
New Insights into Multiple Sclerosis Mechanisms: Lipids on the Track to Control Inflammation and Neurodegeneration




Abstract
:1. Introduction
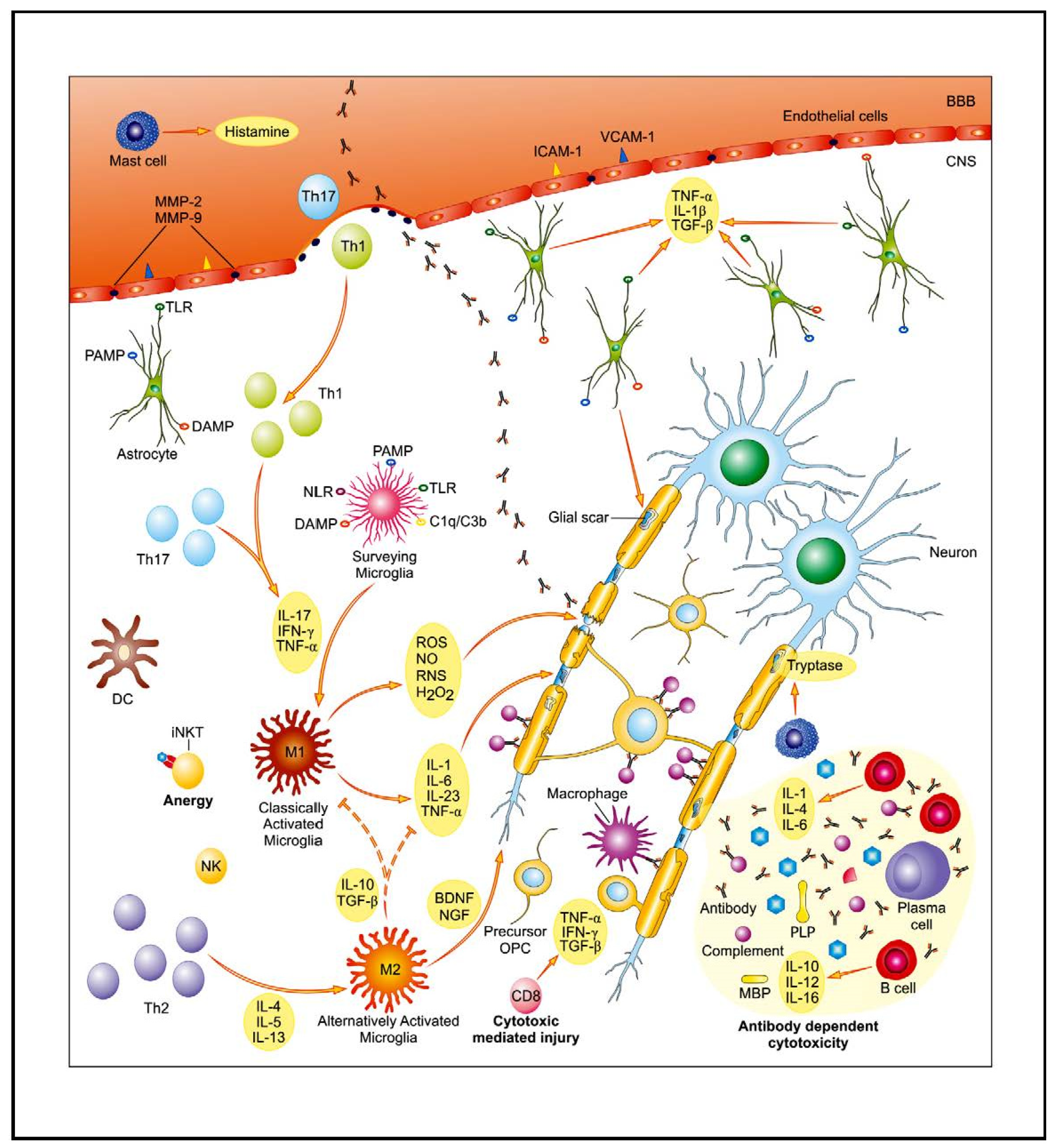
2. MS Relapse
2.1. Th1/Th17 Lymphocyte Populations and the Cellular Immune Response
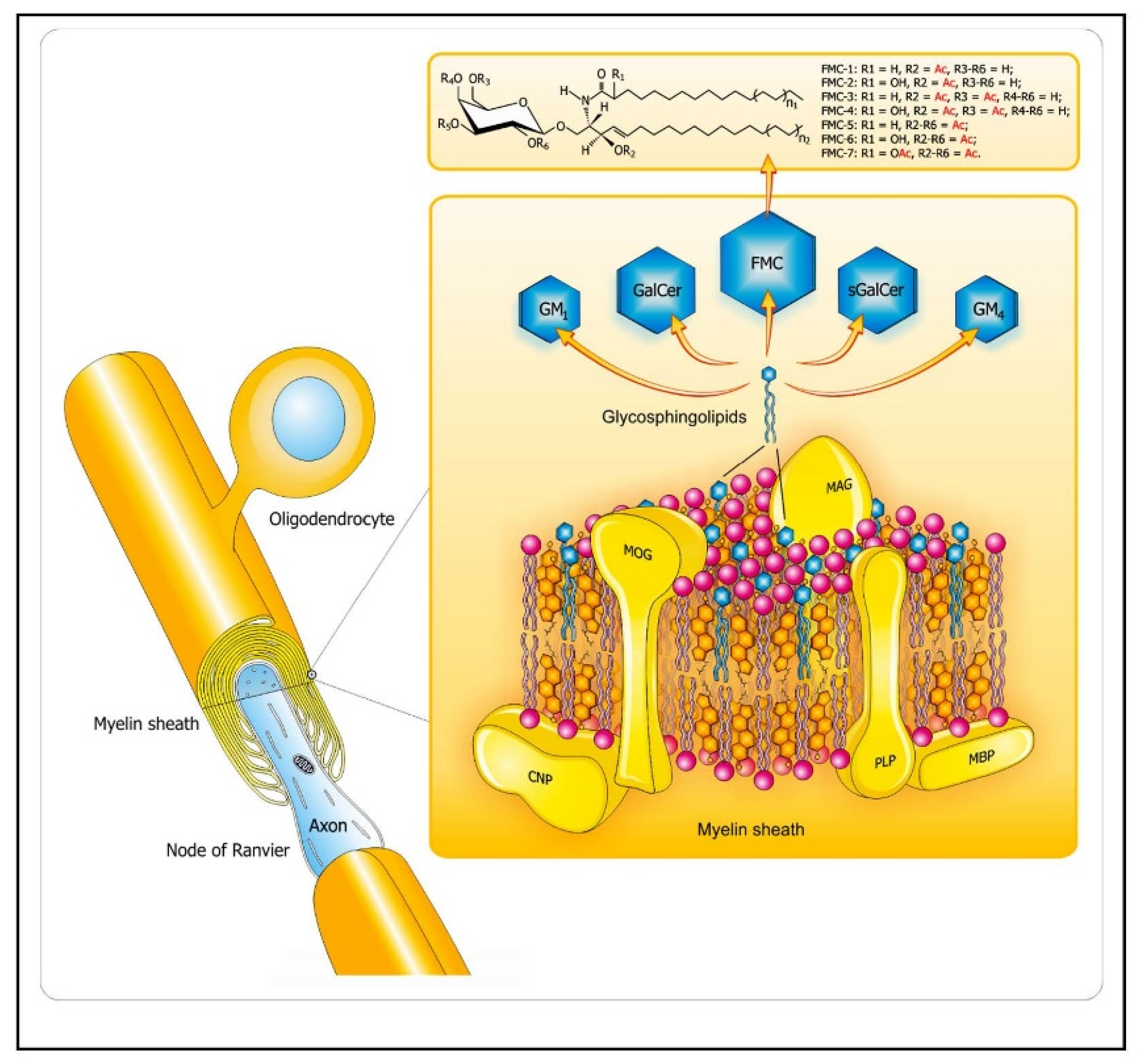
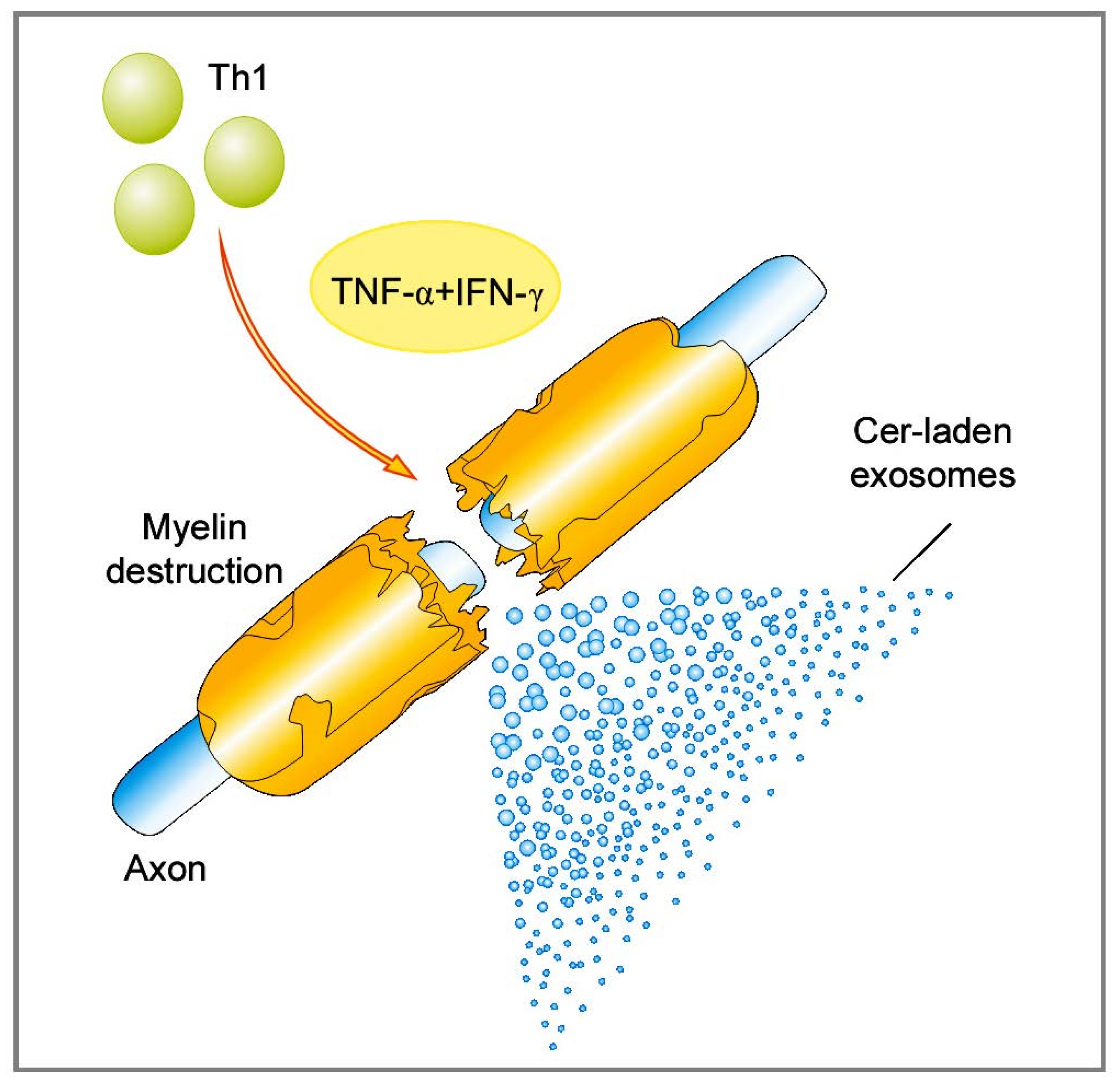
2.1.1. Role of Bioactive Sphingolipid Mediators in the Acute Inflammatory Demyelination in MS
2.1.2. Impact of Sterols on MS Autoimmunity
2.2. Antibody-Dependent Functions of B Cells and the Humoral Immune Response
Molecular Mimicry of Endogenous Myelin Lipids and Pathogenic Antigens as One of the Mechanisms of Initiation of Autoimmune Demyelination in MS
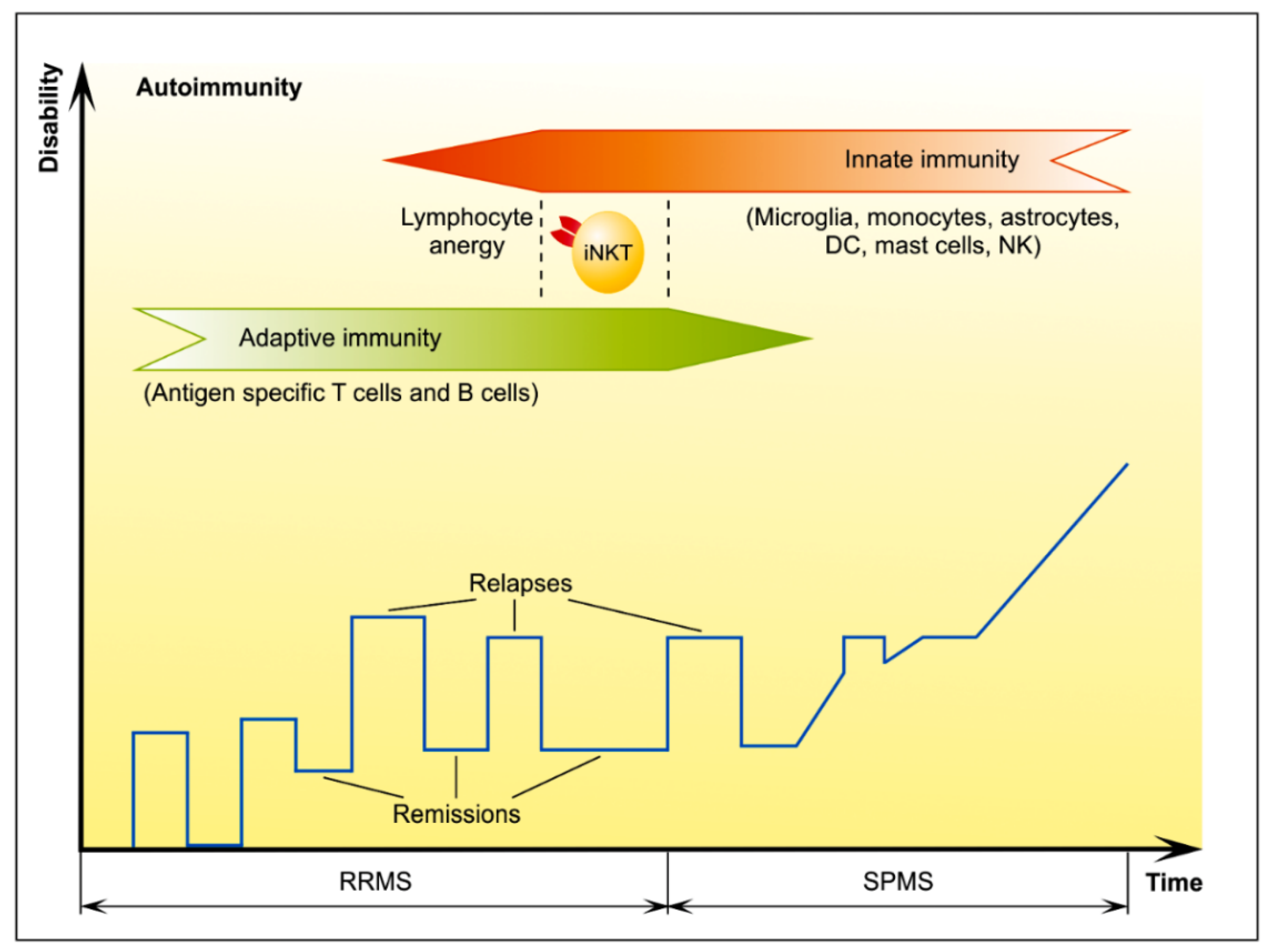
3. MS Remission
3.1. T Cells with Regulatory Features
Endogenous Glycolipid Ligand-Driven iNKT Cell Anergy as a Bridge of Adaptive Immune Response and Innate Immunity in MS
4. MS Progression
4.1. MS Neurodegeneration Background
4.1.1. Impact of Lipids on MS Progression
4.1.2. Lipids as Potential Biomarkers of the MS Course
5. Current and Future Lipids-Based Therapeutical Implications
5.1. Monitoring Therapeutic Response
5.2. Targeting CNS with Therapeutic Agents
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxy-2-nonenal |
| Ac-MS | chronic active multiple sclerosis |
| AD | Alzheimer’s disease |
| ADCC | antibody dependent cellular cytotoxicity |
| APC | antigen presenting cell |
| BBB | blood–brain barrier |
| BDNF | brain-derived neurotrophic factor |
| C1P | ceramide 1-phosphate |
| Cer | ceramide |
| CNS | central nervous system |
| COX | cyclooxygenase |
| cPLA2α | cytosolic phospholipase A2α |
| CSF | cerebrospinal fluid |
| DAMP | danger-associated molecular pattern |
| DC | dendritic cell |
| DG | diacyloglycerol |
| dhCer | dihydroceramide |
| DMF | dimethylfumarate |
| DMT | disease-modifying therapy |
| EAE | experimental autoimmune encephalomyelitis |
| EDSS | expanded disability status scale |
| EV | extracellular vesicles |
| FMC | fast migrating cerebroside |
| Foxp3 | forkhead box protein 3 |
| GalCer | galactosylcermide |
| GL | glycolipid |
| GluCer | glucosylceramide |
| GM | gray matter |
| GPC | glycerophosphatidylcholine |
| GPI | glycosylphosphatidylinositol |
| GSL | glycosphingolipids |
| HDL-C | high-density lipoprotein cholesterol |
| HexCer | hexosylceramide |
| HNE | hydroxynonenal |
| HODE | hydroxy-octadecanoic acid |
| HOG | human oligoendroglioma |
| HOT | hydroxy-alpha-linolenic acid |
| HS | healthy subjects |
| ICAM-1 | intercellular adhesion molecule 1 |
| IFN-β | interferon β |
| IFN-γ | interferon γ |
| Ig | immunoglobulin |
| IL | interleukin |
| iNKT cells | invariant natural killer T cells |
| In-MS | chronic inactive multiple sclerosis |
| I-OND | inflammatory other neurological disease |
| iTCR | invariant T cell receptor |
| iTreg | induced regulatory T cell |
| LacCer | lactosylceramide |
| LDL-C | low-density lipoprotein cholesterol |
| LPA | lysophosphatic acid |
| LPS | lipopolysaccharide |
| LXR | nuclear liver X receptor |
| lysoPC | lysophosphatidylcholine |
| lysoPC-P | putative lysophosphatidylcholine plasmalogen |
| lysoPE | lysophosphatidylethanolamine |
| lysoPI | lysophosphatidylinositol |
| lysoPL | lyso phospholipid |
| MDA | malondialdehyde |
| MHC | major histocompatibility complex |
| MMP | matrix metalloproteinase |
| MRI | magnetic resonance image |
| MS | multiple sclerosis |
| NAWM | normal appearing white matter |
| nCNS | normal central nervous system |
| NCXs | sodium calcium exchangers |
| NFL | neurofilament light chains |
| NGF | nerve growth factor |
| NI-OND | non-inflammatory other neurological disease |
| NKT cells | natural killer T cells |
| NLR | NOD-like receptors |
| NO | nitric oxide |
| Nrf2 | nuclear factor erythroid-derived 2-like 2 |
| nTreg | natural regulatory T cell |
| NZ | natalizumab |
| OCB | oligoclonal bands |
| OHC | hydroxycholesterol |
| OND | other neurological disease |
| PA | phosphatidylanisol |
| PAMP | pathogen-associated molecular pattern |
| PC | phosphatidylcholine |
| PC-P | putative phosphatidylcholine plasmalogen |
| PD | Parkinson’s disease |
| PE | phosphatidylethanolamine |
| PE-NMe | phosphtatidylethanolamine-N-methylethanolamine |
| PE-P | putative phosphatidylethanolamine plasmalogen |
| PG | phosphatidylglycerol |
| PI | phosphatidylinositol |
| PL | phospholipid |
| PPMS | primary progressive multiple sclerosis |
| PRR | pattern recognition receptor |
| PS | phosphatidylserine |
| PUFA | polyunsaturated fatty acid |
| ROS | reactive oxygen species |
| RRMS | relapsing-remitting multiple sclerosis |
| S1P | sphingosine 1-phosphate |
| S1PR | sphingosine 1-phosphate receptor |
| SL | sphingolipid |
| SM | sphingomyelin |
| Sph | sphingosine |
| SphK | sphingosine kinase |
| SPMS | secondary progressive multiple sclerosis |
| SSPE | subacute sclerosing panenecephalitis |
| SQDG | sulfoquinovosyl diacylglycerol |
| SREBPs | sterol regulatory element-binding proteins |
| TCR | T cell receptor |
| TG | triacyloglycerol |
| Th | T helper |
| TLR | Toll-like receptor |
| TNF-α | tumor necrosis factor α |
| Treg | regulatory T cell |
| VCAM-1 | vascular cell adhesion molecule 1 |
| WM | white matter |
References
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Lassmann, H. New concepts on progressive multiple sclerosis. Curr. Neurol. Neurosci. Rep. 2007, 7, 239–244. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front. Immunol. 2018, 9, 3116. [Google Scholar] [CrossRef] [PubMed]
- Podbielska, M.; O’Keeffe, J.; Hogan, E.L. Autoimmunity in multiple sclerosis: Role of sphingolipids, invariant NKT cells and other immune elements in control of inflammation and neurodegeneration. J. Neurol. Sci. 2018, 385, 198–214. [Google Scholar] [CrossRef] [PubMed]
