
A Transcriptome-Wide Isoform Landscape of Melanocytic Nevi and Primary Melanomas Identifies Gene Isoforms Associated with Malignancy

 , ,
, ,  and
and
Abstract
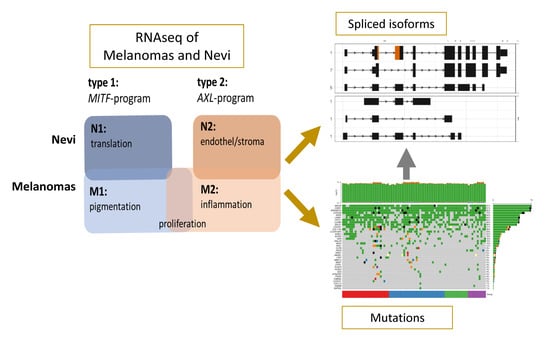
:
1. Introduction
2. Results
2.1. The Transcriptional and Isoform Landscape of Primary Melanomas
2.2. Isoform Switches
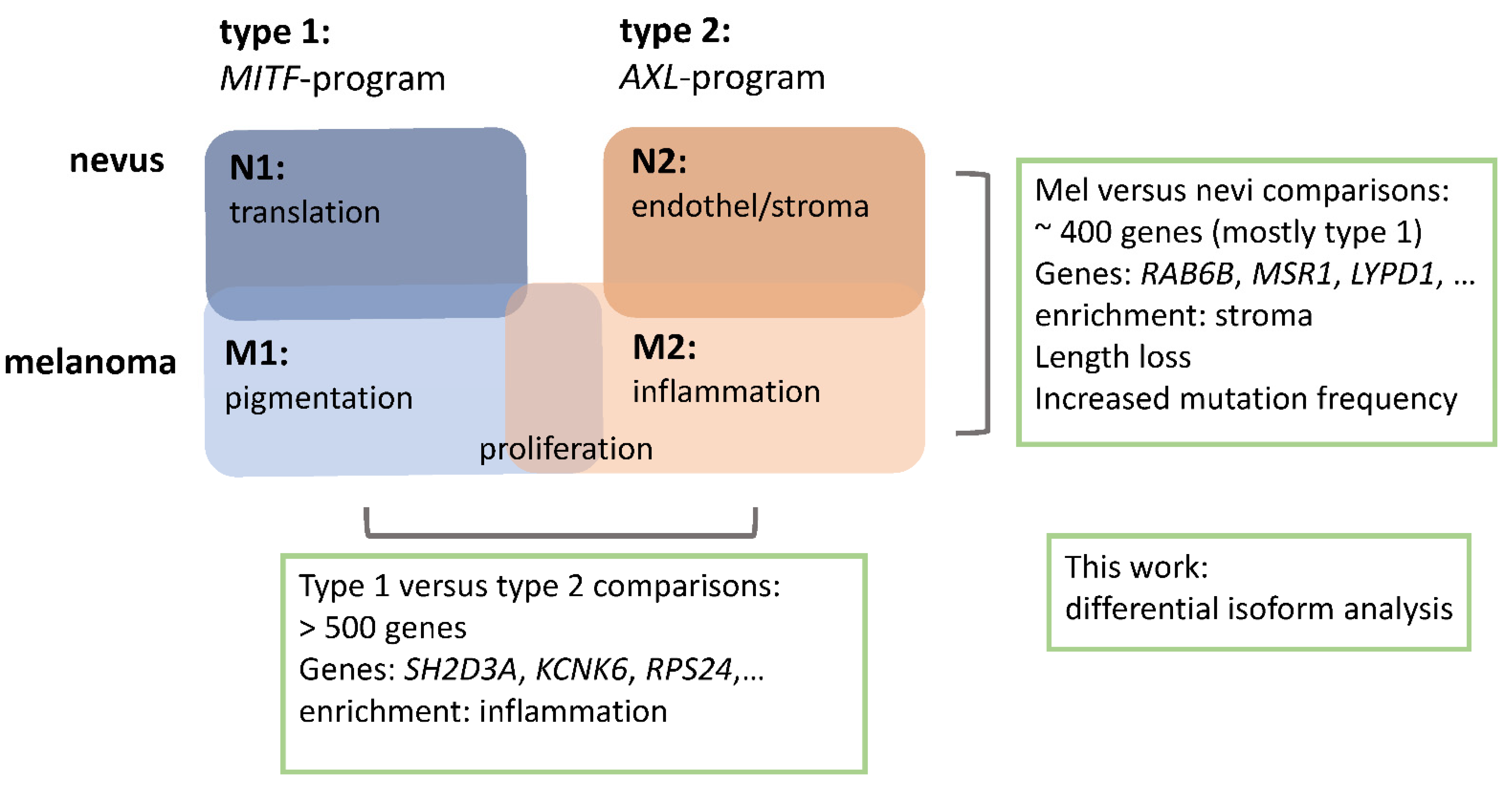
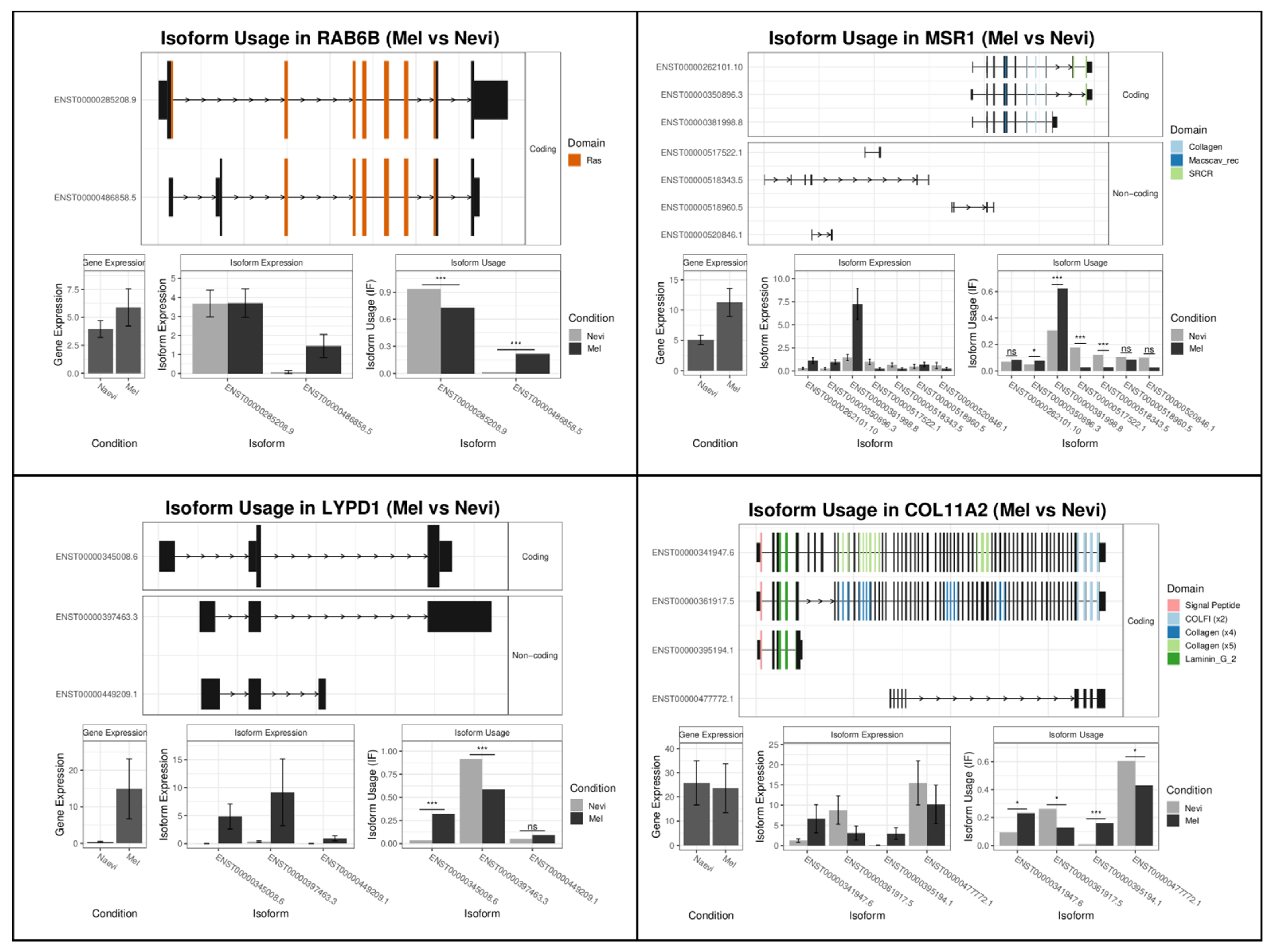
2.2.1. Melanomas vs. Nevi
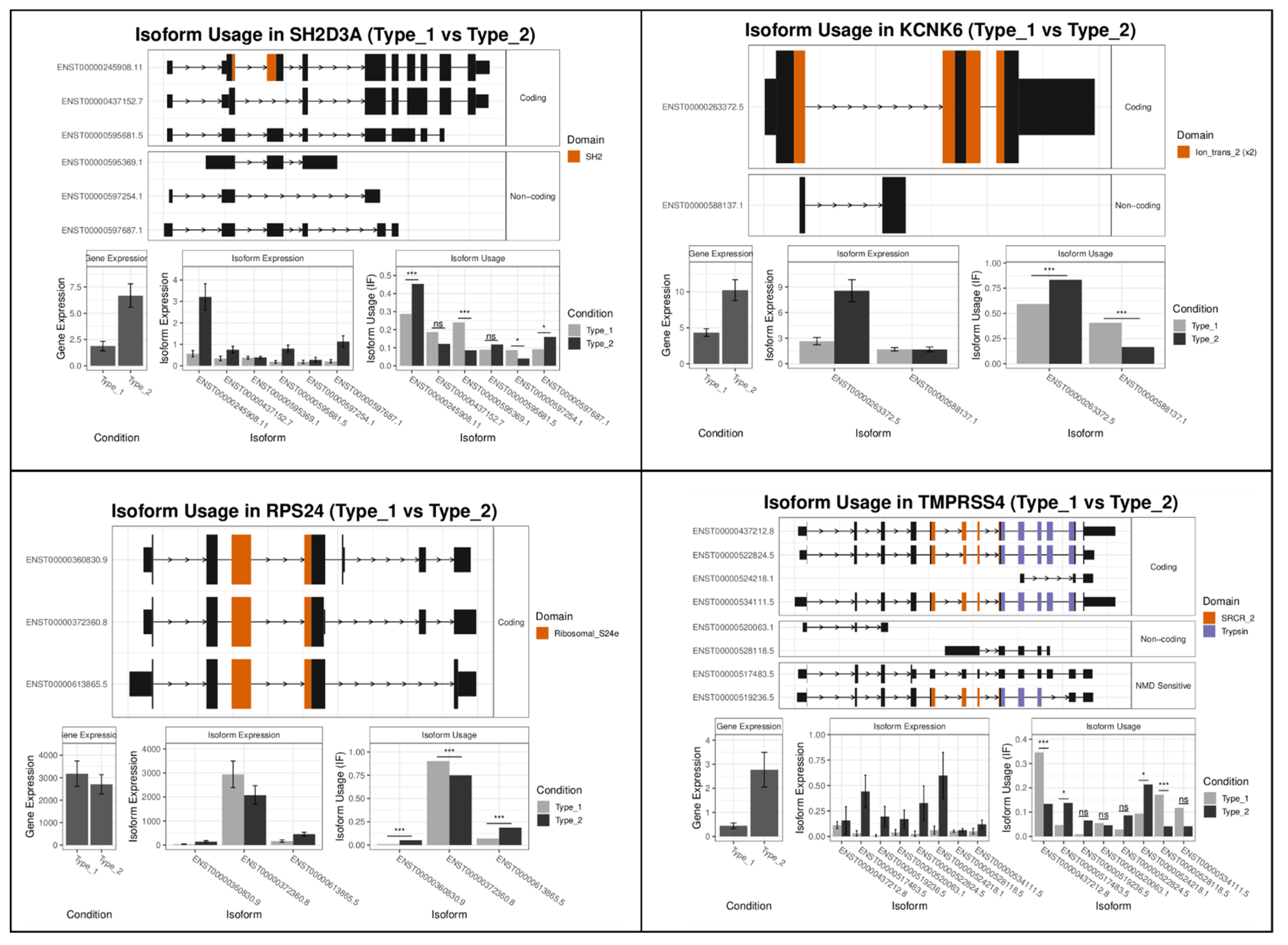
2.2.2. Type 1 vs. Type 2 Lesions
2.3. Functional Categories of Enriched Splice Variants
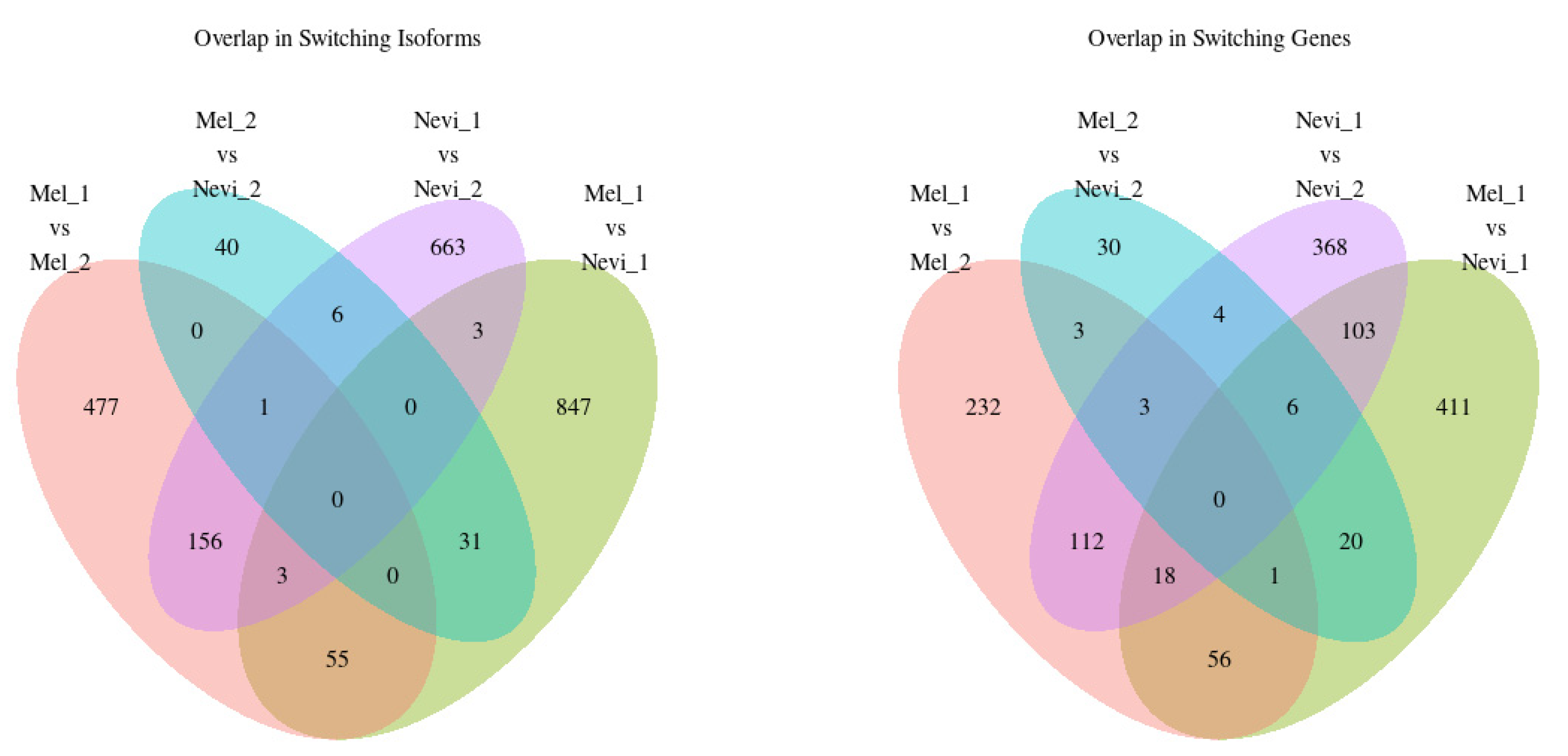
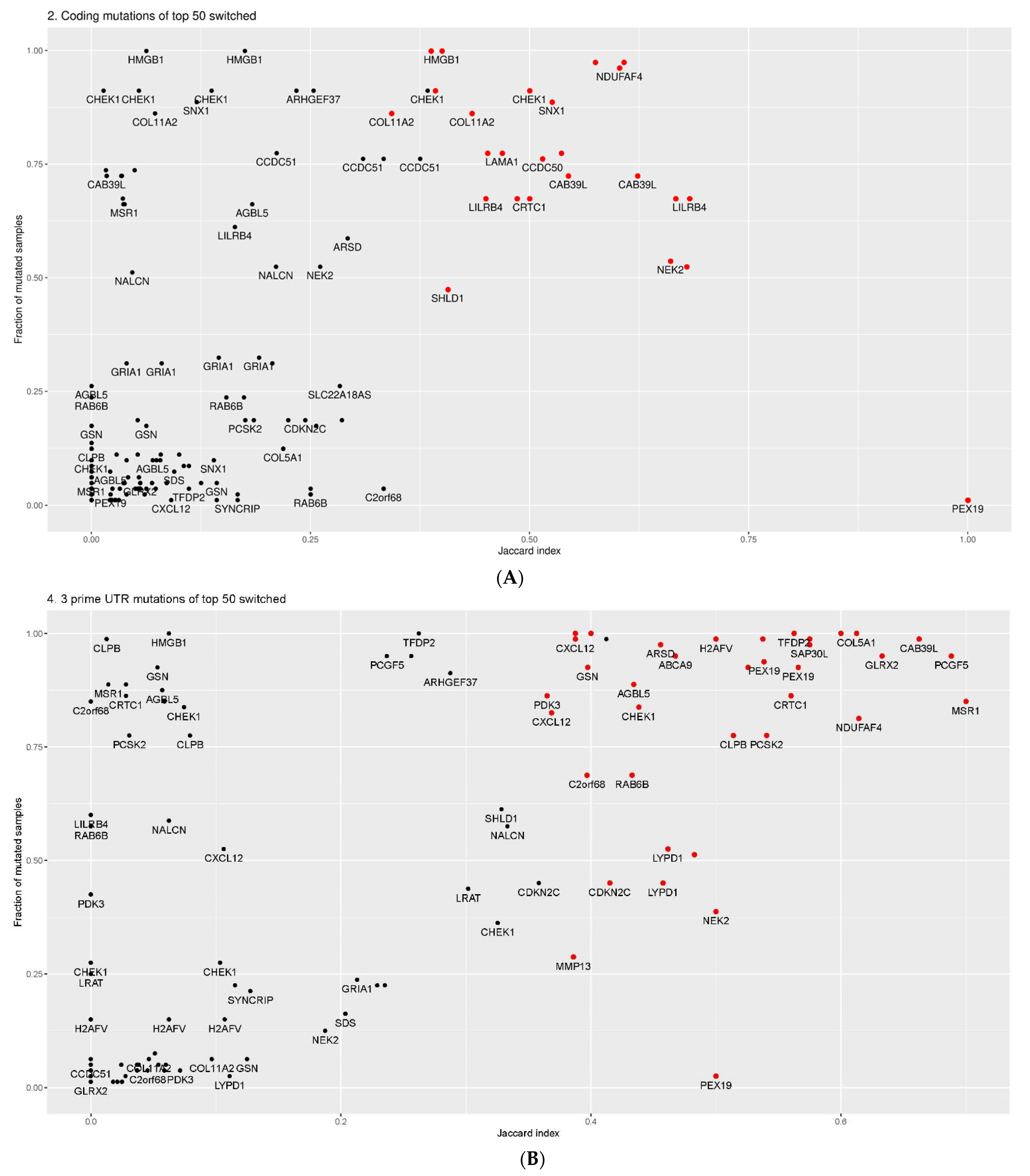
2.4. Overlap between Isoform Switches
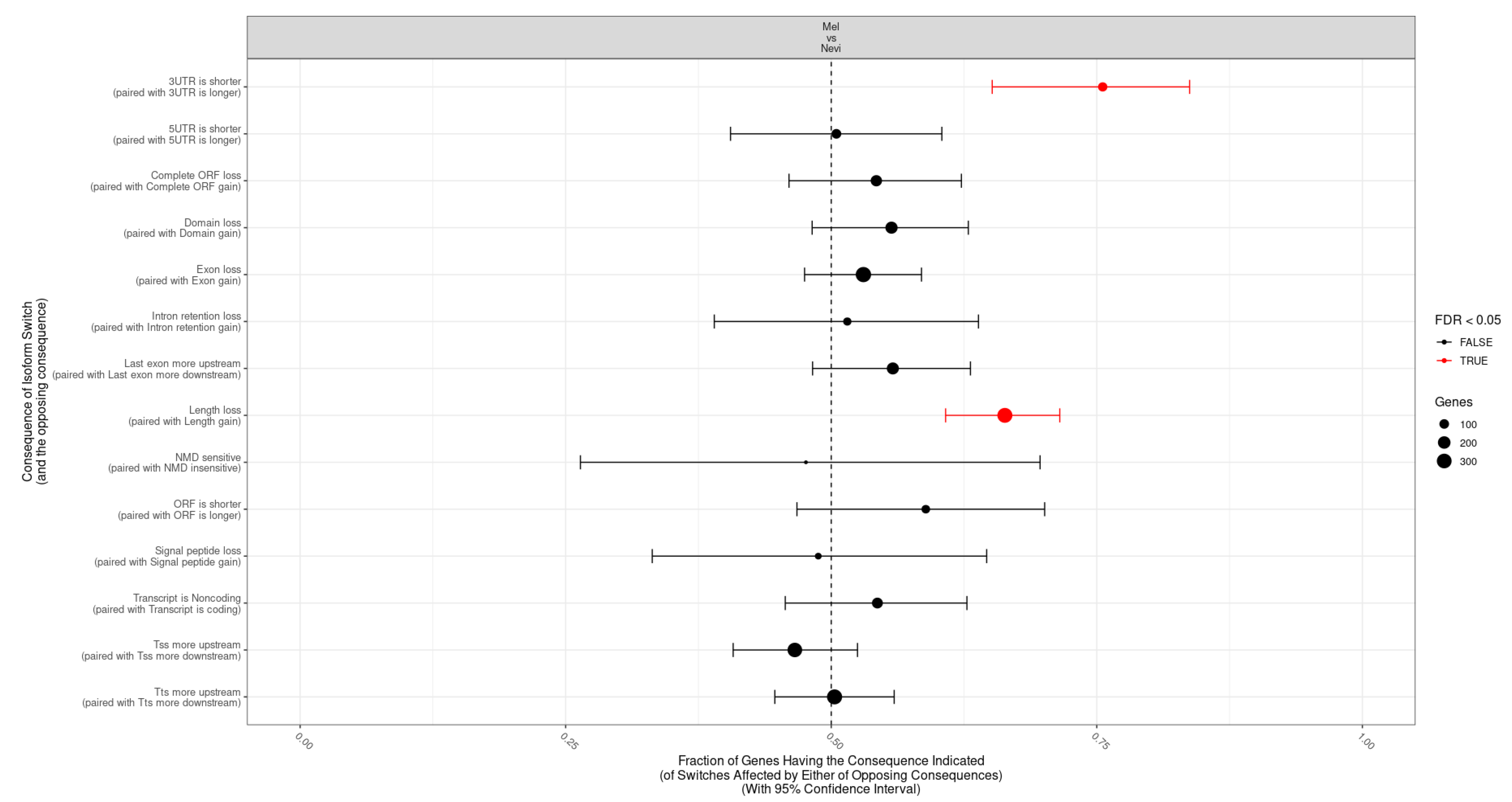
2.5. Categories of Isoform Switching
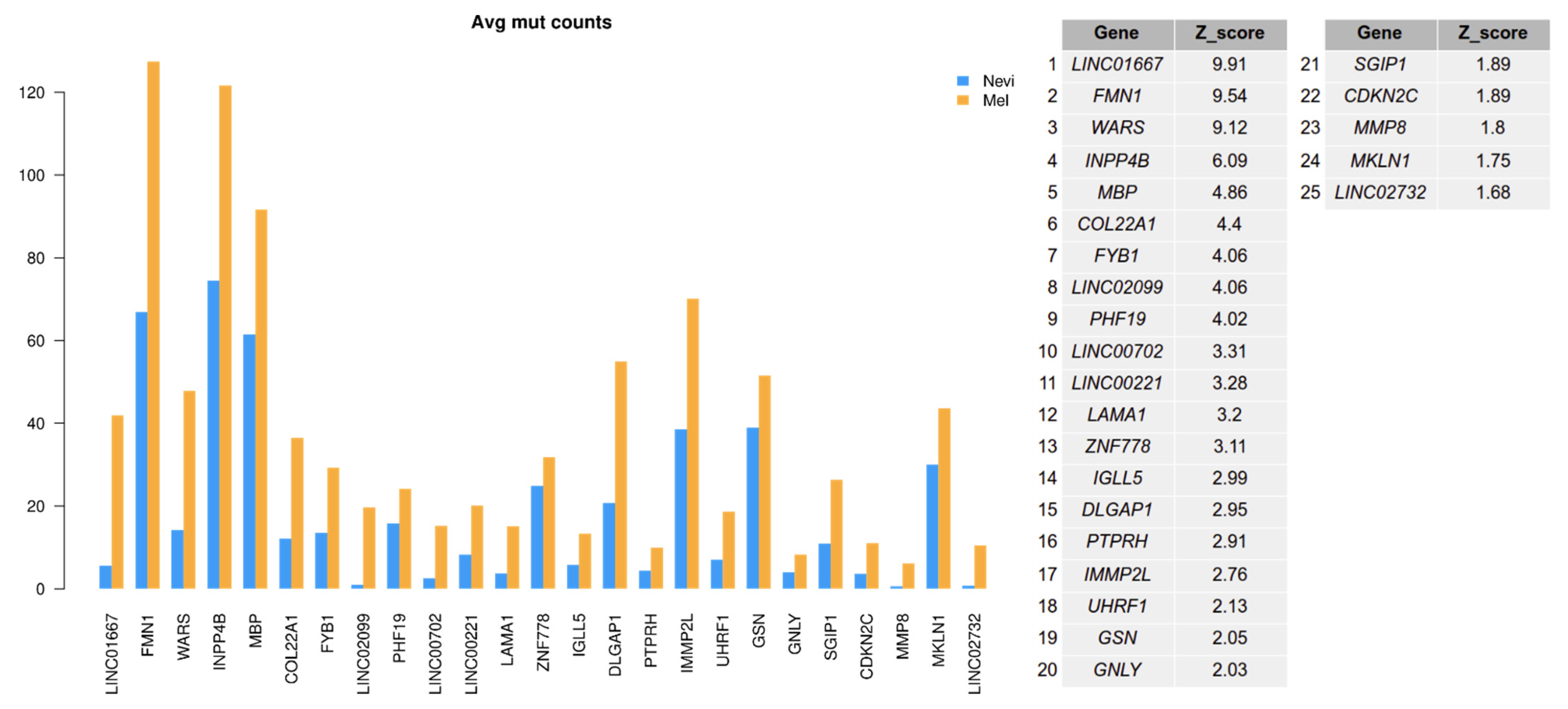
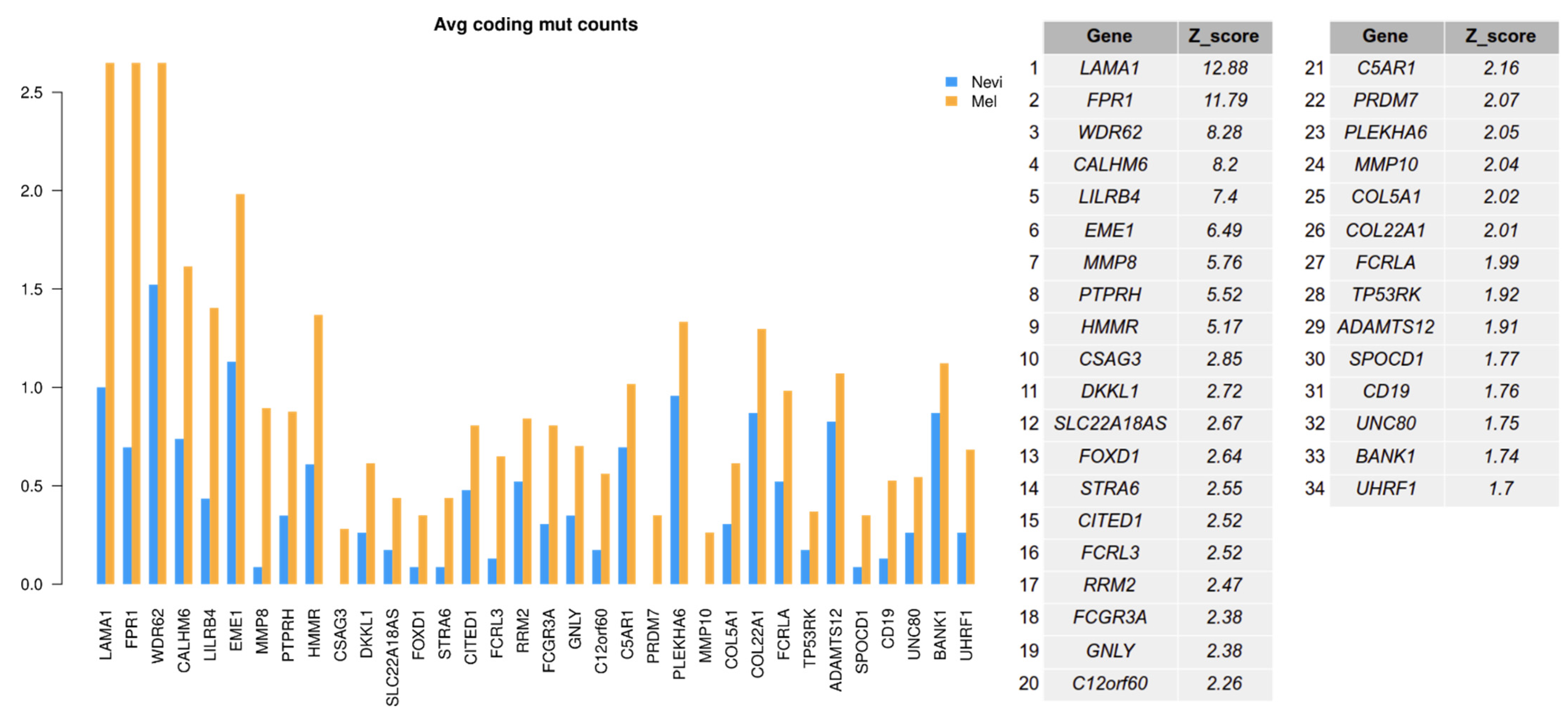
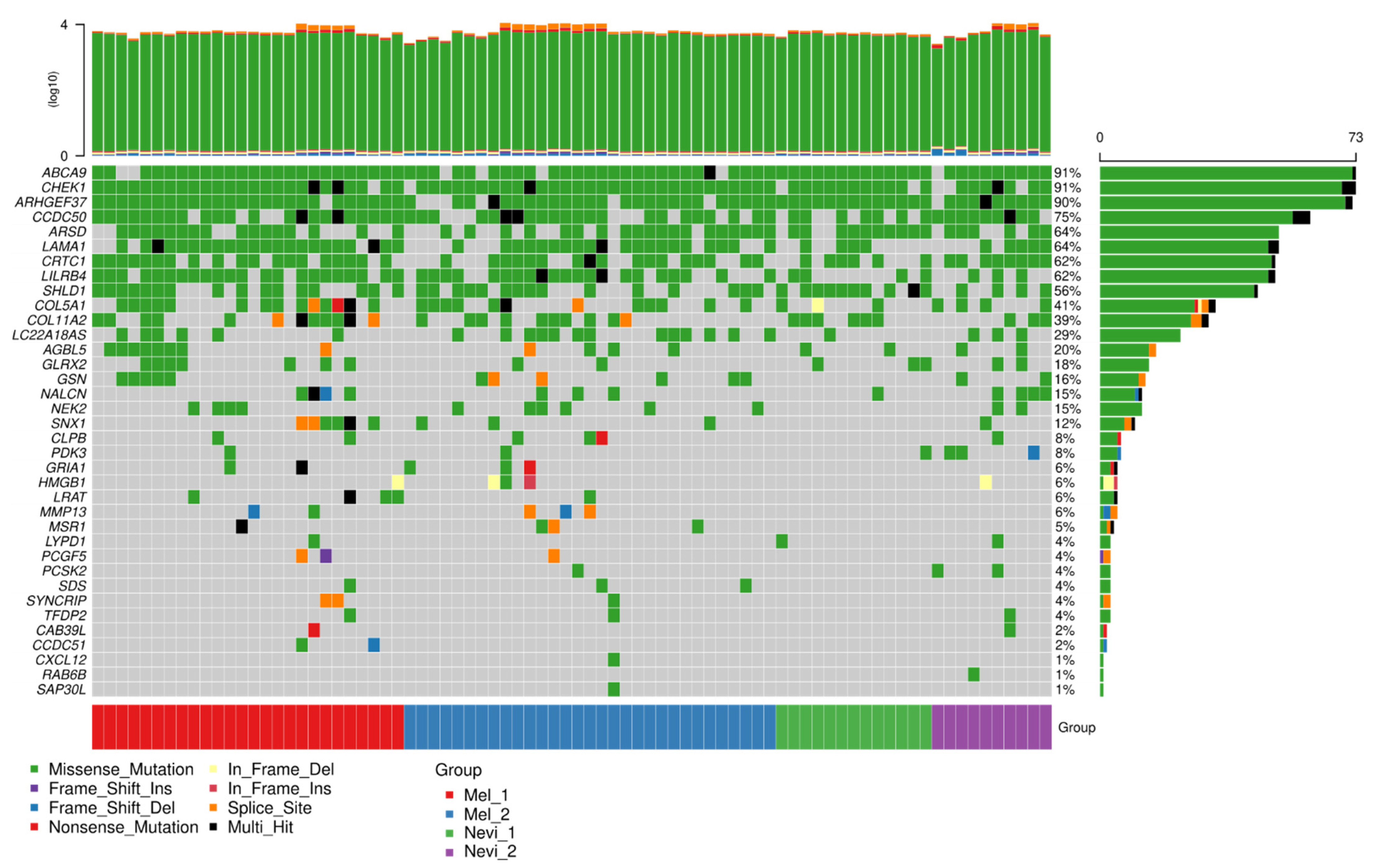
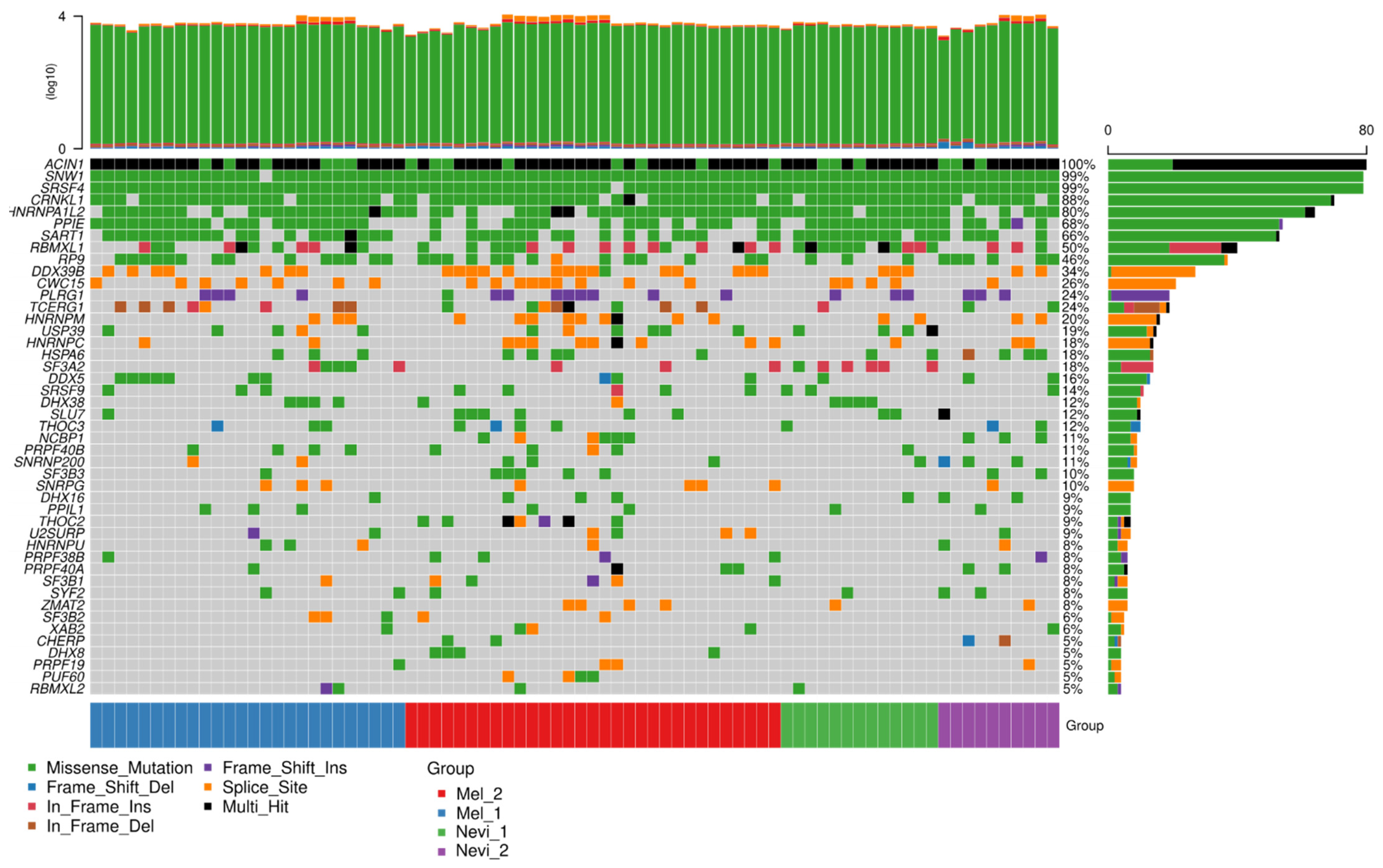
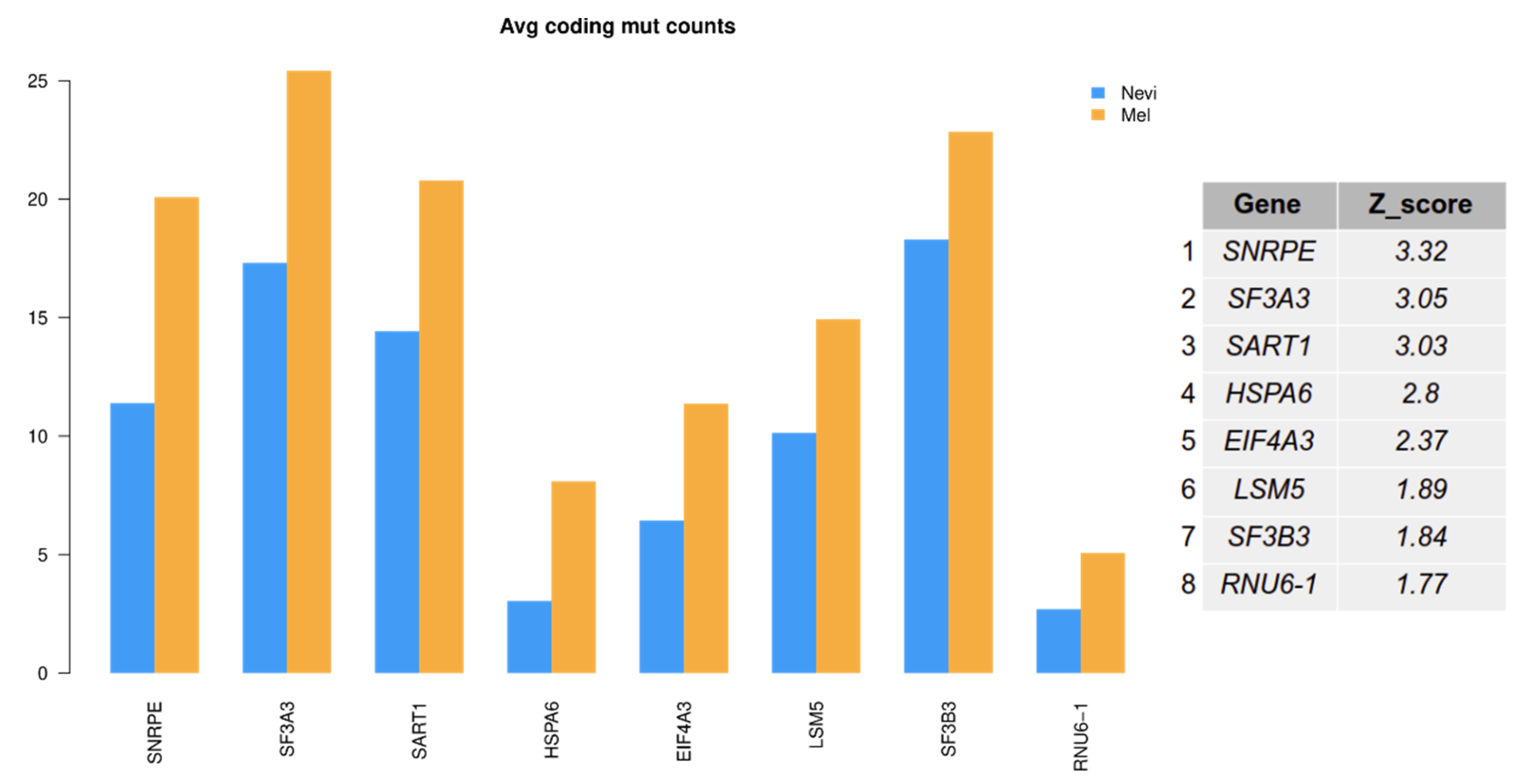
2.6. Mutation Analysis
3. Discussion
4. Materials and Methods
4.1. Nevi and Melanoma Cases and RNA-Seq Data
4.2. Isoform Abundance Quantification Analysis
4.3. Isoform Switches
4.4. Mutation Analysis of RNA-Seq Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Seth, R.; Messersmith, H.; Kaur, V.; Kirkwood, J.M.; Kudchadkar, R.; McQuade, J.L.; Provenzano, A.; Swami, U.; Weber, J.; Alluri, K.C.; et al. Systemic Therapy for Melanoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3947–3970. [Google Scholar] [CrossRef]
- Winnepenninckx, V.; Lazar, V.; Michiels, S.; Dessen, P.; Stas, M.; Alonso, S.R.; Avril, M.-F.; Romero, P.L.O.; Robert, T.; Balacescu, O.; et al. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J. Natl. Cancer Inst. 2006, 98, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Jönsson, G.; Busch, C.; Knappskog, S.; Geisler, J.; Miletic, H.; Ringner, M.; Lillehaug, J.R.; Borg, A.; Lonning, P.E. