
Nuclear Envelope Integrity in Health and Disease: Consequences on Genome Instability and Inflammation



Abstract
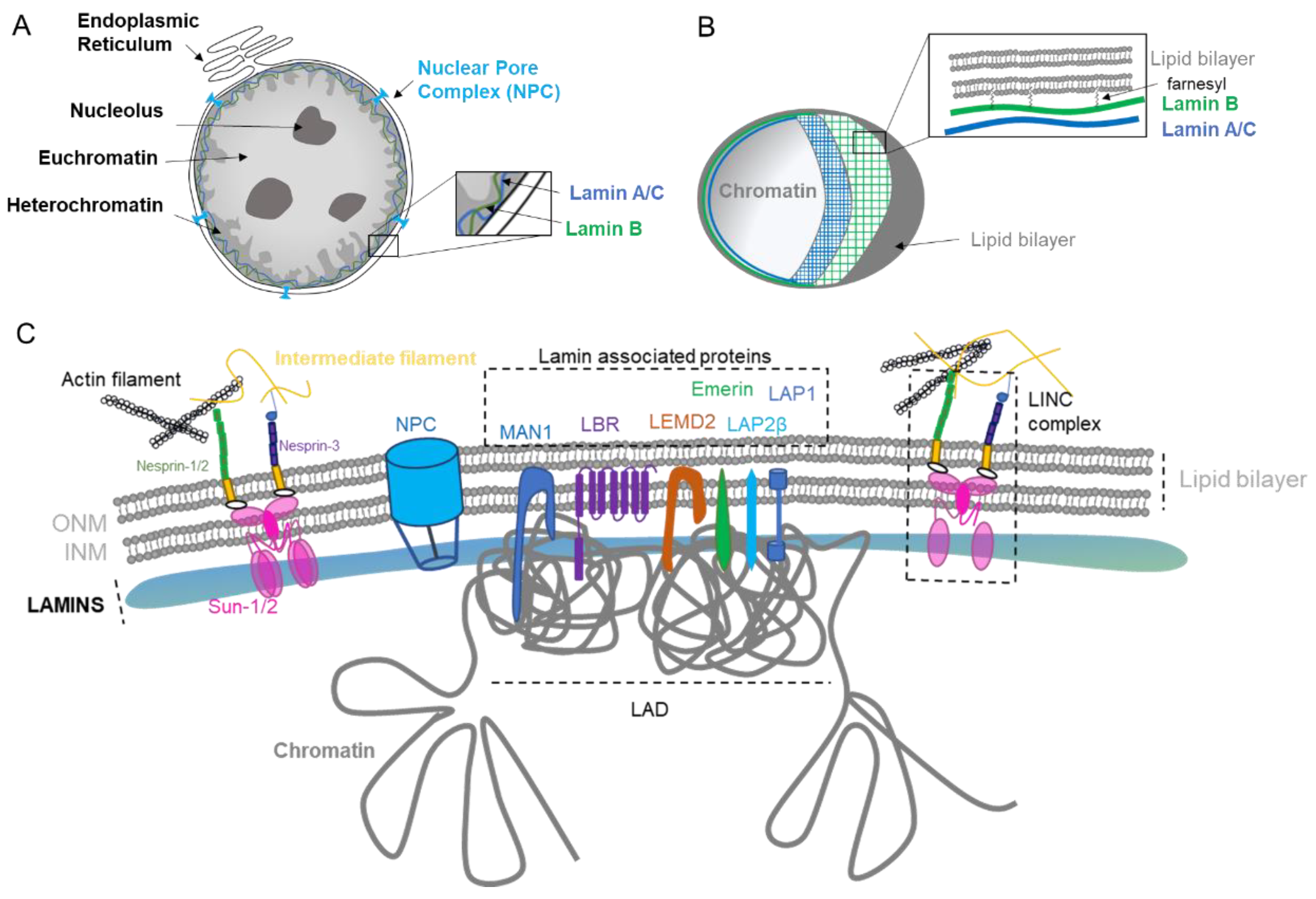
:1. Nuclear Envelope Biology
1.1. Nuclear Lamina
1.2. Nuclear Pore Complex
1.3. Nuclear Lipid Bilayer
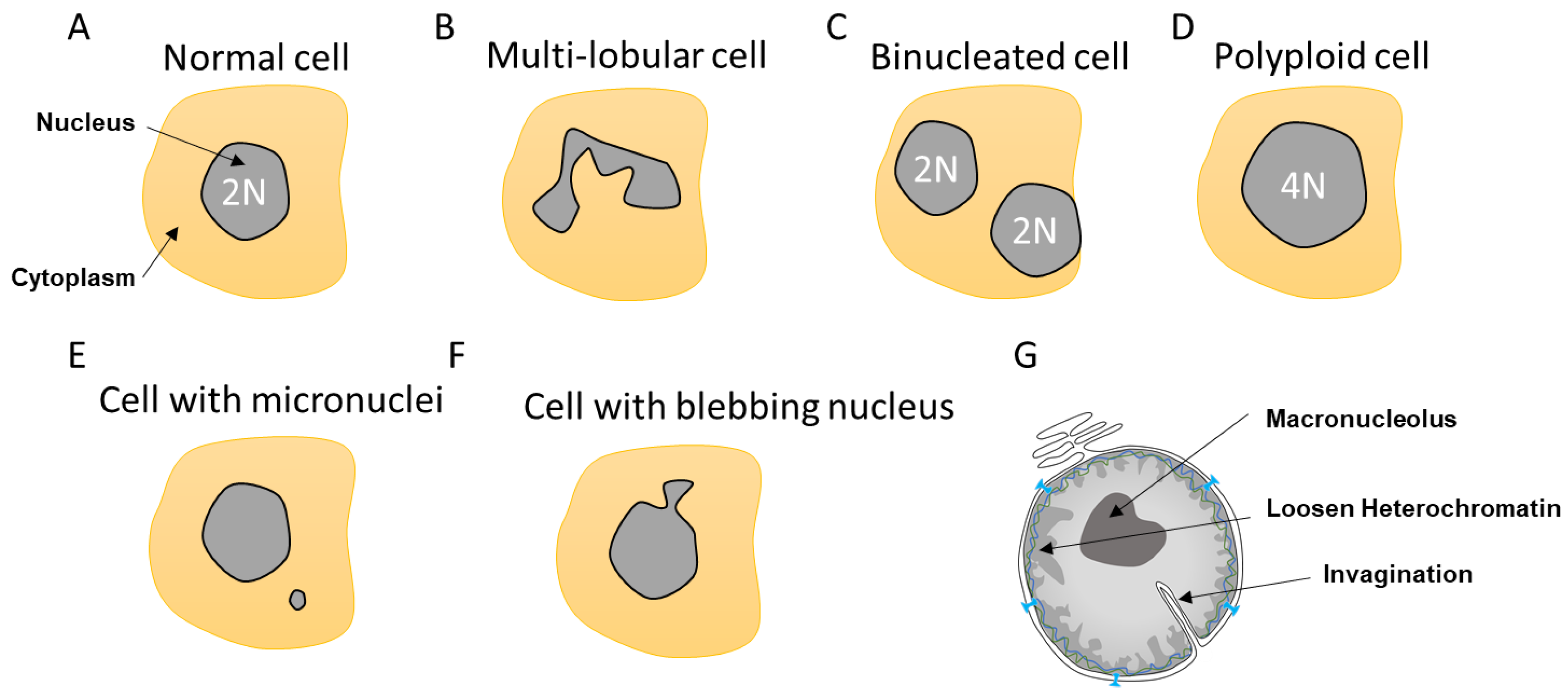
2. Nuclear Envelope Diversity and Abnormalities
2.1. Multi-Lobular Nucleus, Micronuclei, Macronucleoli, and Invagination
2.2. Apoptosis, Senescence, and Aging
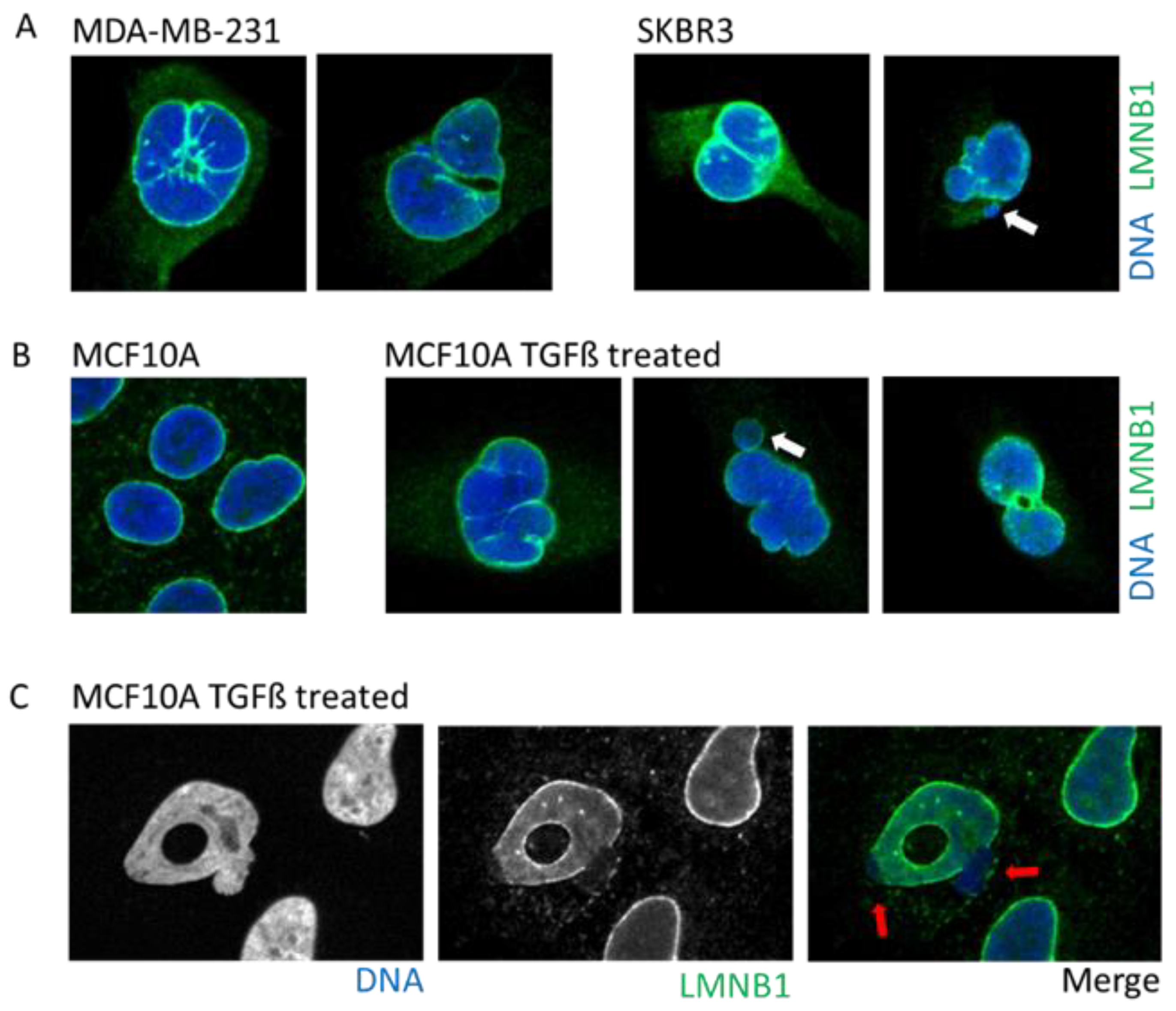
2.3. Cancer
2.4. Cellular Plasticity and Its Effect on Nuclear Envelope
2.5. Envelopathies
3. Nuclear Envelope Disruption
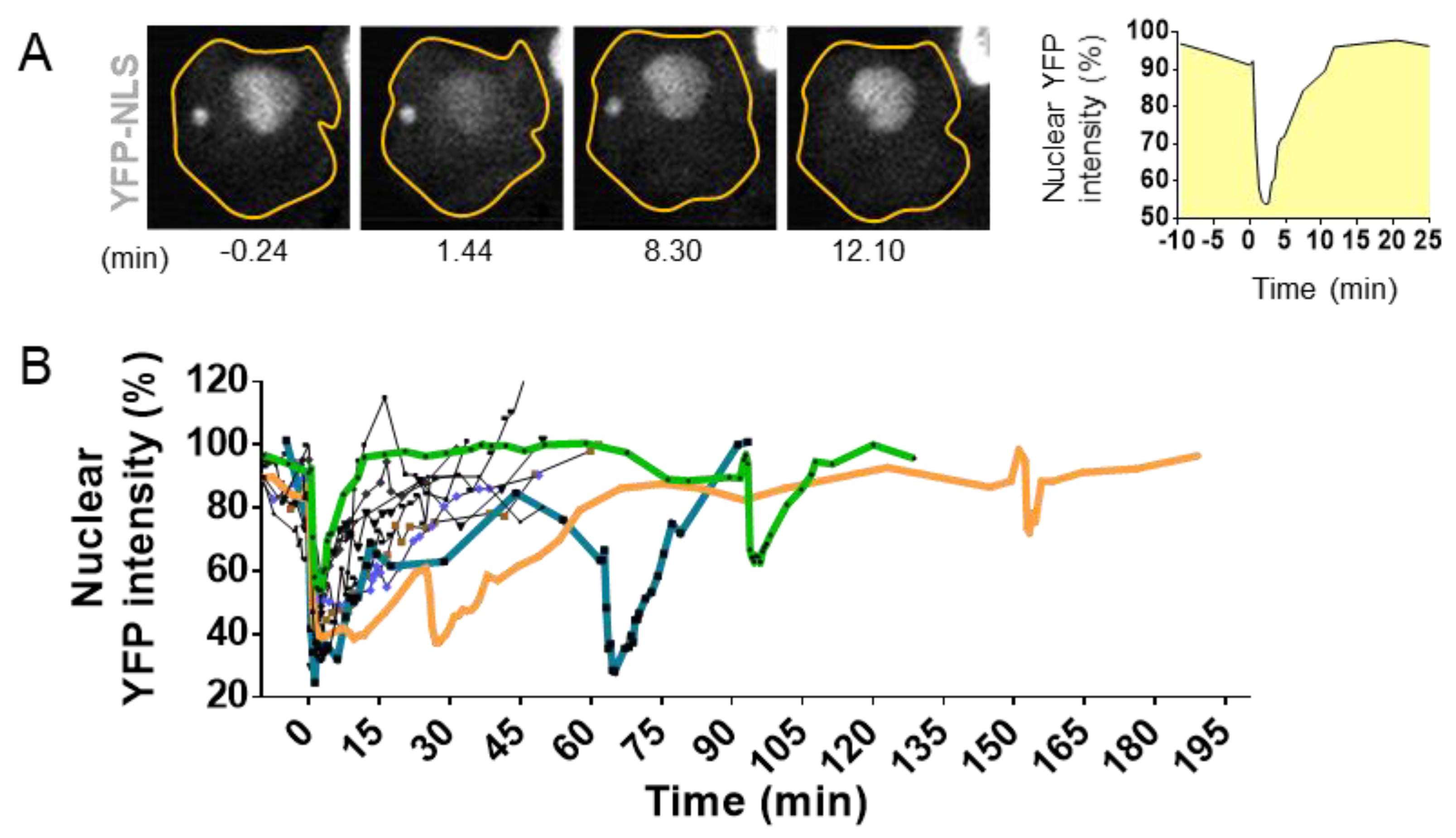
3.1. Nuclear Envelope Disruption and Repair
3.2. Causes of Nuclear Envelope Disruption
3.2.1. Alterations in Expression of Lamins
3.2.2. Migration and Invasion
3.2.3. Micronuclei: NE Extreme Curvation and Improper NE Composition
3.2.4. Telomere Fusion
3.2.5. ATR, RB, and P53 Loss
3.2.6. Heterochromatin Modulation
3.2.7. Virus Infection
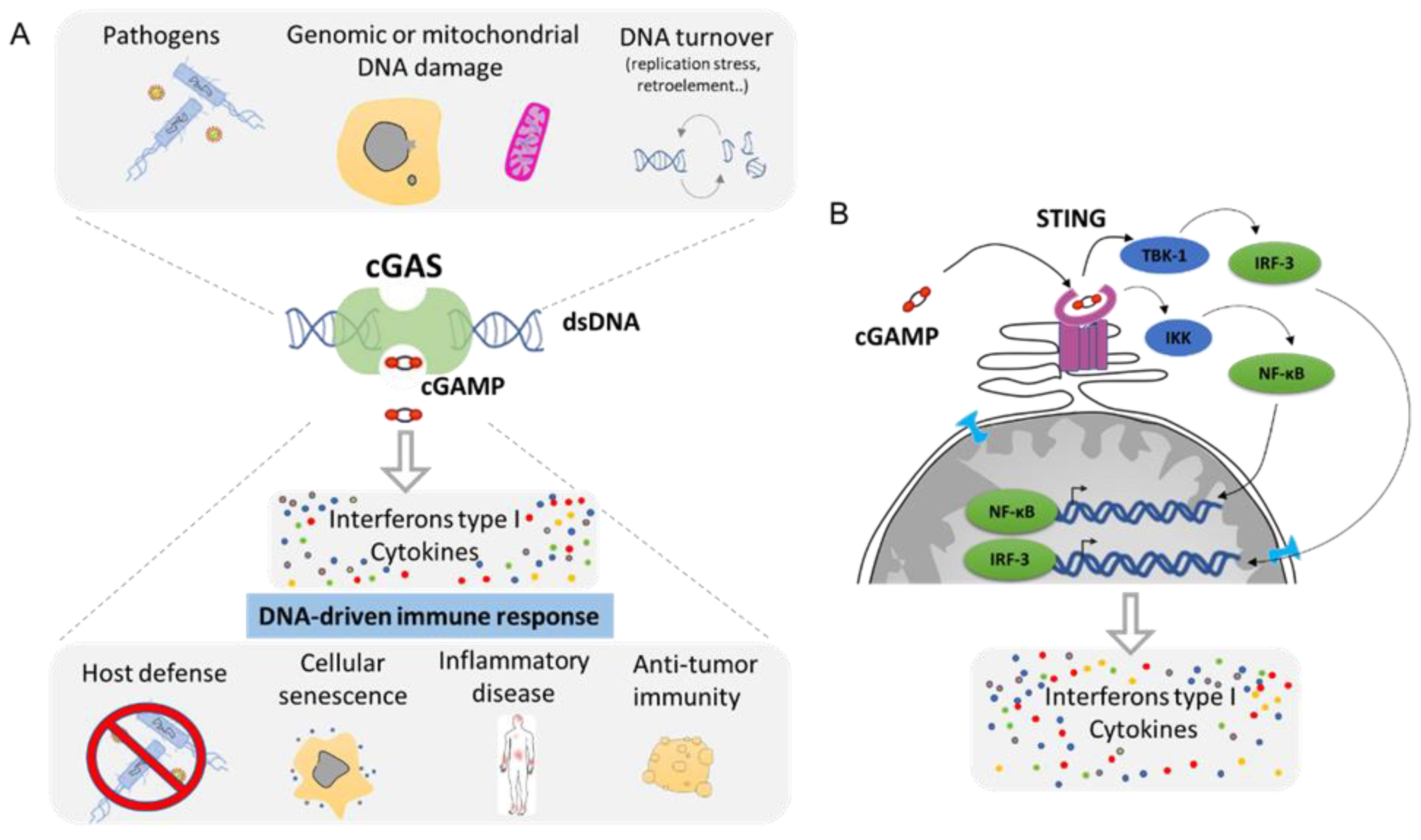
4. Cytoplasmic DNA Induces Inflammation by cGAS/STING Pathway
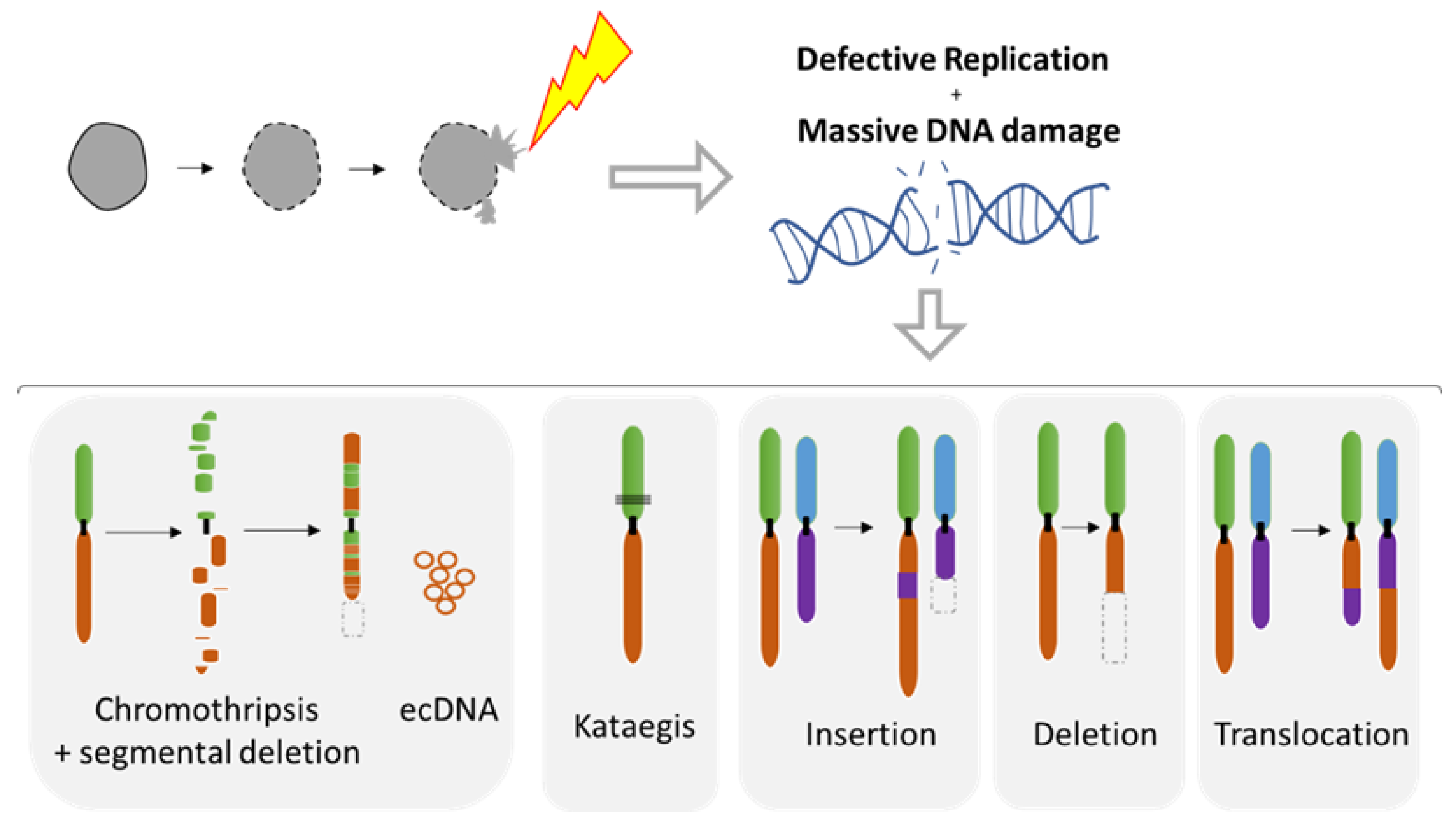
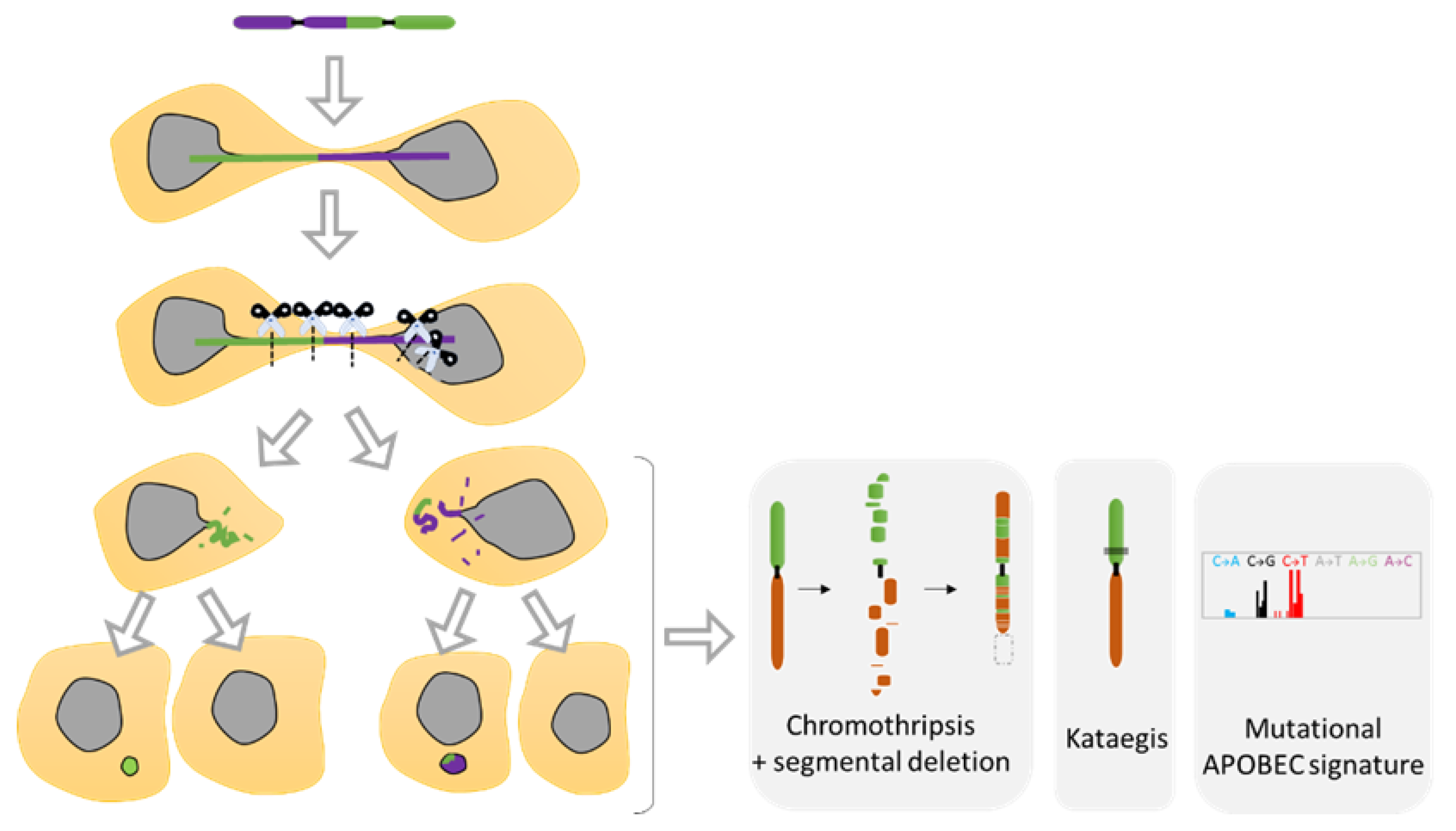
5. Chromosomal Instability Associated to NED
5.1. Diverse Spectrum of Chromosomal Rearrangements Derived from MN Studies
5.2. Telomere Fusion and Chromosomal Bridge
5.3. DNA Damage Arising from NED of the Nucleus
6. Clinical Consequences of Nuclear Envelope Rupture
6.1. Envelopathies and Inflammation
6.1.1. Emery–Dreifuss Muscular Dystrophy: Cell Death Due to Increase DNA Damage
6.1.2. Progeria: DNA Damage, NED, and cGAS Activation
6.1.3. Metabolic Syndrome: A Common Symptom in Envelopathies
6.1.4. Neuropathies, Nuclear Envelope, and Inflammation
6.1.5. Congenital Disease Due to Complex Chromosomal Rearrangement
6.1.6. Cancer: Origin, Evolution, and Survival
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Shin, J.W.; Spinler, K.R.; Swift, J.; Chasis, J.A.; Mohandas, N.; Discher, D.E. Lamins regulate cell trafficking and lineage maturation of adult human hematopoietic cells. Proc. Natl. Acad. Sci. USA 2013, 110, 18892–18897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [Green Version]
- Spann, T.P.; Moir, R.D.; Goldman, A.E.; Stick, R.; Goldman, R.D. Disruption of nuclear lamin organization alters the distribution of replication factors and inhibits DNA synthesis. J. Cell Biol. 1997, 136, 1201–1212. [Google Scholar] [CrossRef]
- Tsai, M.Y.; Wang, S.; Heidinger, J.M.; Shumaker, D.K.; Adam, S.A.; Goldman, R.D.; Zheng, Y. A mitotic lamin B matrix induced by RanGTP required for spindle assembly. Science 2006, 311, 1887–1893. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Sandoval, A.; Gasser, S.M. On TADs and LADs: Spatial Control over Gene Expression. Trends Genet. 2016, 32, 485–495. [Google Scholar] [CrossRef]
- Briand, N.; Collas, P. Lamina-associated domains: Peripheral matters and internal affairs. Genome Biol. 2020, 21, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, M.W. The functional importance of lamins, actin, myosin, spectrin and the LINC complex in DNA repair. Exp. Biol. Med. (Maywood) 2019, 244, 1382–1406. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Chen, S.; Maya-Mendoza, A.; Lovric, J.; Sims, P.F.; Jackson, D.A. Lamin B1 maintains the functional plasticity of nucleoli. J. Cell Sci. 2009, 122, 1551–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bera, M.; Sengupta, K. Nuclear filaments: Role in chromosomal positioning and gene expression. Nucleus 2020, 11, 99–110. [Google Scholar] [CrossRef]
- Comaills, V.; Kabeche, L.; Morris, R.; Buisson, R.; Yu, M.; Madden, M.W.; LiCausi, J.A.; Boukhali, M.; Tajima, K.; Pan, S.; et al. Genomic Instability Is Induced by Persistent Proliferation of Cells Undergoing Epithelial-to-Mesenchymal Transition. Cell Rep. 2016, 17, 2632–2647. [Google Scholar] [CrossRef]
- Liu, S.Y.; Ikegami, K. Nuclear lamin phosphorylation: An emerging role in gene regulation and pathogenesis of laminopathies. Nucleus 2020, 11, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Alcorta-Sevillano, N.; Macias, I.; Rodriguez, C.I.; Infante, A. Crucial Role of Lamin A/C in the Migration and Differentiation of MSCs in Bone. Cells 2020, 9, 1330. [Google Scholar] [CrossRef] [PubMed]