- Popescu, B.F.; Pirko, I.; Lucchinetti, C.F. Pathology of multiple sclerosis: Where do we stand? Continuum. Minneap. Minn. 2013, 19, 901–921. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Fox, R.J.; Comi, G. The window of opportunity for treatment of progressive multiple sclerosis. Curr. Opin. Neurol. 2020, 33, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Pousinis, P.; Ramos, I.R.; Woodroofe, M.N.; Cole, L.M. Lipidomic UPLC-MS/MS profiles of normal-appearing white matter differentiate primary and secondary progressive multiple sclerosis. Metabolites 2020, 10, 366. [Google Scholar] [CrossRef] [PubMed]
- Harlow, D.E.; Honce, J.M.; Miravalle, A.A. Remyelination therapy in multiple sclerosis. Front. Neurol. 2015, 6, 257. [Google Scholar] [CrossRef] [Green Version]
- Zahoor, I.; Rui, B.; Khan, J.; Datta, I.; Giri, S. An emerging potential of metabolomics in multiple sclerosis: A comprehensive overview. Cell. Mol. Life Sci. 2021, 78, 3181–3203. [Google Scholar] [CrossRef]
- Sallusto, F. Heterogeneity of human CD4(+) T cells against microbes. Annu. Rev. Immunol. 2016, 34, 317–334. [Google Scholar] [CrossRef]
- Kunkl, M.; Frascolla, S.; Amormino, C.; Volpe, E.; Tuosto, L. T helper cells: The modulators of inflammation in multiple sclerosis. Cells 2020, 9, 482. [Google Scholar] [CrossRef] [Green Version]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.L.; Stuve, O. Update on inflammation, neurodegeneration, and immunoregulation in multiple sclerosis: Therapeutic implications. Clin. Neuropharmacol. 2009, 32, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Hirahara, K.; Nakayama, T. CD4+ T-cell subsets in inflammatory diseases: Beyond the Th1/Th2 paradigm. Int. Immunol. 2016, 28, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadidi-Niaragh, F.; Mirshafiey, A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand. J. Immunol. 2011, 74, 1–13. [Google Scholar] [CrossRef]
- Babaloo, Z.; Aliparasti, M.R.; Babaiea, F.; Almasi, S.; Baradaran, B.; Farhoudi, M. The role of Th17 cells in patients with relapsing-remitting multiple sclerosis: Interleukin-17A and interleukin-17F serum levels. Immunol. Lett. 2015, 164, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Dos Passos, G.R.; Sato, D.K.; Becker, J.; Fujihara, K. Th17 cells pathways in multiple sclerosis and neuromyelitis optica spectrum disorders: Pathophysiological and therapeutic implications. Mediat. Inflamm. 2016, 2016, 5314541. [Google Scholar] [CrossRef] [PubMed]
- Brucklacher-Waldert, V.; Stuerner, K.; Kolster, M.; Wolthausen, J.; Tolosa, E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 2009, 132, 3329–3341. [Google Scholar] [CrossRef] [PubMed]
- Prajeeth, C.K.; Kronisch, J.; Khorooshi, R.; Knier, B.; Toft-Hansen, H.; Gudi, V.; Floess, S.; Huehn, J.; Owens, T.; Korn, T.; et al. Effectors of Th1 and Th17 cells act on astrocytes and augment their neuroinflammatory properties. J. Neuroinflamm. 2017, 14, 204. [Google Scholar] [CrossRef] [Green Version]
- van Kruining, D.; Luo, Q.; van Echten-Deckert, G.; Mielke, M.M.; Bowman, A.; Ellis, S.; Oliveira, T.G.; Martinez-Martinez, P. Sphingolipids as prognostic biomarkers of neurodegeneration, neuroinflammation, and psychiatric diseases and their emerging role in lipidomic investigation methods. Adv. Drug Deliv. Rev. 2020, 159, 232–244. [Google Scholar] [CrossRef]
- Olsen, A.S.B.; Faergeman, N.J. Sphingolipids: Membrane microdomains in brain development, function and neurological diseases. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Mencarelli, C.; Martinez-Martinez, P. Ceramide function in the brain: When a slight tilt is enough. Cell. Mol. Life Sci. 2013, 70, 181–203. [Google Scholar] [CrossRef] [Green Version]
- Menon, K.K.; Piddlesden, S.J.; Bernard, C.C. Demyelinating antibodies to myelin oligodendrocyte glycoprotein and galactocerebroside induce degradation of myelin basic protein in isolated human myelin. J. Neurochem. 1997, 69, 214–222. [Google Scholar] [CrossRef]
- Mayo, L.; Trauger, S.A.; Blain, M.; Nadeau, M.; Patel, B.; Alvarez, J.I.; Mascanfroni, I.D.; Yeste, A.; Kivisakk, P.; Kallas, K.; et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 2014, 20, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Barthelmes, J.; de Bazo, A.M.; Pewzner-Jung, Y.; Schmitz, K.; Mayer, C.A.; Foerch, C.; Eberle, M.; Tafferner, N.; Ferreiros, N.; Henke, M.; et al. Lack of ceramide synthase 2 suppresses the development of experimental autoimmune encephalomyelitis by impairing the migratory capacity of neutrophils. Brain Behav. Immun. 2015, 46, 280–292. [Google Scholar] [CrossRef]
- Wheeler, D.; Bandaru, V.V.; Calabresi, P.A.; Nath, A.; Haughey, N.J. A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis. Brain 2008, 131, 3092–3102. [Google Scholar] [CrossRef] [Green Version]
- van Doorn, R.; Nijland, P.G.; Dekker, N.; Witte, M.E.; Lopes-Pinheiro, M.A.; van het Hof, B.; Kooij, G.; Reijerkerk, A.; Dijkstra, C.; van van der Valk, P.; et al. Fingolimod attenuates ceramide-induced blood-brain barrier dysfunction in multiple sclerosis by targeting reactive astrocytes. Acta Neuropathol. 2012, 124, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Podbielska, M.; Szulc, Z.M.; Kurowska, E.; Hogan, E.L.; Bielawski, J.; Bielawska, A.; Bhat, N.R. Cytokine-induced release of ceramide-enriched exosomes as a mediator of cell death signaling in an oligodendroglioma cell line. J. Lipid Res. 2016, 57, 2028–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Steelman, A.J.; Zhang, Y.; Kinney, H.C.; Li, J. Aberrant upregulation of astroglial ceramide potentiates oligodendrocyte injury. Brain Pathol. 2012, 22, 41–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurz, J.; Brunkhorst, R.; Foerch, C.; Blum, L.; Henke, M.; Gabriel, L.; Ulshofer, T.; Ferreiros, N.; Parnham, M.J.; Geisslinger, G.; et al. The relevance of ceramides and their synthesizing enzymes for multiple sclerosis. Clin. Sci. 2018, 132, 1963–1976. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Berdyshev, E.; Goya, J.; Natarajan, V.; Dawson, G. Neurons and oligodendrocytes recycle sphingosine 1-phosphate to ceramide: Significance for apoptosis and multiple sclerosis. J. Biol. Chem. 2010, 285, 14134–14143. [Google Scholar] [CrossRef] [Green Version]