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin. Cancer Res. 2010, 16, 3356–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbst, K.; Staaf, J.; Lauss, M.; Karlsson, A.; Måsbäck, A.; Johansson, I.; Bendahl, P.-O.; Vallon-Christersson, J.; Törngren, T.; Ekedahl, H.; et al. Molecular profiling reveals low- and high-grade forms of primary melanoma. Clin. Cancer Res. 2012, 18, 4026–4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerami, P.; Cook, R.W.; Wilkinson, J.; Russell, M.C.; Dhillon, N.; Amaria, R.N.; Gonzalez, R.; Lyle, S.; Johnson, C.E.; Oelschlager, K.M.; et al. Development of a prognostic genetic signature to predict the metastatic risk associated with cutaneous melanoma. Clin. Cancer Res. 2015, 21, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Cirenajwis, H.; Ekedahl, H.; Lauss, M.; Harbst, K.; Carneiro, A.; Enoksson, J.; Rosengren, F.; Werner-Hartman, L.; Törngren, T.; Kvist, A.; et al. Molecular stratification of metastatic melanoma using gene expression profiling: Prediction of survival outcome and benefit from molecular targeted therapy. Oncotarget 2015, 6, 12297–12309. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Kunz, M.; Löffler-Wirth, H.; Dannemann, M.; Willscher, E.; Doose, G.; Kelso, J.; Kottek, T.; Nickel, B.; Hopp, L.; Landsberg, J.; et al. RNA-seq analysis identifies different transcriptomic types and developmental trajectories of primary melanomas. Oncogene 2018, 37, 6136–6151. [Google Scholar] [CrossRef] [PubMed]
- Sebestyén, E.; Singh, B.; Miñana, B.; Pagès, A.; Mateo, F.; Pujana, M.A. Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks. Genome Res. 2016, 26, 732–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebestyén, E.; Singh, B.; Miñana, B.; Pagès, A.; Mateo, F.; Pujana, M.A.; Valcarcel, J.; Eyras, E. Corrigendum: Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks. Genome Res. 2018, 28, 1426. [Google Scholar] [CrossRef]