- Donnaloja, F.; Carnevali, F.; Jacchetti, E.; Raimondi, M.T. Lamin A/C Mechanotransduction in Laminopathies. Cells 2020, 9, 1306. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Chojnowski, A.; Boudier, T.; Lim, J.S.; Ahmed, S.; Ser, Z.; Stewart, C.; Burke, B. A-type Lamins Form Distinct Filamentous Networks with Differential Nuclear Pore Complex Associations. Curr. Biol. 2016, 26, 2651–2658. [Google Scholar] [CrossRef] [Green Version]
- Nmezi, B.; Xu, J.; Fu, R.; Armiger, T.J.; Rodriguez-Bey, G.; Powell, J.S.; Ma, H.; Sullivan, M.; Tu, Y.; Chen, N.Y.; et al. Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc. Natl. Acad. Sci. USA 2019, 116, 4307–4315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Vashisth, M.; Abbas, A.; Majkut, S.; Vogel, K.; Xia, Y.; Ivanovska, I.L.; Irianto, J.; Tewari, M.; Zhu, K.; et al. Mechanosensing by the Lamina Protects against Nuclear Rupture, DNA Damage, and Cell-Cycle Arrest. Dev. Cell 2019, 49, 920–935.e925. [Google Scholar] [CrossRef] [PubMed]
- Karoutas, A.; Akhtar, A. Functional mechanisms and abnormalities of the nuclear lamina. Nat. Cell Biol. 2021, 23, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Torvaldson, E.; Kochin, V.; Eriksson, J.E. Phosphorylation of lamins determine their structural properties and signaling functions. Nucleus 2015, 6, 166–171. [Google Scholar] [CrossRef] [Green Version]
- Shevelyov, Y.Y.; Ulianov, S.V. The Nuclear Lamina as an Organizer of Chromosome Architecture. Cells 2019, 8, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammerding, J.; Hsiao, J.; Schulze, P.C.; Kozlov, S.; Stewart, C.L.; Lee, R.T. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J. Cell Biol. 2005, 170, 781–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D.; Estevez-Salmeron, L.D.; Stroschein, S.L.; Zhu, X.; He, J.; Zhou, S.; Luo, K. The integral inner nuclear membrane protein MAN1 physically interacts with the R-Smad proteins to repress signaling by the transforming growth factor-{beta} superfamily of cytokines. J. Biol. Chem. 2005, 280, 15992–16001. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.H.; Hoelz, A. The Structure of the Nuclear Pore Complex (An Update). Annu. Rev. Biochem. 2019, 88, 725–783. [Google Scholar] [CrossRef] [Green Version]
- Pascual-Garcia, P.; Capelson, M. Nuclear pores in genome architecture and enhancer function. Curr. Opin. Cell Biol. 2019, 58, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Haider, A.; Wei, Y.C.; Lim, K.; Barbosa, A.D.; Liu, C.H.; Weber, U.; Mlodzik, M.; Oras, K.; Collier, S.; Hussain, M.M.; et al. PCYT1A Regulates Phosphatidylcholine Homeostasis from the Inner Nuclear Membrane in Response to Membrane Stored Curvature Elastic Stress. Dev. Cell 2018, 45, 481–495.e488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanauska, A.; Kohler, A. The Inner Nuclear Membrane Is a Metabolically Active Territory that Generates Nuclear Lipid Droplets. Cell 2018, 174, 700–715.e718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiam, A.R.; Ikonen, E. Lipid Droplet Nucleation. Trends Cell Biol. 2021, 31, 108–118. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Schlieker, C. Lipid and protein dynamics that shape nuclear envelope identity. Mol. Biol. Cell 2020, 31, 1315–1323. [Google Scholar] [CrossRef]
- Fischer, E.G. Nuclear Morphology and the Biology of Cancer Cells. Acta Cytol. 2020, 64, 511–519. [Google Scholar] [CrossRef]
- Skinner, B.M.; Johnson, E.E. Nuclear morphologies: Their diversity and functional relevance. Chromosoma 2017, 126, 195–212. [Google Scholar] [CrossRef] [Green Version]
- Ganem, N.J.; Storchova, Z.; Pellman, D. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 2007, 17, 157–162. [Google Scholar] [CrossRef]
- Ovrebo, J.I.; Edgar, B.A. Polyploidy in tissue homeostasis and regeneration. Development 2018, 145, dev156034. [Google Scholar] [CrossRef] [Green Version]