- Piccinini, M.; Scandroglio, F.; Prioni, S.; Buccinna, B.; Loberto, N.; Aureli, M.; Chigorno, V.; Lupino, E.; DeMarco, G.; Lomartire, A.; et al. Deregulated sphingolipid metabolism and membrane organization in neurodegenerative disorders. Mol. Neurobiol. 2010, 41, 314–340. [Google Scholar] [CrossRef]
- El Alwani, M.; Wu, B.X.; Obeid, L.M.; Hannun, Y.A. Bioactive sphingolipids in the modulation of the inflammatory response. Pharmacol. Ther. 2006, 112, 171–183. [Google Scholar] [CrossRef]
- Scurlock, B.; Dawson, G. Differential responses of oligodendrocytes to tumor necrosis factor and other pro-apoptotic agents: Role of ceramide in apoptosis. J. Neurosci. Res. 1999, 55, 514–522. [Google Scholar] [CrossRef]
- Podbielska, M. New pathomechanisms related to autoimmune demyelination in multiple sclerosis. In Annual Report 2017; Communications and Science Promotion Department Polish Academy of Sciences: Warsaw, Poland, 2017; pp. 64–65. [Google Scholar]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orth, M.; Bellosta, S. Cholesterol: Its regulation and role in central nervous system disorders. Cholesterol 2012, 2012, 292598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkhem, I.; Cedazo-Minguez, A.; Leoni, V.; Meaney, S. Oxysterols and neurodegenerative diseases. Mol. Aspects Med. 2009, 30, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Nury, T.; Zarrouk, A.; Mackrill, J.J.; Samadi, M.; Durand, P.; Riedinger, J.M.; Doria, M.; Vejux, A.; Limagne, E.; Delmas, D.; et al. Induction of oxiapoptophagy on 158N murine oligodendrocytes treated by 7-ketocholesterol-, 7beta-hydroxycholesterol-, or 24(S)-hydroxycholesterol: Protective effects of alpha-tocopherol and docosahexaenoic acid (DHA.; C22:6 n-3). Steroids 2015, 99, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Poitelon, Y.; Kopec, A.M.; Belin, S. Myelin fat facts: An overview of lipids and fatty acid metabolism. Cells 2020, 9, 812. [Google Scholar] [CrossRef] [Green Version]
- Janowski, B.A.; Grogan, M.J.; Jones, S.A.; Wisely, G.B.; Kliewer, S.A.; Corey, E.J.; Mangelsdorf, D.J. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc. Natl. Acad. Sci. USA 1999, 96, 266–271. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Qin, X.; Wu, L.; Zhang, Y.; Sheng, X.; Yu, Q.; Sheng, H.; Xi, B.; Zhang, J.Z.; Zang, Y.Q. Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J. Clin. Investig. 2011, 121, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wagoner, G.; Douglas, J.C.; Drew, P.D. Liver X receptor agonist regulation of Th17 lymphocyte function in autoimmunity. J. Leukoc. Biol. 2009, 86, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Leoni, V.; Caccia, C. Oxysterols as biomarkers in neurodegenerative diseases. Chem. Phys. Lipids 2011, 164, 515–524. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Floris, S.; Sonke, M.; Dijkstra, C.D.; De Vries, H.E.; Lutjohann, D. 24S-hydroxycholesterol in relation to disease manifestations of acute experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2007, 85, 1499–1505. [Google Scholar] [CrossRef]
- Leoni, V.; Masterman, T.; Mousavi, F.S.; Wretlind, B.; Wahlund, L.O.; Diczfalusy, U.; Hillert, J.; Bjorkhem, I. Diagnostic use of cerebral and extracerebral oxysterols. Clin. Chem. Lab. Med. 2004, 42, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Caccia, C. 24S-hydroxycholesterol in plasma: A marker of cholesterol turnover in neurodegenerative diseases. Biochimie 2013, 95, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Vejux, A.; Ghzaiel, I.; Nury, T.; Schneider, V.; Charriere, K.; Sghaier, R.; Zarrouk, A.; Leoni, V.; Moreau, T.; Lizard, G. Oxysterols and multiple sclerosis: Physiopathology, evolutive biomarkers and therapeutic strategy. J. Steroid Biochem. Mol. Biol. 2021, 210, 105870. [Google Scholar] [CrossRef] [PubMed]
- Duc, D.; Vigne, S.; Pot, C. Oxysterols in autoimmunity. Int. J. Mol. Sci. 2019, 20, 4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wekerle, H. B cells in multiple sclerosis. Autoimmunity 2017, 50, 57–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Budingen, H.C.; Kuo, T.C.; Sirota, M.; van Belle, C.J.; Apeltsin, L.; Glanville, J.; Cree, B.A.; Gourraud, P.A.; Schwartzburg, A.; Huerta, G.; et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J. Clin. Investig. 2012, 122, 4533–4543. [Google Scholar] [CrossRef]
- Lisak, R.P.; Benjamins, J.A.; Nedelkoska, L.; Barger, J.L.; Ragheb, S.; Fan, B.; Ouamara, N.; Johnson, T.A.; Rajasekharan, S.; Bar-Or, A. Secretory products of multiple sclerosis B cells are cytotoxic to oligodendroglia in vitro. J. Neuroimmunol. 2012, 246, 85–95. [Google Scholar] [CrossRef]
- Jelcic, I.; Al Nimer, F.; Wang, J.; Lentsch, V.; Planas, R.; Jelcic, I.; Madjovski, A.; Ruhrmann, S.; Faigle, W.; Frauenknecht, K.; et al. Memory B cells activate brain-homing, autoreactive CD4(+) T cells in multiple sclerosis. Cell 2018, 175, 85–100.e23. [Google Scholar] [CrossRef] [Green Version]
- Wootla, B.; Denic, A.; Keegan, B.M.; Winters, J.L.; Astapenko, D.; Warrington, A.E.; Bieber, A.J.; Rodriguez, M. Evidence for the role of B cells and immunoglobulins in the pathogenesis of multiple sclerosis. Neurol. Res. Int. 2011, 2011, 780712. [Google Scholar] [CrossRef]
