konishi, H.; Okamoto, T.; Hara, Y.; Komine, O.; Tamada, H.; Maeda, M.; Osako, F.; Kobayashi, M.; Nishiyama, A.; Kataoka, Y.; et al. Astrocytic phagocytosis is a compensatory mechanism for microglial dysfunction. EMBO J. 2020, 39, e104464. [Google Scholar] [CrossRef]
- Martínez, A.; Mas, A.; Heras, V.D.L.; Arroyo, R.; Fernández-Arquero, M.; De La Concha, E.G.; Urcelay, E. Early B-cell Factor gene association with multiple sclerosis in the Spanish population. BMC Neurol. 2005, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Swarup, V.; Phaneuf, D.; Dupré, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.-P. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB-mediated pathogenic pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Haidet-Phillips, A.M.; Hester, M.; Miranda, C.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [Green Version]
- Crosio, C.; Valle, C.; Casciati, A.; Iaccarino, C.; Carrì, M.T. Astroglial Inhibition of NF-κB Does Not Ameliorate Disease Onset and Progression in a Mouse Model for Amyotrophic Lateral Sclerosis (ALS). PLoS ONE 2011, 6, e17187. [Google Scholar] [CrossRef] [Green Version]
- Pollari, E.; Goldsteins, G.; Bart, G.; Koistinaho, J.; Giniatullin, R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 131. [Google Scholar] [CrossRef] [Green Version]
- Blasco, H.; Garcon, G.; Patin, F.; Veyrat-Durebex, C.; Boyer, J.; Devos, D.; Vourc’H, P.; Andres, C.; Corcia, P. Panel of Oxidative Stress and Inflammatory Biomarkers in ALS: A Pilot Study. Can. J. Neurol. Sci. 2017, 44, 90–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.-M. Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochem. Res. 2015, 40, 2570–2582. [Google Scholar] [CrossRef]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta—Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Von Neuhoff, N.; Dengler, R.; Petri, S. Nuclear Erythroid 2-Related Factor 2-Antioxidative Response Element Signaling Pathway in Motor Cortex and Spinal Cord in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Petri, S.; Körner, S.; Kiaei, M. Nrf2/ARE Signaling Pathway: Key Mediator in Oxidative Stress and Potential Therapeutic Target in ALS. Neurol. Res. Int. 2012, 2012, 1–7. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Yadav, S. The glutathione cycle: Glutathione metabolism beyond the γ-glutamyl cycle. IUBMB Life 2018, 70, 585–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofyanovich, O.A.; Nishiuchi, H.; Yamagishi, K.; Matrosova, E.V.; Serebrianyi, V.A. Multiple pathways for the formation of the γ-glutamyl peptides γ-glutamyl-valine and γ- glutamyl-valyl-glycine in Saccharomyces cerevisiae. PLoS ONE 2019, 14, e0216622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.; Singh, R.; Kalita, J.; Misra, U. Oxidant–antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochem. Int. 2008, 52, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Kuźma, M.; Jamrozik, Z.; Barańczyk-Kuźma, A. Activity and expression of glutathione S-transferase pi in patients with amyotrophic lateral sclerosis. Clin. Chim. Acta 2006, 364, 217–221. [Google Scholar] [CrossRef]
- Cha, S.J.; Han, Y.J.; Choi, H.-J.; Kim, H.-J.; Kim, K. Glutathione S-Transferase Rescues Motor Neuronal Toxicity in Fly Model of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 615. [Google Scholar] [CrossRef] [PubMed]
- Filho, A.D.B.C.; Pinto, I.F.D.; Dantas, L.S.; Xavier, A.M.; Inague, A.; Faria, R.L.; Medeiros, M.H.G.; Glezer, I.; Yoshinaga, M.Y.; Miyamoto, S. Alterations in lipid metabolism of spinal cord linked to amyotrophic lateral sclerosis. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Tracey, T.; Kirk, S.; Steyn, F.; Ngo, S. The role of lipids in the central nervous system and their pathological implications in amyotrophic lateral sclerosis. Semin. Cell Dev. Biol. 2020, 112, 69–81. [Google Scholar] [CrossRef]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal Lipid Metabolism: Multiple Pathways Driving Functional Outcomes in Health and Disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.-N.; Lee, J.; Seo, K.; Lee, H. Astrocytic Regulation of Neural Circuits Underlying Behaviors. Cells 2021, 10, 296. [Google Scholar] [CrossRef]