- Duncan, A.W.; Taylor, M.H.; Hickey, R.D.; Hanlon Newell, A.E.; Lenzi, M.L.; Olson, S.B.; Finegold, M.J.; Grompe, M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010, 467, 707–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejowski, J.; Hatch, E.M. Nuclear Membrane Rupture and Its Consequences. Annu. Rev. Cell Dev. Biol. 2020, 36, 85–114. [Google Scholar] [CrossRef] [PubMed]
- Stephens, A.D.; Liu, P.Z.; Banigan, E.J.; Almassalha, L.M.; Backman, V.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol. Biol. Cell 2018, 29, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Dumenil, A.M.; Manel, N.; et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016, 352, 359–362. [Google Scholar] [CrossRef]
- Malhas, A.N.; Vaux, D.J. Nuclear envelope invaginations and cancer. Adv. Exp. Med. Biol. 2014, 773, 523–535. [Google Scholar] [CrossRef]
- Jorgens, D.M.; Inman, J.L.; Wojcik, M.; Robertson, C.; Palsdottir, H.; Tsai, W.T.; Huang, H.; Bruni-Cardoso, A.; Lopez, C.S.; Bissell, M.J.; et al. Deep nuclear invaginations are linked to cytoskeletal filaments—Integrated bioimaging of epithelial cells in 3D culture. J. Cell Sci. 2017, 130, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenboim, L.; Zohar, H.; Worman, H.J.; Stein, R. The nuclear envelope: Target and mediator of the apoptotic process. Cell Death Discov. 2020, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Pathak, R.U.; Soujanya, M.; Mishra, R.K. Deterioration of nuclear morphology and architecture: A hallmark of senescence and aging. Ageing Res. Rev. 2021, 67, 101264. [Google Scholar] [CrossRef]
- Dreesen, O.; Chojnowski, A.; Ong, P.F.; Zhao, T.Y.; Common, J.E.; Lunny, D.; Lane, E.B.; Lee, S.J.; Vardy, L.A.; Stewart, C.L.; et al. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J. Cell Biol. 2013, 200, 605–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, F.; Sousa, J.; Pereira, C.D.; da Cruz, E.S.O.A.B.; Rebelo, S. Nuclear envelope dysfunction and its contribution to the aging process. Aging Cell 2020, 19, e13143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, M.A.; Raices, M.; Panowski, S.H.; Hetzer, M.W. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 2009, 136, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Burla, R.; La Torre, M.; Saggio, I. Mammalian telomeres and their partnership with lamins. Nucleus 2016, 7, 187–202. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.R.; George, S.H.; Kobetz, E.; Xu, X.X. New biological research and understanding of Papanicolaou’s test. Diagn. Cytopathol. 2018, 46, 507–515. [Google Scholar] [CrossRef]
- Ren, B.; Liu, X.; Suriawinata, A.A. Pancreatic Ductal Adenocarcinoma and Its Precursor Lesions: Histopathology, Cytopathology, and Molecular Pathology. Am. J. Pathol. 2019, 189, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Hruban, R.H.; Maitra, A.; Goggins, M. Update on pancreatic intraepithelial neoplasia. Int. J. Clin. Exp. Pathol. 2008, 1, 306–316. [Google Scholar]
- Tang, Y.; Chen, Y.; Jiang, H.; Nie, D. Promotion of tumor development in prostate cancer by progerin. Cancer Cell Int. 2010, 10, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef] [Green Version]
- Merrell, A.J.; Stanger, B.Z. Adult cell plasticity in vivo: De-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol. 2016, 17, 413–425. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Guerrero-Martinez, J.A.; Ceballos-Chavez, M.; Koehler, F.; Peiro, S.; Reyes, J.C. TGFbeta promotes widespread enhancer chromatin opening and operates on genomic regulatory domains. Nat. Commun. 2020, 11, 6196. [Google Scholar] [CrossRef]
- Verstraeten, V.L.; Peckham, L.A.; Olive, M.; Capell, B.C.; Collins, F.S.; Nabel, E.G.; Young, S.G.; Fong, L.G.; Lammerding, J. Protein farnesylation inhibitors cause donut-shaped cell nuclei attributable to a centrosome separation defect. Proc. Natl. Acad. Sci. USA 2011, 108, 4997–5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janin, A.; Bauer, D.; Ratti, F.; Millat, G.; Mejat, A. Nuclear envelopathies: A complex LINC between nuclear envelope and pathology. Orphanet J. Rare Dis. 2017, 12, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galant, D.; Gaborit, B.; Desgrouas, C.; Abdesselam, I.; Bernard, M.; Levy, N.; Merono, F.; Coirault, C.; Roll, P.; Lagarde, A.; et al. A Heterozygous ZMPSTE24 Mutation Associated with Severe Metabolic Syndrome, Ectopic Fat Accumulation, and Dilated Cardiomyopathy. Cells 2016, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Kreienkamp, R.; Gonzalo, S. Metabolic Dysfunction in Hutchinson-Gilford Progeria Syndrome. Cells 2020, 9, 395. [Google Scholar] [CrossRef] [Green Version]