- Petzold, A. Intrathecal oligoclonal IgG synthesis in multiple sclerosis. J. Neuroimmunol. 2013, 262, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fitzner, B.; Hecker, M.; Zettl, U.K. Molecular biomarkers in cerebrospinal fluid of multiple sclerosis patients. Autoimmun. Rev. 2015, 14, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Walvoort, M.T.; Testa, C.; Eilam, R.; Aharoni, R.; Nuti, F.; Rossi, G.; Real-Fernandez, F.; Lanzillo, R.; Brescia Morra, V.; Lolli, F.; et al. Antibodies from multiple sclerosis patients preferentially recognize hyperglucosylated adhesin of non-typeable Haemophilus influenzae. Sci. Rep. 2016, 6, 39430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vartdal, F.; Vandvik, B.; Norrby, E. Viral and bacterial antibody responses in multiple sclerosis. Ann. Neurol. 1980, 8, 248–255. [Google Scholar] [CrossRef]
- Hughes, L.E.; Smith, P.A.; Bonell, S.; Natt, R.S.; Wilson, C.; Rashid, T.; Amor, S.; Thompson, E.J.; Croker, J.; Ebringer, A. Cross-reactivity between related sequences found in Acinetobacter sp., Pseudomonas aeruginosa, myelin basic protein and myelin oligodendrocyte glycoprotein in multiple sclerosis. J. Neuroimmunol. 2003, 144, 105–115. [Google Scholar] [CrossRef]
- Bagos, P.G.; Nikolopoulos, G.; Ioannidis, A. Chlamydia pneumoniae infection and the risk of multiple sclerosis: A meta-analysis. Mult. Scler. J. 2006, 12, 397–411. [Google Scholar] [CrossRef]
- Bahar, M.; Ashtari, F.; Aghaei, M.; Akbari, M.; Salari, M.; Ghalamkari, S. Mycoplasma pneumonia seroposivity in Iranian patients with relapsing-remitting multipl sclerosis: A randomized case-control study. J. Pak. Med. Assoc. 2012, 62, S6–S8. [Google Scholar]
- Bray, P.F.; Luka, J.; Bray, P.F.; Culp, K.W.; Schlight, J.P. Antibodies against Epstein-Barr nuclear antigen (EBNA) in multiple sclerosis CSF, and two pentapeptide sequence identities between EBNA and myelin basic protein. Neurology 1992, 42, 1798–1804. [Google Scholar] [CrossRef]
- Felgenhauer, K.; Schadlich, H.J.; Nekic, M.; Ackermann, R. Cerebrospinal fluid virus antibodies. A diagnostic indicator for multiple sclerosis? J. Neurol. Sci. 1985, 71, 291–299. [Google Scholar] [CrossRef]
- Schubert, J.; Weissbrich, B. Detection of virus-specific intrathecally synthesised immunoglobulin G with a fully automated enzyme immunoassay system. BMC Neurol. 2007, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandberg-Wollheim, M.; Vandvik, B.; Nadj, C.; Norrby, E. The intrathecal immune response in the early stage of multiple sclerosis. J. Neurol. Sci. 1987, 81, 45–53. [Google Scholar] [CrossRef]
- Wucherpfennig, K.W. Mechanisms for the induction of autoimmunity by infectious agents. J. Clin. Investig. 2001, 108, 1097–1104. [Google Scholar] [CrossRef]
- Oldstone, M.B. Molecular mimicry, microbial infection, and autoimmune disease: Evolution of the concept. Curr. Top. Microbiol. Immunol. 2005, 296, 1–17. [Google Scholar]
- Cross, A.H.; Trotter, J.L.; Lyons, J. B cells and antibodies in CNS demyelinating disease. J. Neuroimmunol. 2001, 112, 1–14. [Google Scholar] [CrossRef]
- Dharmasaroja, P. Specificity of autoantibodies to epitopes of myelin proteins in multiple sclerosis. J. Neurol. Sci. 2003, 206, 7–16. [Google Scholar] [CrossRef]
- Schmidt, S. Candidate autoantigens in multiple sclerosis. Mult. Scler. J. 1999, 5, 147–160. [Google Scholar] [CrossRef]
- Acarin, N.; Rio, J.; Fernandez, A.L.; Tintore, M.; Duran, I.; Galan, I.; Montalban, X. Different antiganglioside antibody pattern between relapsing-remitting and progressive multiple sclerosis. Acta Neurol. Scand. 1996, 93, 99–103. [Google Scholar] [CrossRef]
- Mata, S.; Lolli, F.; Soderstrom, M.; Pinto, F.; Link, H. Multiple sclerosis is associated with enhanced B cell responses to the ganglioside GD1a. Mult. Scler. J. 1999, 5, 379–388. [Google Scholar] [CrossRef]
- Sadatipour, B.T.; Greer, J.M.; Pender, M.P. Increased circulating antiganglioside antibodies in primary and secondary progressive multiple sclerosis. Ann. Neurol. 1998, 44, 980–983. [Google Scholar] [CrossRef] [Green Version]
- Pender, M.P.; Csurhes, P.A.; Wolfe, N.P.; Hooper, K.D.; Good, M.F.; McCombe, P.A.; Greer, J.M. Increased circulating T cell reactivity to GM3 and GQ1b gangliosides in primary progressive multiple sclerosis. J. Clin. Neurosci. 2003, 10, 63–66. [Google Scholar] [CrossRef] [Green Version]
- Ryberg, B. Multiple specificities of antibrain antibodies in multiple sclerosis and chronic myelopathy. J. Neurol. Sci. 1978, 38, 357–382. [Google Scholar] [CrossRef]
- Ilyas, A.A.; Chen, Z.W.; Cook, S.D. Antibodies to sulfatide in cerebrospinal fluid of patients with multiple sclerosis. J. Neuroimmunol. 2003, 139, 76–80. [Google Scholar] [CrossRef]
- Brennan, K.M.; Galban-Horcajo, F.; Rinaldi, S.; O’Leary, C.P.; Goodyear, C.S.; Kalna, G.; Arthur, A.; Elliot, C.; Barnett, S.; Linington, C.; et al. Lipid arrays identify myelin-derived lipids and lipid complexes as prominent targets for oligoclonal band antibodies in multiple sclerosis. J. Neuroimmunol. 2011, 238, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, Y.; Yamamoto, T. Characterization of serum anti-phospholipid antibodies in patients with multiple sclerosis. Tohoku J. Exp. Med. 1996, 178, 203–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanter, J.L.; Narayana, S.; Ho, P.P.; Catz, I.; Warren, K.G.; Sobel, R.A.; Steinman, L.; Robinson, W.H. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat. Med. 2006, 12, 138–143. [Google Scholar] [CrossRef]
- Quintana, F.J.; Farez, M.F.; Viglietta, V.; Iglesias, A.H.; Merbl, Y.; Izquierdo, G.; Lucas, M.; Basso, A.S.; Khoury, S.J.; Lucchinetti, C.F.; et al. Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 18889–18894. [Google Scholar] [CrossRef] [Green Version]