- Trincado, J.L.; Sebestyén, E.; Pagés, A.; Eyras, E. The prognostic potential of alternative transcript isoforms across human tumors. Genome Med. 2016, 8, 85. [Google Scholar] [CrossRef] [Green Version]
- Sebestyén, E.; Zawisza, M.; Eyras, E. Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Res. 2015, 43, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Wan, Q.; Sang, X.; Jin, L.; Wang, Z. Alternative Splicing Events as Indicators for the Prognosis of Uveal Melanoma. Genes 2020, 11, 227. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; He, R.; Lin, P.; Zhong, J.; Ma, J.; Yang, H.; Hu, X.; Chen, G. Profiling of prognostic alternative splicing in melanoma. Oncol Lett. 2019, 18, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Mei, C.; Song, P.-Y.; Zhang, W.; Zhou, H.-H.; Li, X.; Liu, Z.-Q. Aberrant RNA Splicing Events Driven by Mutations of RNA-Binding Proteins as Indicators for Skin Cutaneous Melanoma Prognosis. Front. Oncol. 2020, 10, 568469. [Google Scholar] [CrossRef]
- Faridi, P.; Woods, K.; Ostrouska, S.; Deceneux, C.; Aranha, R.; Duscharla, D.; Wong, S.Q.; Chen, W.; Ramarathinam, S.H.; Sian, T.C.L.K.; et al. Spliced Peptides and Cytokine-Driven Changes in the Immunopeptidome of Melanoma. Cancer Immunol. Res. 2020, 8, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Vitting-Seerup, K.; Sandelin, A. The Landscape of Isoform Switches in Human Cancers. Mol. Cancer Res. 2017, 15, 1206–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Zhao, L.; Xue, M.; Zhang, L.; Guo, B.; Qin, Y.; Jiang, Q.; Sun, R.; Yang, J.; Wang, L.; Liu, L.; et al. MicroRNA-4268 inhibits cell proliferation via AKT/JNK signalling pathways by targeting Rab6B in human gastric cancer. Cancer Gene Ther. 2020, 27, 461–472. [Google Scholar] [CrossRef]