- Desgrouas, C.; Varlet, A.A.; Dutour, A.; Galant, D.; Merono, F.; Bonello-Palot, N.; Bourgeois, P.; Lasbleiz, A.; Petitjean, C.; Ancel, P.; et al. Unraveling LMNA Mutations in Metabolic Syndrome: Cellular Phenotype and Clinical Pitfalls. Cells 2020, 9, 310. [Google Scholar] [CrossRef] [Green Version]
- Charar, C.; Gruenbaum, Y. Lamins and metabolism. Clin. Sci. (Lond.) 2017, 131, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Dutour, A.; Roll, P.; Gaborit, B.; Courrier, S.; Alessi, M.C.; Tregouet, D.A.; Angelis, F.; Robaglia-Schlupp, A.; Lesavre, N.; Cau, P.; et al. High prevalence of laminopathies among patients with metabolic syndrome. Hum. Mol. Genet. 2011, 20, 3779–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillin-Amarelle, C.; Fernandez-Pombo, A.; Sanchez-Iglesias, S.; Araujo-Vilar, D. Lipodystrophic laminopathies: Diagnostic clues. Nucleus 2018, 9, 249–260. [Google Scholar] [CrossRef] [Green Version]
- van Tienen, F.H.J.; Lindsey, P.J.; Kamps, M.A.F.; Krapels, I.P.; Ramaekers, F.C.S.; Brunner, H.G.; van den Wijngaard, A.; Broers, J.L.V. Assessment of fibroblast nuclear morphology aids interpretation of LMNA variants. Eur. J. Hum. Genet. 2019, 27, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Dahl, K.N.; Scaffidi, P.; Islam, M.F.; Yodh, A.G.; Wilson, K.L.; Misteli, T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 10271–10276. [Google Scholar] [CrossRef] [Green Version]
- De Vos, W.H.; Houben, F.; Kamps, M.; Malhas, A.; Verheyen, F.; Cox, J.; Manders, E.M.; Verstraeten, V.L.; van Steensel, M.A.; Marcelis, C.L.; et al. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum. Mol. Genet. 2011, 20, 4175–4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, J.D.; Hatch, E.M.; Anderson, D.J.; Hetzer, M.W. Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus 2012, 3, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.Y.; Yang, Y.; Weston, T.A.; Belling, J.N.; Heizer, P.; Tu, Y.; Kim, P.; Edillo, L.; Jonas, S.J.; Weiss, P.S.; et al. An absence of lamin B1 in migrating neurons causes nuclear membrane ruptures and cell death. Proc. Natl. Acad. Sci. USA 2019, 116, 25870–25879. [Google Scholar] [CrossRef]
- Vietri, M.; Schultz, S.W.; Bellanger, A.; Jones, C.M.; Petersen, L.I.; Raiborg, C.; Skarpen, E.; Pedurupillay, C.R.J.; Kjos, I.; Kip, E.; et al. Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat. Cell Biol. 2020, 22, 856–867. [Google Scholar] [CrossRef]
- Beier, K.T.; Samson, M.E.; Matsuda, T.; Cepko, C.L. Conditional expression of the TVA receptor allows clonal analysis of descendents from Cre-expressing progenitor cells. Dev. Biol. 2011, 353, 309–320. [Google Scholar] [CrossRef] [Green Version]
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 2011, 18, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Nava, M.M.; Miroshnikova, Y.A.; Biggs, L.C.; Whitefield, D.B.; Metge, F.; Boucas, J.; Vihinen, H.; Jokitalo, E.; Li, X.; Garcia Arcos, J.M.; et al. Heterochromatin-Driven Nuclear Softening Protects the Genome against Mechanical Stress-Induced Damage. Cell 2020, 181, 800–817.e822. [Google Scholar] [CrossRef] [PubMed]
- Khatau, S.B.; Kim, D.H.; Hale, C.M.; Bloom, R.J.; Wirtz, D. The perinuclear actin cap in health and disease. Nucleus 2010, 1, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroud, M.J.; Banerjee, I.; Veevers, J.; Chen, J. Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circ. Res. 2014, 114, 538–548. [Google Scholar] [CrossRef] [Green Version]
- Lomakin, A.J.; Cattin, C.J.; Cuvelier, D.; Alraies, Z.; Molina, M.; Nader, G.P.F.; Srivastava, N.; Saez, P.J.; Garcia-Arcos, J.M.; Zhitnyak, I.Y.; et al. The nucleus acts as a ruler tailoring cell responses to spatial constraints. Science 2020, 370, eaba2894. [Google Scholar] [CrossRef] [PubMed]