- Menge, T.; Lalive, P.H.; von Budingen, H.C.; Cree, B.; Hauser, S.L.; Genain, C.P. Antibody responses against galactocerebroside are potential stage-specific biomarkers in multiple sclerosis. J. Allergy Clin. Immunol. 2005, 116, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Podbielska, M.; Dasgupta, S.; Levery, S.B.; Tourtellotte, W.W.; Annuk, H.; Moran, A.P.; Hogan, E.L. Novel myelin penta- and hexa-acetyl-galactosyl-ceramides: Structural characterization and immunoreactivity in cerebrospinal fluid. J. Lipid Res. 2010, 51, 1394–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahringer, U.; Wagner, F.; Rietschel, E.T.; Ben-Menachem, G.; Deutsch, J.; Rottem, S. Primary structure of a new phosphocholine-containing glycoglycerolipid of Mycoplasma fermentans. J. Biol. Chem. 1997, 272, 26262–26270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sospedra, M.; Martin, R. Molecular mimicry in multiple sclerosis. Autoimmunity 2006, 39, 3–8. [Google Scholar] [CrossRef]
- McCoy, L.; Tsunoda, I.; Fujinami, R.S. Multiple sclerosis and virus induced immune responses: Autoimmunity can be primed by molecular mimicry and augmented by bystander activation. Autoimmunity 2006, 39, 9–19. [Google Scholar] [CrossRef]
- Barnett, L.A.; Fujinami, R.S. Molecular mimicry: A mechanism for autoimmune injury. FASEB J. 1992, 6, 840–844. [Google Scholar] [CrossRef]
- Hogan, E.L.; Podbielska, M.; O’Keeffe, J. Implications of lymphocyte anergy to glycolipids in multiple sclerosis (MS): iNKT cells may mediate the MS infectious trigger. J. Clin. Cell Immunol. 2013, 4, 144. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Ono, M.; Setoguchi, R.; Yagi, H.; Hori, S.; Fehervari, Z.; Shimizu, J.; Takahashi, T.; Nomura, T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 2006, 212, 8–27. [Google Scholar] [CrossRef]
- Grant, C.R.; Liberal, R.; Mieli-Vergani, G.; Vergani, D.; Longhi, M.S. Regulatory T-cells in autoimmune diseases: Challenges, controversies and--yet--unanswered questions. Autoimmun. Rev. 2015, 14, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Curotto de Lafaille, M.A.; Lafaille, J.J. Natural and adaptive foxp3+ regulatory T cells: More of the same or a division of labor? Immunity 2009, 30, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Correale, J.; Farez, M. Association between parasite infection and immune responses in multiple sclerosis. Ann. Neurol. 2007, 61, 97–108. [Google Scholar] [CrossRef]
- Endharti, A.T.; Rifa, I.M.; Shi, Z.; Fukuoka, Y.; Nakahara, Y.; Kawamoto, Y.; Takeda, K.; Isobe, K.; Suzuki, H. Cutting edge: CD8+CD122+ regulatory T cells produce IL-10 to suppress IFN-gamma production and proliferation of CD8+ T cells. J. Immunol. 2005, 175, 7093–7097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, M.; Tashiro, T.; Dashtsoodol, N.; Hongo, N.; Watarai, H. The specialized iNKT cell system recognizes glycolipid antigens and bridges the innate and acquired immune systems with potential applications for cancer therapy. Int. Immunol. 2010, 22, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Keeffe, J.; Podbielska, M.; Hogan, E.L. Invariant natural killer T cells and their ligands: Focus on multiple sclerosis. Immunology 2015, 145, 468–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illes, Z.; Kondo, T.; Newcombe, J.; Oka, N.; Tabira, T.; Yamamura, T. Differential expression of NK T cell V alpha 24J alpha Q invariant TCR chain in the lesions of multiple sclerosis and chronic inflammatory demyelinating polyneuropathy. J. Immunol. 2000, 164, 4375–4381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demoulins, T.; Gachelin, G.; Bequet, D.; Dormont, D. A biased Valpha24+ T-cell repertoire leads to circulating NKT-cell defects in a multiple sclerosis patient at the onset of his disease. Immunol. Lett. 2003, 90, 223–228. [Google Scholar] [CrossRef]
- Araki, M.; Kondo, T.; Gumperz, J.E.; Brenner, M.B.; Miyake, S.; Yamamura, T. Th2 bias of CD4+ NKT cells derived from multiple sclerosis in remission. Int. Immunol. 2003, 15, 279–288. [Google Scholar] [CrossRef] [Green Version]
- O’Keeffe, J.; Gately, C.M.; Counihan, T.; Hennessy, M.; Leahy, T.; Moran, A.P.; Hogan, E.L. T-cells expressing natural killer (NK) receptors are altered in multiple sclerosis and responses to alpha-galactosylceramide are impaired. J. Neurol. Sci. 2008, 275, 22–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gately, C.M.; Podbielska, M.; Counihan, T.; Hennessy, M.; Leahy, T.; Moran, A.P.; Hogan, E.L.; O’Keeffe, J. Invariant Natural Killer T-cell anergy to endogenous myelin acetyl-glycolipids in multiple sclerosis. J. Neuroimmunol. 2013, 259, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jahng, A.W.; Maricic, I.; Pedersen, B.; Burdin, N.; Naidenko, O.; Kronenberg, M.; Koezuka, Y.; Kumar, V. Activation of natural killer T cells potentiates or prevents experimental autoimmune encephalomyelitis. J. Exp. Med. 2001, 194, 1789–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mycko, M.P.; Sliwinska, B.; Cichalewska, M.; Cwiklinska, H.; Raine, C.S.; Selmaj, K.W. Brain glycolipids suppress T helper cells and inhibit autoimmune demyelination. J. Neurosci. 2014, 34, 8646–8658. [Google Scholar] [CrossRef] [Green Version]
- Correale, J. The role of microglial activation in disease progression. Mult. Scler. J. 2014, 20, 1288–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramaniam, B.; Carter, D.A.; Mayer, E.J.; Dick, A.D. Microglia derived IL-6 suppresses neurosphere generation from adult human retinal cell suspensions. Exp. Eye Res. 2009, 89, 757–766. [Google Scholar] [CrossRef]
- Dheen, S.T.; Jun, Y.; Yan, Z.; Tay, S.S.; Ling, E.A. Retinoic acid inhibits expression of TNF-alpha and iNOS in activated rat microglia. Glia 2005, 50, 21–31. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, S.S.; Cho, J.J.; Choi, D.H.; Hwang, O.; Shin, D.H.; Chun, H.S.; Beal, M.F.; Joh, T.H. Matrix metalloproteinase-3: A novel signaling proteinase from apoptotic neuronal cells that activates microglia. J. Neurosci. 2005, 25, 3701–3711. [Google Scholar] [CrossRef] [Green Version]