- Qian, J.; Yi, H.; Guo, C.; Yu, X.; Zuo, D.; Chen, X.; Kane, J.M.; Repasky, E.A.; Subjeck, J.R.; Wang, X.-Y. CD204 suppresses large heat shock protein-facilitated priming of tumor antigen gp100-specific T cells and chaperone vaccine activity against mouse melanoma. J. Immunol. 2011, 187, 2905–2914. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-C.; Chao, Y.-J.; Tung, H.-L.; Wang, H.-C.; Shan, Y.-S. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer 2014, 120, 2766–2777. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zheng, S.L.; Komiya, A.; Mychaleckyj, J.C.; Isaacs, S.D.; Hu, J.J.; Sterling, D.; Lange, E.M.; Hawkins, G.A.; Turner, A.R.; et al. Germline mutations and sequence variants of the macrophage scavenger receptor 1 gene are associated with prostate cancer risk. Nat. Genet. 2002, 32, 321–325. [Google Scholar] [CrossRef]
- Ichimura, T.; Abe, H.; Morikawa, T.; Yamashita, H.; Ishikawa, S.; Ushiku, T. Low density of CD204-positive M2-type tumor-associated macrophages in Epstein-Barr virus-associated gastric cancer: A clinicopathologic study with digital image analysis. Hum. Pathol. 2016, 56, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.A.; Johnston, J.; Li, J.; Mandikian, D.; Hristopoulos, M.; Clark, R.; Nickles, D.; Liang, W.-C.; Hötzel, K.; Dunlap, D.; et al. Anti-LYPD1/CD3 T-Cell-Dependent Bispecific Antibody for the Treatment of Ovarian Cancer. Mol. Cancer Ther. 2021, 20, 716–725. [Google Scholar] [CrossRef]
- Roos, L.; Van Dongen, J.; Bell, C.G.; Burri, A.; Deloukas, P.; Boomsma, D.I.; Spector, T.D.; Bell, J.T. Integrative DNA methylome analysis of pan-cancer biomarkers in cancer discordant monozygotic twin-pairs. Clin. Epigenetics 2016, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Moreno, V.; Gadea, G.; Ahn JPaterson, H.; Marra, P.; Pinner, S. Rac activation and inactivation control plasticity of tumor cell movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [Green Version]