- Earle, A.J.; Kirby, T.J.; Fedorchak, G.R.; Isermann, P.; Patel, J.; Iruvanti, S.; Moore, S.A.; Bonne, G.; Wallrath, L.L.; Lammerding, J. Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat. Mater. 2020, 19, 464–473. [Google Scholar] [CrossRef]
- Xia, Y.; Pfeifer, C.R.; Zhu, K.; Irianto, J.; Liu, D.; Pannell, K.; Chen, E.J.; Dooling, L.J.; Tobin, M.P.; Wang, M.; et al. Rescue of DNA damage after constricted migration reveals a mechano-regulated threshold for cell cycle. J. Cell Biol. 2019, 218, 2545–2563. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kwon, M.; Mannino, M.; Yang, N.; Renda, F.; Khodjakov, A.; Pellman, D. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 2018, 561, 551–555. [Google Scholar] [CrossRef]
- Kneissig, M.; Keuper, K.; de Pagter, M.S.; van Roosmalen, M.J.; Martin, J.; Otto, H.; Passerini, V.; Campos Sparr, A.; Renkens, I.; Kropveld, F.; et al. Micronuclei-based model system reveals functional consequences of chromothripsis in human cells. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, D.; LaBella, K.A.; DePinho, R.A. Telomeres: History, health, and hallmarks of aging. Cell 2021, 184, 306–322. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazinski, S.A.; Zou, L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mazzanti, M.; Mistrik, M.; Kosar, M.; Beznoussenko, G.V.; Mironov, A.A.; Garre, M.; Parazzoli, D.; Shivashankar, G.V.; Scita, G.; et al. ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell 2014, 158, 633–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidiyoor, G.R.; Li, Q.; Bastianello, G.; Bruhn, C.; Giovannetti, I.; Mohamood, A.; Beznoussenko, G.V.; Mironov, A.; Raab, M.; Piel, M.; et al. ATR is essential for preservation of cell mechanics and nuclear integrity during interstitial migration. Nat. Commun. 2020, 11, 4828. [Google Scholar] [CrossRef]
- Yang, Z.; Maciejowski, J.; de Lange, T. Nuclear Envelope Rupture Is Enhanced by Loss of p53 or Rb. Mol. Cancer Res. 2017, 15, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Furusawa, T.; Rochman, M.; Taher, L.; Dimitriadis, E.K.; Nagashima, K.; Anderson, S.; Bustin, M. Chromatin decompaction by the nucleosomal binding protein HMGN5 impairs nuclear sturdiness. Nat. Commun. 2015, 6, 6138. [Google Scholar] [CrossRef] [Green Version]
- Mettenleiter, T.C. Breaching the Barrier-The Nuclear Envelope in Virus Infection. J. Mol. Biol. 2016, 428, 1949–1961. [Google Scholar] [CrossRef]
- Fay, N.; Pante, N. Nuclear entry of DNA viruses. Front. Microbiol. 2015, 6, 467. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Marr, A.K.; Garcin, P.; Pante, N. Nuclear envelope disruption involving host caspases plays a role in the parvovirus replication cycle. J. Virol. 2011, 85, 4863–4874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.; Behzad, A.R.; Carroll, J.B.; Pante, N. Parvoviral nuclear import: Bypassing the host nuclear-transport machinery. J. Gen. Virol. 2006, 87, 3209–3213. [Google Scholar] [CrossRef]
- Fan, Y.; Sanyal, S.; Bruzzone, R. Breaking Bad: How Viruses Subvert the Cell Cycle. Front. Cell Infect. Microbiol. 2018, 8, 396. [Google Scholar] [CrossRef] [PubMed]
- Martin-Lluesma, S.; Schaeffer, C.; Robert, E.I.; van Breugel, P.C.; Leupin, O.; Hantz, O.; Strubin, M. Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 2008, 48, 1467–1476. [Google Scholar] [CrossRef]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Simpson, S.R.; Hemphill, W.O.; Hudson, T.; Perrino, F.W. TREX1—Apex predator of cytosolic DNA metabolism. DNA Repair (Amst.) 2020, 94, 102894. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363. [Google Scholar] [CrossRef]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Gluck, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 1–22. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Ly, P.; Brunner, S.F.; Shoshani, O.; Kim, D.H.; Lan, W.; Pyntikova, T.; Flanagan, A.M.; Behjati, S.; Page, D.C.; Campbell, P.J.; et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat. Genet. 2019, 51, 705–715. [Google Scholar] [CrossRef]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef]
- Umbreit, N.T.; Zhang, C.Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368, eaba0712. [Google Scholar] [CrossRef]