- Levesque, S.; Wilson, B.; Gregoria, V.; Thorpe, L.B.; Dallas, S.; Polikov, V.S.; Hong, J.S.; Block, M.L. Reactive microgliosis: Extracellular micro-calpain and microglia-mediated dopaminergic neurotoxicity. Brain 2010, 133, 808–821. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Liu, B.; Gao, H.M.; Wang, J.Y.; Jeohn, G.H.; Cooper, C.L.; Hong, J.S. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann. N. Y. Acad. Sci. 2002, 962, 318–331. [Google Scholar] [CrossRef]
- Sghaier, R.; Zarrouk, A.; Nury, T.; Badreddine, I.; O’Brien, N.; Mackrill, J.J.; Vejux, A.; Samadi, M.; Nasser, B.; Caccia, C.; et al. Biotin attenuation of oxidative stress, mitochondrial dysfunction, lipid metabolism alteration and 7beta-hydroxycholesterol-induced cell death in 158N murine oligodendrocytes. Free Radic Res. 2019, 53, 535–561. [Google Scholar] [CrossRef] [Green Version]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, S.A.; Suarez-Pozos, E.; Escalante, M.; Myo, Y.P.; Fuss, B. Sodium-Calcium Exchangers of the SLC8 Family in Oligodendrocytes: Functional Properties in Health and Disease. Neurochem. Res.. 2020, 45, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Giladi, M.; Shor, R.; Lisnyansky, M.; Khananshvili, D. Structure-Functional Basis of Ion Transport in Sodium-Calcium Exchanger (NCX) Proteins. Int. J. Mol. Sci. 2016, 17, 1949. [Google Scholar] [CrossRef] [PubMed]
- DiPolo, R.; Beauge, L. Sodium/calcium exchanger: Influence of metabolic regulation on ion carrier interactions. Physiol. Rev. 2006, 86, 155–203. [Google Scholar] [CrossRef] [Green Version]
- Cousido-Siah, A.; Ayoub, D.; Berberian, G.; Bollo, M.; Van Dorsselaer, A.; Debaene, F.; DiPolo, R.; Petrova, T.; Schulze-Briese, C.; Olieric, V.; et al. Structural and functional studies of ReP1-NCXSQ, a protein regulating the squid nerve Na+/Ca2+ exchanger. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 1098–1107. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Kintner, D.B.; Jones, M.; Matsuda, T.; Baba, A.; Kiedrowski, L.; Sun, D. AMPA-mediated excitotoxicity in oligodendrocytes: Role for Na(+)-K(+)-Cl(-) co-transport and reversal of Na(+)/Ca(2+) exchanger. J. Neurochem. 2007, 102, 1783–1795. [Google Scholar] [CrossRef]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Yeung, M.S.Y.; Djelloul, M.; Steiner, E.; Bernard, S.; Salehpour, M.; Possnert, G.; Brundin, L.; Frisen, J. Dynamics of oligodendrocyte generation in multiple sclerosis. Nature 2019, 566, 538–542. [Google Scholar] [CrossRef]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchetti, S.; Fransen, N.L.; van Eden, C.G.; Ramaglia, V.; Mason, M.; Huitinga, I. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: A retrospective autopsy cohort analysis. Acta Neuropathol. 2018, 135, 511–528. [Google Scholar] [CrossRef] [Green Version]
- Dal-Bianco, A.; Grabner, G.; Kronnerwetter, C.; Weber, M.; Hoftberger, R.; Berger, T.; Auff, E.; Leutmezer, F.; Trattnig, S.; Lassmann, H.; et al. Slow expansion of multiple sclerosis iron rim lesions: Pathology and 7 T magnetic resonance imaging. Acta Neuropathol. 2017, 133, 25–42. [Google Scholar] [CrossRef] [Green Version]
- Absinta, M.; Lassmann, H.; Trapp, B.D. Mechanisms underlying progression in multiple sclerosis. Curr. Opin. Neurol. 2020, 33, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Reynolds, R.; Calabrese, M. MRI of cortical lesions and its use in studying their role in MS pathogenesis and disease course. Brain Pathol. 2018, 28, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Podbielska, M.; Szulc, Z.M.; Ariga, T.; Pokryszko-Dragan, A.; Fortuna, W.; Bilinska, M.; Podemski, R.; Jaskiewicz, E.; Kurowska, E.; Yu, R.K.; et al. Distinctive sphingolipid patterns in chronic multiple sclerosis lesions. J. Lipid Res. 2020, 61, 1464–1479. [Google Scholar] [CrossRef] [PubMed]
- Podbielska, M.; Krotkiewski, H.; Hogan, E.L. Signaling and regulatory functions of bioactive sphingolipids as therapeutic targets in multiple sclerosis. Neurochem. Res. 2012, 37, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Vidaurre, O.G.; Haines, J.D.; Katz Sand, I.; Adula, K.P.; Huynh, J.L.; McGraw, C.A.; Zhang, F.; Varghese, M.; Sotirchos, E.; Bhargava, P.; et al. Cerebrospinal fluid ceramides from patients with multiple sclerosis impair neuronal bioenergetics. Brain 2014, 137, 2271–2286. [Google Scholar] [CrossRef]
- Pettus, B.J.; Bielawska, A.; Spiegel, S.; Roddy, P.; Hannun, Y.A.; Chalfant, C.E. Ceramide kinase mediates cytokine- and calcium ionophore-induced arachidonic acid release. J. Biol. Chem. 2003, 278, 38206–38213. [Google Scholar] [CrossRef] [Green Version]
- Pettus, B.J.; Bielawska, A.; Subramanian, P.; Wijesinghe, D.S.; Maceyka, M.; Leslie, C.C.; Evans, J.H.; Freiberg, J.; Roddy, P.; Hannun, Y.A.; et al. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J. Biol. Chem. 2004, 279, 11320–11326. [Google Scholar] [CrossRef] [Green Version]
- Simanshu, D.K.; Kamlekar, R.K.; Wijesinghe, D.S.; Zou, X.; Zhai, X.; Mishra, S.K.; Molotkovsky, J.G.; Malinina, L.; Hinchcliffe, E.H.; Chalfant, C.E.; et al. Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature 2013, 500, 463–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoessel, D.; Stellmann, J.P.; Willing, A.; Behrens, B.; Rosenkranz, S.C.; Hodecker, S.C.; Sturner, K.H.; Reinhardt, S.; Fleischer, S.; Deuschle, C.; et al. Metabolomic Profiles for Primary Progressive Multiple Sclerosis Stratification and Disease Course Monitoring. Front. Hum. Neurosci. 2018, 12, 226. [Google Scholar] [CrossRef]
- Pieragostino, D.; D’Alessandro, M.; di Ioia, M.; Rossi, C.; Zucchelli, M.; Urbani, A.; Di Ilio, C.; Lugaresi, A.; Sacchetta, P.; Del Boccio, P. An integrated metabolomics approach for the research of new cerebrospinal fluid biomarkers of multiple sclerosis. Mol. Biosyst. 2015, 11, 1563–1572. [Google Scholar] [CrossRef] [Green Version]