- Dookeran, K.A.; Zhang, W.; Stayner, L.; Argos, M. Associations of two-pore domain potassium channels and triple negative breast cancer subtype in The Cancer Genome Atlas: Systematic evaluation of gene expression and methylation. BMC Res. Notes 2017, 10, 475. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, Y.; Xiao, K.; Zhao, Y.; Lv, G.; Xu, S.; Wu, F. RPS24c Isoform Facilitates Tumor Angiogenesis Via Promoting the Stability of MVIH in Colorectal Cancer. Curr. Mol. Med. 2020, 20, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Huang, W.; Zhang, J.; Wang, X.; Tao, T.; Yang, L. TMPRSS4 Promotes Cell Proliferation and Inhibits Apoptosis in Pancreatic Ductal Adenocarcinoma by Activating ERK1/2 Signaling Pathway. Front. Oncol. 2021, 11, 628353. [Google Scholar] [CrossRef] [PubMed]
- Min, H.-J.; Lee, M.K.; Lee, J.W.; Kim, S. TMPRSS4 induces cancer cell invasion through pro-uPA processing. Biochem. Biophys. Res. Commun. 2014, 446, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Masamha, C.P.; Xia, Z.; Yang, J.; Albrecht, T.R.; Li, M.; Shyu, A.-B.; Li, W.; Wagner, E.J. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 2014, 510, 412–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef] [Green Version]
- Palavalli, L.H.; Prickett, T.D.; Wunderlich, J.R.; Wei, X.; Burrell, A.S.; Porter-Gill, P. Analysis of the matrix metalloproteinase family reveals that MMP8 is often mutated in melanoma. Nat. Genet. 2009, 41, 518–520. [Google Scholar] [CrossRef] [Green Version]
- Gardberg, M.; Heuser, V.D.; Koskivuo, I.; Koivisto, M.; Carpén, O. FMNL2/FMNL3 formins are linked with oncogenic pathways and predict melanoma outcome. J. Pathol. Clin. Res. 2016, 2, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Kage, F.; Winterhoff, M.; Dimchev, V.; Mueller, J.; Thalheim, T.; Freise, A. FMNL formins boost lamellipodial force generation. Nat. Commun. 2017, 8, 14832. [Google Scholar] [CrossRef]
- Domogatskaya, A.; Rodin, S.; Tryggvason, K. Functional Diversity of Laminins. Annu. Rev. Cell Dev. Biol. 2012, 28, 523–555. [Google Scholar] [CrossRef]