- Kato, H.; Sandberg, A.A. Chromosome pulverization in human cells with micronuclei. J. Natl. Cancer Inst. 1968, 40, 165–179. [Google Scholar]
- Kwon, M.; Leibowitz, M.L.; Lee, J.H. Small but mighty: The causes and consequences of micronucleus rupture. Exp. Mol. Med. 2020, 52, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Teitz, L.S.; Kim, D.H.; Shoshani, O.; Skaletsky, H.; Fachinetti, D.; Page, D.C.; Cleveland, D.W. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol. 2017, 19, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021, 53, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Shoshani, O.; Brunner, S.F.; Yaeger, R.; Ly, P.; Nechemia-Arbely, Y.; Kim, D.H.; Fang, R.; Castillon, G.A.; Yu, M.; Li, J.S.Z.; et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2021, 591, 137–141. [Google Scholar] [CrossRef]
- Turner, K.M.; Deshpande, V.; Beyter, D.; Koga, T.; Rusert, J.; Lee, C.; Li, B.; Arden, K.; Ren, B.; Nathanson, D.A.; et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017, 543, 122–125. [Google Scholar] [CrossRef]
- Maciejowski, J.; Chatzipli, A.; Dananberg, A.; Chu, K.; Toufektchan, E.; Klimczak, L.J.; Gordenin, D.A.; Campbell, P.J.; de Lange, T. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat. Genet. 2020, 52, 884–890. [Google Scholar] [CrossRef]
- Zou, J.; Wang, C.; Ma, X.; Wang, E.; Peng, G. APOBEC3B, a molecular driver of mutagenesis in human cancers. Cell Biosci. 2017, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, R.; Hu, Q.; Ly, P. Mechanistic origins of diverse genome rearrangements in cancer. Semin. Cell Dev. Biol. 2021. [Google Scholar] [CrossRef]
- Pfeifer, C.R.; Xia, Y.; Zhu, K.; Liu, D.; Irianto, J.; Garcia, V.M.M.; Millan, L.M.S.; Niese, B.; Harding, S.; Deviri, D.; et al. Constricted migration increases DNA damage and independently represses cell cycle. Mol. Biol. Cell 2018, 29, 1948–1962. [Google Scholar] [CrossRef]
- Eldarov, C.M.; Vangely, I.M.; Vays, V.B.; Sheval, E.V.; Holtze, S.; Hildebrandt, T.B.; Kolosova, N.G.; Popkov, V.A.; Plotnikov, E.Y.; Zorov, D.B.; et al. Mitochondria in the Nuclei of Rat Myocardial Cells. Cells 2020, 9, 712. [Google Scholar] [CrossRef] [Green Version]
- Sur, I.; Neumann, S.; Noegel, A.A. Nesprin-1 role in DNA damage response. Nucleus 2014, 5, 173–191. [Google Scholar] [CrossRef] [Green Version]
- Lei, K.; Zhu, X.; Xu, R.; Shao, C.; Xu, T.; Zhuang, Y.; Han, M. Inner nuclear envelope proteins SUN1 and SUN2 play a prominent role in the DNA damage response. Curr. Biol. 2012, 22, 1609–1615. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.D.; Martins, F.; Santos, M.; Mueller, T.; da Cruz, E.S.O.A.B.; Rebelo, S. Nuclear Accumulation of LAP1:TRF2 Complex during DNA Damage Response Uncovers a Novel Role for LAP1. Cells 2020, 9, 1804. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Li, C.; Zhou, B.; Sun, H.; Koullourou, V.; Holt, I.; Puckelwartz, M.J.; Warren, D.T.; Hayward, R.; Lin, Z.; et al. Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis. Hum. Mol. Genet. 2017, 26, 2258–2276. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Lu, G.F.; Chen, S.C.; Xia, Y.P.; Ye, Z.M.; Cao, F.; Hu, B. Synergistic inflammatory signaling by cGAS may be involved in the development of atherosclerosis. Aging (Albany NY) 2021, 13, 5650–5673. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.H.; Luu, J.; Heizer, P.; Tu, Y.; Weston, T.A.; Chen, N.; Lim, C.; Li, R.L.; Lin, P.Y.; Dunn, J.C.Y.; et al. Disrupting the LINC complex in smooth muscle cells reduces aortic disease in a mouse model of Hutchinson-Gilford progeria syndrome. Sci. Transl. Med. 2018, 10, eaat7163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graziano, S.; Kreienkamp, R.; Coll-Bonfill, N.; Gonzalo, S. Causes and consequences of genomic instability in laminopathies: Replication stress and interferon response. Nucleus 2018, 9, 258–275. [Google Scholar] [CrossRef] [Green Version]