- Nogueras, L.; Gonzalo, H.; Jove, M.; Sol, J.; Gil-Sanchez, A.; Hervas, J.V.; Valcheva, P.; Gonzalez-Mingot, C.; Solana, M.J.; Peralta, S.; et al. Lipid profile of cerebrospinal fluid in multiple sclerosis patients: A potential tool for diagnosis. Sci. Rep. 2019, 9, 11313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatruda, M.; Petracca, M.; Wentling, M.; Inbar, B.; Castro, K.; Chen, E.Y.; Kiebish, M.A.; Edwards, K.; Inglese, M.; Casaccia, P. Retrospective unbiased plasma lipidomic of progressive multiple sclerosis patients-identifies lipids discriminating those with faster clinical deterioration. Sci. Rep. 2020, 10, 15644. [Google Scholar] [CrossRef]
- Yung, Y.C.; Stoddard, N.C.; Mirendil, H.; Chun, J. Lysophosphatidic Acid signaling in the nervous system. Neuron 2015, 85, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Foster, C.A.; Howard, L.M.; Schweitzer, A.; Persohn, E.; Hiestand, P.C.; Balatoni, B.; Reuschel, R.; Beerli, C.; Schwartz, M.; Billich, A. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: Consequences for mode of action in multiple sclerosis. J. Pharmacol. Exp. Ther. 2007, 323, 469–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Doorn, R.; Van Horssen, J.; Verzijl, D.; Witte, M.; Ronken, E.; Van Het Hof, B.; Lakeman, K.; Dijkstra, C.D.; Van Der Valk, P.; Reijerkerk, A.; et al. Sphingosine 1-phosphate receptor 1 and 3 are upregulated in multiple sclerosis lesions. Glia 2010, 58, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Rothhammer, V.; Kenison, J.E.; Tjon, E.; Takenaka, M.C.; de Lima, K.A.; Borucki, D.M.; Chao, C.C.; Wilz, A.; Blain, M.; Healy, L.; et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, 2012–2017. [Google Scholar] [CrossRef] [Green Version]
- Hagen, N.; Van Veldhoven, P.P.; Proia, R.L.; Park, H.; Merrill, A.H., Jr.; van Echten-Deckert, G. Subcellular origin of sphingosine 1-phosphate is essential for its toxic effect in lyase-deficient neurons. J. Biol. Chem. 2009, 284, 11346–11353. [Google Scholar] [CrossRef] [Green Version]
- Kulakowska, A.; Zendzian-Piotrowska, M.; Baranowski, M.; Kononczuk, T.; Drozdowski, W.; Gorski, J.; Bucki, R. Intrathecal increase of sphingosine 1-phosphate at early stage multiple sclerosis. Neurosci. Lett. 2010, 477, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Pebay, A.; Toutant, M.; Premont, J.; Calvo, C.F.; Venance, L.; Cordier, J.; Glowinski, J.; Tence, M. Sphingosine-1-phosphate induces proliferation of astrocytes: Regulation by intracellular signalling cascades. Eur. J. Neurosci. 2001, 13, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V. FTY720 (fingolimod) in Multiple Sclerosis: Therapeutic effects in the immune and the central nervous system. Br. J. Pharmacol. 2009, 158, 1173–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamczyk, B.; Niedziela, N.; Adamczyk-Sowa, M. Novel approaches of oxidative stress mechanisms in the multiple sclerosis pathophysiology and therapy. In Multiple Sclerosis: Perspectives in Treatment and Pathogenesis; Zagon, I.S., McLaughlin, P.J., Eds.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Nakazawa, T.; Miyanoki, Y.; Urano, Y.; Uehara, M.; Saito, Y.; Noguchi, N. Effect of vitamin E on 24(S)-hydroxycholesterol-induced necroptosis-like cell death and apoptosis. J. Steroid Biochem. Mol. Biol. 2017, 169, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, Y.; Meaney, S.; Heverin, M.; Ekstrom, L.; Brafman, A.; Shafir, M.; Andersson, U.; Olin, M.; Eggertsen, G.; Diczfalusy, U.; et al. Studies on the transcriptional regulation of cholesterol 24-hydroxylase (CYP46A1): Marked insensitivity toward different regulatory axes. J. Biol. Chem. 2006, 281, 3810–3820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, G.G.; Pacheco-Moises, F.P.; Bitzer-Quintero, O.K.; Ramirez-Anguiano, A.C.; Flores-Alvarado, L.J.; Ramirez-Ramirez, V.; Macias-Islas, M.A.; Torres-Sanchez, E.D. Immunology and oxidative stress in multiple sclerosis: Clinical and basic approach. Clin. Dev. Immunol. 2013, 2013, 708659. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Umeno, A.; Shichiri, M. Lipid peroxidation biomarkers for evaluating oxidative stress and assessing antioxidant capacity in vivo. J. Clin. Biochem. Nutr. 2013, 52, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Ibitoye, R.; Kemp, K.; Rice, C.; Hares, K.; Scolding, N.; Wilkins, A. Oxidative stress-related biomarkers in multiple sclerosis: A review. Biomark. Med. 2016, 10, 889–902. [Google Scholar] [CrossRef] [Green Version]
- Adamczyk, B.; Adamczyk-Sowa, M. New insights into the role of oxidative stress mechanisms in the pathophysiology and treatment of multiple sclerosis. Oxid. Med. Cell. Longev. 2016, 2016, 1973834. [Google Scholar] [CrossRef] [Green Version]
- Sghaier, R.; Nury, T.; Leoni, V.; Caccia, C.; Pais De Barros, J.P.; Cherif, A.; Vejux, A.; Moreau, T.; Limem, K.; Samadi, M.; et al. Dimethyl fumarate and monomethyl fumarate attenuate oxidative stress and mitochondrial alterations leading to oxiapoptophagy in 158N murine oligodendrocytes treated with 7beta-hydroxycholesterol. J. Steroid Biochem. Mol. Biol. 2019, 194, 105432. [Google Scholar] [CrossRef]
- Checa, A.; Khademi, M.; Sar, D.G.; Haeggstrom, J.Z.; Lundberg, J.O.; Piehl, F.; Olsson, T.; Wheelock, C.E. Hexosylceramides as intrathecal markers of worsening disability in multiple sclerosis. Mult. Scler. J. 2015, 21, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Novakova, L.; Axelsson, M.; Malmestrom, C.; Zetterberg, H.; Bjorkhem, I.; Karrenbauer, V.D.; Lycke, J. Reduced cerebrospinal fluid concentrations of oxysterols in response to natalizumab treatment of relapsing remitting multiple sclerosis. J. Neurol. Sci. 2015, 358, 201–206. [Google Scholar] [CrossRef] [PubMed]