- Cox, T.R. The matrix in cancer. Nat. Rev. Cancer 2021, 21, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Rondepierre, F.; Bouchon, B.; Papon, J.; Bonnet, M.; Kintossou, R.; Moins, N.; Maublant, J.; Madelmont, J.C.; D’Incan, M.; Degoul, F. Proteomic studies of B16 lines: Involvement of annexin A1 in melanoma dissemination. Biochim. Biophys. Acta 2009, 1794, 61–69. [Google Scholar] [CrossRef]
- Gerber, T.; Willscher, E.; Loeffler-Wirth, H.; Hopp, L.; Schadendorf, D.; Schartl, M.; Anderegg, U.; Camp, G.; Treutlein, B.; Binder, H.; et al. Mapping heterogeneity in patient-derived melanoma cultures by single-cell RNA-seq. Oncotarget 2017, 8, 846–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Manley, J.L.; Krainer, A.R. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev. 2010, 24, 1073–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Gong, Q.; Lin, Z.; Wang, Y.; Li, M.; Wang, L.; Ding, H.; Li, P. Emerging Roles of SRSF3 as a Therapeutic Target for Cancer. Front. Oncol. 2020, 10, 577636. [Google Scholar] [CrossRef]
- Karp, C.M.; Shukla, M.N.; Buckley, D.J.; Buckley, A.R. HRPAP20: A novel calmodulin-binding protein that increases breast cancer cell invasion. Oncogene 2007, 26, 1780–1788. [Google Scholar] [CrossRef] [PubMed]
- Kokuryo, T.; Yokoyama, Y.; Yamaguchi, J.; Tsunoda, N.; Ebata, T.; Nagino, M. NEK2 Is an Effective Target for Cancer Therapy with Potential to Induce Regression of Multiple Human Malignancies. Anticancer Res. 2019, 39, 2251–2258. [Google Scholar] [CrossRef]
- De Faria Poloni, J.; Bonatto, D. Influence of transcriptional variants on metastasis. RNA Biol. 2018, 15, 1006–1024. [Google Scholar] [CrossRef]
- Lee, S.C.-W.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Opdam, F.J.; Echard, A.; Croes, H.J.; van den Hurk, J.A.; van de Vorstenbosch, R.A.; Ginsel, L.A. The small GTPase Rab6B, a novel Rab6 subfamily member, is cell-type specifically expressed and localised to the Golgi apparatus. J. Cell Sci. 2000, 113, 2725–2735. [Google Scholar] [CrossRef]