- Kreienkamp, R.; Graziano, S.; Coll-Bonfill, N.; Bedia-Diaz, G.; Cybulla, E.; Vindigni, A.; Dorsett, D.; Kubben, N.; Batista, L.F.Z.; Gonzalo, S. A Cell-Intrinsic Interferon-like Response Links Replication Stress to Cellular Aging Caused by Progerin. Cell Rep. 2018, 22, 2006–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidault, G.; Garcia, M.; Capeau, J.; Morichon, R.; Vigouroux, C.; Bereziat, V. Progerin Expression Induces Inflammation, Oxidative Stress and Senescence in Human Coronary Endothelial Cells. Cells 2020, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Lanktree, M.B.; Johansen, C.T.; Joy, T.R.; Hegele, R.A. A translational view of the genetics of lipodystrophy and ectopic fat deposition. Prog. Mol. Biol. Transl. Sci. 2010, 94, 159–196. [Google Scholar] [CrossRef]
- Misra, A.; Peethambaram, A.; Garg, A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: Report of 35 cases and review of the literature. Medicine (Baltimore) 2004, 83, 18–34. [Google Scholar] [CrossRef]
- Salo, V.T.; Li, S.; Vihinen, H.; Holtta-Vuori, M.; Szkalisity, A.; Horvath, P.; Belevich, I.; Peranen, J.; Thiele, C.; Somerharju, P.; et al. Seipin Facilitates Triglyceride Flow to Lipid Droplet and Counteracts Droplet Ripening via Endoplasmic Reticulum Contact. Dev. Cell 2019, 50, 478–493.e479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Tan, Y.; Zhao, P.; Ren, Z. SEIPIN: A Key Factor for Nuclear Lipid Droplet Generation and Lipid Homeostasis. Int. J. Mol. Sci. 2020, 21, 8208. [Google Scholar] [CrossRef]
- Zhukovsky, M.A.; Filograna, A.; Luini, A.; Corda, D.; Valente, C. Phosphatidic acid in membrane rearrangements. FEBS Lett. 2019, 593, 2428–2451. [Google Scholar] [CrossRef] [Green Version]
- Padiath, Q.S.; Saigoh, K.; Schiffmann, R.; Asahara, H.; Yamada, T.; Koeppen, A.; Hogan, K.; Ptacek, L.J.; Fu, Y.H. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 2006, 38, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- De Sandre-Giovannoli, A.; Chaouch, M.; Kozlov, S.; Vallat, J.M.; Tazir, M.; Kassouri, N.; Szepetowski, P.; Hammadouche, T.; Vandenberghe, A.; Stewart, C.L.; et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet. 2002, 70, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Parry, D.A.; Martin, C.A.; Greene, P.; Marsh, J.A.; Genomics England Research, C.; Blyth, M.; Cox, H.; Donnelly, D.; Greenhalgh, L.; Greville-Heygate, S.; et al. Heterozygous lamin B1 and lamin B2 variants cause primary microcephaly and define a novel laminopathy. Genet. Med. 2021, 23, 408–414. [Google Scholar] [CrossRef]
- Cristofoli, F.; Moss, T.; Moore, H.W.; Devriendt, K.; Flanagan-Steet, H.; May, M.; Jones, J.; Roelens, F.; Fons, C.; Fernandez, A.; et al. De Novo Variants in LMNB1 Cause Pronounced Syndromic Microcephaly and Disruption of Nuclear Envelope Integrity. Am. J. Hum. Genet. 2020, 107, 753–762. [Google Scholar] [CrossRef]
- Gao, Y.L.; Wang, N.; Sun, F.R.; Cao, X.P.; Zhang, W.; Yu, J.T. Tau in neurodegenerative disease. Ann. Transl. Med. 2018, 6, 175. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Bardai, F.H.; Feany, M.B. Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr. Biol. 2016, 26, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paonessa, F.; Evans, L.D.; Solanki, R.; Larrieu, D.; Wray, S.; Hardy, J.; Jackson, S.P.; Livesey, F.J. Microtubules Deform the Nuclear Membrane and Disrupt Nucleocytoplasmic Transport in Tau-Mediated Frontotemporal Dementia. Cell Rep. 2019, 26, 582–593.e585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lagier-Tourenne, C. Nuclear pores: The gate to neurodegeneration. Nat. Neurosci. 2018, 21, 156–158. [Google Scholar] [CrossRef]
- Gasset-Rosa, F.; Chillon-Marinas, C.; Goginashvili, A.; Atwal, R.S.; Artates, J.W.; Tabet, R.; Wheeler, V.C.; Bang, A.G.; Cleveland, D.W.; Lagier-Tourenne, C. Polyglutamine-Expanded Huntingtin Exacerbates Age-Related Disruption of Nuclear Integrity and Nucleocytoplasmic Transport. Neuron 2017, 94, 48–57.e44. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [Green Version]
- Chin, A.C. Neuroinflammation and the cGAS-STING pathway. J. Neurophysiol. 2019, 121, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Fryer, A.L.; Abdullah, A.; Taylor, J.M.; Crack, P.J. The Complexity of the cGAS-STING Pathway in CNS Pathologies. Front. Neurosci. 2021, 15, 621501. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H.; Bohr, V.A. Signaling by cGAS-STING in Neurodegeneration, Neuroinflammation, and Aging. Trends Neurosci. 2021, 44, 83–96. [Google Scholar] [CrossRef]