- Zhu, Y.-B.; Gao, W.; Zhang, Y.; Jia, F.; Zhang, H.-L.; Liu, Y.-Z.; Sun, X.-F.; Yin, Y.; Yin, D.-M. Astrocyte-derived phosphatidic acid promotes dendritic branching. Sci. Rep. 2016, 6, 21096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P. The Glutamate Hypothesis in ALS: Pathophysiology and Drug Development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef]
- Dal Canto, M.C.; Gurney, M.E. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu,Zn SOD, and in mice overexpressing wild type human SOD: A model of familial amyotrophic lateral sclerosis (FALS). Brain Res. 1995, 676, 25–40. [Google Scholar] [CrossRef]
- Norante, R.P.; Massimino, M.L.; Lorenzon, P.; De Mario, A.; Peggion, C.; Vicario, M.; Albiero, M.; Sorgato, M.C.; Lopreiato, R.; Bertoli, A. Generation and validation of novel adeno-associated viral vectors for the analysis of Ca2 + homeostasis in motor neurons. Sci. Rep. 2017, 7, 6521. [Google Scholar] [CrossRef] [Green Version]
- Gomez, E.; Muñoz, M.; Simó, C.; Ibáñez, C.; Carrocera, S.; Martín-González, D.; Cifuentes, A. Non-invasive metabolomics for improved determination of embryonic sex markers in chemically defined culture medium. J. Chromatogr. A 2016, 1474, 138–144. [Google Scholar] [CrossRef]
- Stella, R.; Bovo, D.; Mastrorilli, E.; Manuali, E.; Pezzolato, M.; Bozzetta, E.; Lega, F.; Angeletti, R.; Biancotto, G. A novel tool to screen for treatments with clenbuterol in bovine: Identification of two hepatic markers by metabolomics investigation. Food Chem. 2021, 353, 129366. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Biancotto, G.; Bovo, D.; Mastrorilli, E.; Manuali, E.; Angeletti, R.; Stella, R. TMT-Based Proteomics Profiling of Bovine Liver Underscores Protein Markers of Anabolic Treatments. Proteomics 2019, 19, e1800422. [Google Scholar] [CrossRef]
- Peggion, C.; Massimino, M.L.; Stella, R.; Bortolotto, R.; Agostini, J.; Maldi, A.; Sartori, G.; Tonello, F.; Bertoli, A.; Lopreiato, R. Nucleolin Rescues TDP-43 Toxicity in Yeast and Human Cell Models. Front. Cell. Neurosci. 2021, 15, 115. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Moritz, C.P. Tubulin or Not Tubulin: Heading Toward Total Protein Staining as Loading Control in Western Blots. Proteomics 2017, 17, 1600189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, A.T.; Arenillas, D.J.; Worsley Hunt, R.; Wasserman, W.W. oPOSSUM-3: Advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 2012, 2, 987–1002. [Google Scholar] [CrossRef]
- Zambelli, F.; Pesole, G.; Pavesi, G. Pscan: Finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res. 2009, 37, W247–W252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defrance, M.; Touzet, H. Predicting transcription factor binding sites using local over-representation and comparative genomics. BMC Bioinform. 2006, 7, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandelin, A.; Alkema, W.; Engström, P.; Wasserman, W.; Lenhard, B. JASPAR: An open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004, 32, 91D–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveros, J.C. Venny. An interactive tool for comparing lists with Venn Diagrams. 2007. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 28 June 2021).






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stella, R.; Bonadio, R.S.; Cagnin, S.; Massimino, M.L.; Bertoli, A.; Peggion, C. Perturbations of the Proteome and of Secreted Metabolites in Primary Astrocytes from the hSOD1(G93A) ALS Mouse Model. Int. J. Mol. Sci. 2021, 22, 7028. https://doi.org/10.3390/ijms22137028
Stella R, Bonadio RS, Cagnin S, Massimino ML, Bertoli A, Peggion C. Perturbations of the Proteome and of Secreted Metabolites in Primary Astrocytes from the hSOD1(G93A) ALS Mouse Model. International Journal of Molecular Sciences. 2021; 22(13):7028. https://doi.org/10.3390/ijms22137028
Chicago/Turabian StyleStella, Roberto, Raphael Severino Bonadio, Stefano Cagnin, Maria Lina Massimino, Alessandro Bertoli, and Caterina Peggion. 2021. "Perturbations of the Proteome and of Secreted Metabolites in Primary Astrocytes from the hSOD1(G93A) ALS Mouse Model" International Journal of Molecular Sciences 22, no. 13: 7028. https://doi.org/10.3390/ijms22137028
APA StyleStella, R., Bonadio, R. S., Cagnin, S., Massimino, M. L., Bertoli, A., & Peggion, C. (2021). Perturbations of the Proteome and of Secreted Metabolites in Primary Astrocytes from the hSOD1(G93A) ALS Mouse Model. International Journal of Molecular Sciences, 22(13), 7028. https://doi.org/10.3390/ijms22137028