- Brandt, A.C.; McNally, L.; Lorimer, E.L.; Unger, B.; Koehn, O.J.; Suazo, K.F. Splice switching an oncogenic ratio of SmgGDS isoforms as a strategy to diminish malignancy. Proc. Natl. Acad. Sci. USA 2020, 117, 3627–3636. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Thanawala, R.; Bon, G.; Falcioni, R.; Prasad, G.L. Resensitization of breast cancer cells to anoikis by tropomyosin-1: Role of Rho kinase-dependent cytoskeleton and adhesion. Oncogene 2005, 24, 8291–8303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allami, R.H.; Graf, C.; Martchenko, K.; Voss, B.; Becker, M.; Berger, M.R.; Galle, P.R.; Theobald, M.; Wehler, T.C.; Schimanski, C.C. Analysis of the expression of SDF-1 splicing variants in human colorectal cancer and normal mucosa tissues. Oncol. Lett. 2016, 11, 1873–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sonderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.; Punta, M.; Qureshi, M.; Sangrador, A.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Gene Name Mel-vs-Nevi | Gene Switch_q_Value | Gene Name Type1-vs-Type2 | Gene Switch_q_Value |
|---|---|---|---|---|
| 1 | RAB6B | 2.02 × 10−14 | SH2D3A | 2.49 × 10−19 |
| 2 | MSR1 | 1.69 × 10−12 | KCNK6 | 5.28 × 10−17 |
| 3 | LYPD1 | 3.07 × 10−12 | RPS24 | 3.40 × 10−13 |
| 4 | COL11A2 | 1.36 × 10−11 | TMPRSS4 | 3.09 × 10−12 |
| 5 | GRIA1 | 1.06 × 10−10 | NEBL | 4.83 × 10−12 |
| 6 | TFDP2 | 1.67 × 10−10 | CYSLTR1 | 4.83 × 10−12 |
| 7 | CHEK1 | 1.07 × 10−9 | PRKCH | 5.34 × 10−12 |
| 8 | NALCN | 1.94 × 10−9 | ICOSLG | 5.89 × 10−12 |
| 9 | SYNCRIP | 2.86 × 10−9 | SMAGP | 6.09 × 10−12 |
| 10 | C2orf68 | 3.57 × 10−9 | LFNG | 8.28 × 10−12 |
| Type 1 vs. Type 2 Filtered (Performed on Subset of Genes Which Had a Functional Isoform Switch) | |||||
|---|---|---|---|---|---|
| Gene Set | Biological Process | Enrichment Ratio | p Value | FDR | userId |