- Mathur, V.; Burai, R.; Vest, R.T.; Bonanno, L.N.; Lehallier, B.; Zardeneta, M.E.; Mistry, K.N.; Do, D.; Marsh, S.E.; Abud, E.M.; et al. Activation of the STING-Dependent Type I Interferon Response Reduces Microglial Reactivity and Neuroinflammation. Neuron 2017, 96, 1290–1302.e1296. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Rajendrarao, S.; Shahani, N.; Ramirez-Jarquin, U.N.; Subramaniam, S. Cyclic GMP-AMP synthase promotes the inflammatory and autophagy responses in Huntington disease. Proc. Natl. Acad. Sci. USA 2020, 117, 15989–15999. [Google Scholar] [CrossRef] [PubMed]
- Benkhalifa, M.; Kasakyan, S.; Clement, P.; Baldi, M.; Tachdjian, G.; Demirol, A.; Gurgan, T.; Fiorentino, F.; Mohammed, M.; Qumsiyeh, M.B. Array comparative genomic hybridization profiling of first-trimester spontaneous abortions that fail to grow in vitro. Prenat. Diagn. 2005, 25, 894–900. [Google Scholar] [CrossRef]
- Cavazza, T.; Takeda, Y.; Politi, A.Z.; Aushev, M.; Aldag, P.; Baker, C.; Choudhary, M.; Bucevicius, J.; Lukinavicius, G.; Elder, K.; et al. Parental genome unification is highly error-prone in mammalian embryos. Cell 2021, 184, 2860–2877.e2822. [Google Scholar] [CrossRef] [PubMed]
- Zepeda-Mendoza, C.J.; Morton, C.C. The Iceberg under Water: Unexplored Complexity of Chromoanagenesis in Congenital Disorders. Am. J. Hum. Genet. 2019, 104, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelsen, B.; Nazaryan-Petersen, L.; Sun, W.; Mehrjouy, M.M.; Xie, G.; Chen, W.; Hjermind, L.E.; Taschner, P.E.; Tumer, Z. A germline chromothripsis event stably segregating in 11 individuals through three generations. Genet. Med. 2016, 18, 494–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolkestein, M.; Wong, J.K.L.; Thewes, V.; Korber, V.; Hlevnjak, M.; Elgaafary, S.; Schulze, M.; Kommoss, F.K.F.; Sinn, H.P.; Anzeneder, T.; et al. Chromothripsis in Human Breast Cancer. Cancer Res. 2020, 80, 4918–4931. [Google Scholar] [CrossRef]
- Cortes-Ciriano, I.; Lee, J.J.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Voronina, N.; Wong, J.K.L.; Hubschmann, D.; Hlevnjak, M.; Uhrig, S.; Heilig, C.E.; Horak, P.; Kreutzfeldt, S.; Mock, A.; Stenzinger, A.; et al. The landscape of chromothripsis across adult cancer types. Nat. Commun. 2020, 11, 2320. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015, 7, 283ra252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.O.; Li, C.W.; Xia, W.; Cha, J.H.; Chan, L.C.; Wu, Y.; Chang, S.S.; Lin, W.C.; Hsu, J.M.; Hsu, Y.H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheridan, C. Drug developers switch gears to inhibit STING. Nat. Biotechnol. 2019, 37, 199–201. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutated Gene | Name Syndrome | Clinical Pathology | OMIM ID |
|---|---|---|---|
| Lamin B2 | Barraquer–Simons syndrome (APL) | Lipodystrophy | 608709 |
| Lamin A/C | FPLD2, LDHCP, MADA, HGPS, WRN | Lipoatrophic diabetes/Lipodystrophy | 151660, 608056, 248370, 176670, 277700 |
| ZMPSTE24 | MADB | Lipodystrophy | 608612 |
| AGPAT2 or BSCL2 | Berardinelli–Seip congenital lipodystrophy type 2 | Lipodystrophy/Insulin resistance | 603100, 269700, 615924 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gauthier, B.R.; Comaills, V. Nuclear Envelope Integrity in Health and Disease: Consequences on Genome Instability and Inflammation. Int. J. Mol. Sci. 2021, 22, 7281. https://doi.org/10.3390/ijms22147281
Gauthier BR, Comaills V. Nuclear Envelope Integrity in Health and Disease: Consequences on Genome Instability and Inflammation. International Journal of Molecular Sciences. 2021; 22(14):7281. https://doi.org/10.3390/ijms22147281
Chicago/Turabian StyleGauthier, Benoit R., and Valentine Comaills. 2021. "Nuclear Envelope Integrity in Health and Disease: Consequences on Genome Instability and Inflammation" International Journal of Molecular Sciences 22, no. 14: 7281. https://doi.org/10.3390/ijms22147281
APA StyleGauthier, B. R., & Comaills, V. (2021). Nuclear Envelope Integrity in Health and Disease: Consequences on Genome Instability and Inflammation. International Journal of Molecular Sciences, 22(14), 7281. https://doi.org/10.3390/ijms22147281