| GO: 0006955 | Immune response | 1.77 | 7.30 × 10−6 | 0.036 | AOC1; MMP25; CD6; PRKCH; TNFRSF17; TXK; ICAM3; CEACAM6; HAMP; LFNG; CRTAM; CCR6; ITK; ARG1; LRP1; ZBP1; F12; GCH1; CHI3L1; KLRD1; KLRC1; BLK; IL18BP; MMP7; KLRG1; RHOF; IGSF6; VAV1; PRKACB; FCGR2A; IL2RG; FCRL3; CD300LG; INAVA; PYHIN1; AIM2; DNASE1L3; CLEC4E; NOD2; RAB4B; IL7R; ADORA2B; FPR1; CXCR6; CTSW; KLHL6; CYSLTR1; XCR1; TLR5; CARD9; ZP3; SEMA4A; C5AR1; SPN; MBP; RAET1G; LIME1; PSMB10; CFI; PVRIG |
| GO: 0031349 | Positive regulation of defense response | 2.91 | 7.94 × 10−6 | 0.036 | CD6; TXK; ICAM3; CRTAM; ARG1; ZBP1; LDLR; F12; VAV1; PRKACB; FCRL3; INAVA; PYHIN1; AIM2; CLEC4E; NOD2; ADORA2B; TLR5; CARD9; ZP3; TGM2; RAET1G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hakobyan, S.; Loeffler-Wirth, H.; Arakelyan, A.; Binder, H.; Kunz, M. A Transcriptome-Wide Isoform Landscape of Melanocytic Nevi and Primary Melanomas Identifies Gene Isoforms Associated with Malignancy. Int. J. Mol. Sci. 2021, 22, 7165. https://doi.org/10.3390/ijms22137165
Hakobyan S, Loeffler-Wirth H, Arakelyan A, Binder H, Kunz M. A Transcriptome-Wide Isoform Landscape of Melanocytic Nevi and Primary Melanomas Identifies Gene Isoforms Associated with Malignancy. International Journal of Molecular Sciences. 2021; 22(13):7165. https://doi.org/10.3390/ijms22137165
Chicago/Turabian StyleHakobyan, Siras, Henry Loeffler-Wirth, Arsen Arakelyan, Hans Binder, and Manfred Kunz. 2021. "A Transcriptome-Wide Isoform Landscape of Melanocytic Nevi and Primary Melanomas Identifies Gene Isoforms Associated with Malignancy" International Journal of Molecular Sciences 22, no. 13: 7165. https://doi.org/10.3390/ijms22137165
APA StyleHakobyan, S., Loeffler-Wirth, H., Arakelyan, A., Binder, H., & Kunz, M. (2021). A Transcriptome-Wide Isoform Landscape of Melanocytic Nevi and Primary Melanomas Identifies Gene Isoforms Associated with Malignancy. International Journal of Molecular Sciences, 22(13), 7165. https://doi.org/10.3390/ijms22137165