- van de Kraats, C.; Killestein, J.; Popescu, V.; Rijkers, E.; Vrenken, H.; Lutjohann, D.; Barkhof, F.; Polman, C.H.; Teunissen, C.E. Oxysterols and cholesterol precursors correlate to magnetic resonance imaging measures of neurodegeneration in multiple sclerosis. Mult. Scler. J. 2014, 20, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Murali, N.; Browne, R.W.; Fellows Maxwell, K.; Bodziak, M.L.; Jakimovski, D.; Hagemeier, J.; Bergsland, N.; Weinstock-Guttman, B.; Zivadinov, R.; Ramanathan, M. Cholesterol and neurodegeneration: Longitudinal changes in serum cholesterol biomarkers are associated with new lesions and gray matter atrophy in multiple sclerosis over 5 years of follow-up. Eur. J. Neurol. 2020, 27, 188-e4. [Google Scholar] [CrossRef]
- Villoslada, P.; Alonso, C.; Agirrezabal, I.; Kotelnikova, E.; Zubizarreta, I.; Pulido-Valdeolivas, I.; Saiz, A.; Comabella, M.; Montalban, X.; Villar, L.; et al. Metabolomic signatures associated with disease severity in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McComb, M.; Parambi, R.; Browne, R.W.; Bodziak, M.L.; Jakimovski, D.; Bergsland, N.; Maceski, A.; Weinstock-Guttman, B.; Kuhle, J.; Zivadinov, R.; et al. Apolipoproteins AI and E are associated with neuroaxonal injury to gray matter in multiple sclerosis. Mult. Scler. Relat. Disord. 2020, 45, 102389. [Google Scholar] [CrossRef]
- Moore, G.R. Current concepts in the neuropathology and pathogenesis of multiple sclerosis. Can. J. Neurol. Sci. 2010, 37 (Suppl. S2), S5–S15. [Google Scholar] [CrossRef] [Green Version]
- Behrangi, N.; Fischbach, F.; Kipp, M. Mechanism of siponimod: Anti-inflammatory and neuroprotective mode of action. Cells 2019, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Zou, Q.; Chen, Y.; Liao, X.; Chen, R.; Ma, L.; Zhang, D.; Li, Q. Efficacy and acceptability of the S1P receptor in the treatment of multiple sclerosis: A meta-analysis. Neurol. Sci. 2021, 42, 1687–1695. [Google Scholar] [CrossRef]
- Bross, M.; Hackett, M.; Bernitsas, E. Approved and emerging disease modifying therapies on neurodegeneration in multiple sclerosis. Int. J. Mol. Sci. 2020, 21, 4312. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Yadav, S.K.; Soin, D.; Ito, K.; Dhib-Jalbut, S. Insight into the mechanism of action of dimethyl fumarate in multiple sclerosis. J. Mol. Med. 2019, 97, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, C.H.; Cohen, J.A. The rise and fall of high-dose biotin to treat progressive multiple sclerosis. Neurotherapeutics 2020, 17, 968–970. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Gutierrez-Vazquez, C.; Rothhammer, V.; Mayo, L.; Wheeler, M.A.; Tjon, E.C.; Zandee, S.E.J.; Blain, M.; de Lima, K.A.; Takenaka, M.C.; et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 2019, 179, 1483–1498.e22. [Google Scholar] [CrossRef] [PubMed]
- Alaamery, M.; Albesher, N.; Aljawini, N.; Alsuwailm, M.; Massadeh, S.; Wheeler, M.A.; Chao, C.C.; Quintana, F.J. Role of sphingolipid metabolism in neurodegeneration. J. Neurochem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Poisson, L.M.; Suhail, H.; Singh, J.; Datta, I.; Denic, A.; Labuzek, K.; Hoda, M.N.; Shankar, A.; Kumar, A.; Cerghet, M.; et al. Untargeted plasma metabolomics identifies endogenous metabolite with drug-like properties in chronic animal model of multiple sclerosis. J. Biol. Chem. 2015, 290, 30697–30712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooij, G.; Troletti, C.D.; Leuti, A.; Norris, P.C.; Riley, I.; Albanese, M.; Ruggieri, S.; Libreros, S.; van der Pol, S.M.A.; van Het Hof, B.; et al. Specialized pro-resolving lipid mediators are differentially altered in peripheral blood of patients with multiple sclerosis and attenuate monocyte and blood-brain barrier dysfunction. Haematologica 2020, 105, 2056–2070. [Google Scholar] [CrossRef] [Green Version]
- Sturner, K.H.; Werz, O.; Koeberle, A.; Otto, M.; Pless, O.; Leypoldt, F.; Paul, F.; Heesen, C. Lipid mediator profiles predict response to therapy with an oral frankincense extract in relapsing-remitting multiple sclerosis. Sci. Rep. 2020, 10, 8776. [Google Scholar] [CrossRef]
- Bhargava, P.; Fitzgerald, K.C.; Venkata, S.L.V.; Smith, M.D.; Kornberg, M.D.; Mowry, E.M.; Haughey, N.J.; Calabresi, P.A. Dimethyl fumarate treatment induces lipid metabolism alterations that are linked to immunological changes. Ann. Clin. Transl. Neurol. 2019, 6, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottenlinger, F.M.; Mayer, C.A.; Ferreiros, N.; Schreiber, Y.; Schwiebs, A.; Schmidt, K.G.; Ackermann, H.; Pfeilschifter, J.M.; Radeke, H.H. Interferon-beta increases plasma ceramides of specific chain length in multiple sclerosis patients, unlike fingolimod or natalizumab. Front. Pharmacol. 2016, 7, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauma, I.; Huhtala, H.; Soilu-Hanninen, M.; Kuusisto, H. Lipid Profile Alterations during Fingolimod Treatment in Multiple Sclerosis Patients. J. Neuroimmune Pharmacol. 2020, 15, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Vanherle, S.; Haidar, M.; Irobi, J.; Bogie, J.F.J.; Hendriks, J.J.A. Extracellular vesicle-associated lipids in central nervous system disorders. Adv. Drug Deliv. Rev. 2020, 159, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.I.; Lopes, C.M.; Amaral, M.H.; Costa, P.C. Current insights on lipid nanocarrier-assisted drug delivery in the treatment of neurodegenerative diseases. Eur. J. Pharm. Biopharm. 2020, 149, 192–217. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proposed Lipid Subspecies | Altered in/Compared to | Material | Reference |
|---|---|---|---|
| C18:0-dhCer↑, C18:1-dhCer↑, C24-dhCer↑, C24:1-dhCer↑ | Ac-MS/nCNS and Ac-MS/In-MS | Plaque | [128] |
| C18:0-SM↓, C18:1-SM↓, C24-SM↓, C24:1-SM↓ | In-MS/nCNS and In-MS/Ac-MS | Plaque | [128] |
| C16:0-HexCer↑, C18:0-HexCer↑, C18:1-HexCer↑, C24:0-HexCer↑, C24:1-HexCer↑ | In-MS/nCNS and In-MS/Ac-MS | Plaque | [128] |
| C16:0-C1P↑, C18:0-C1P↑, C18:1-C1P↑, C24:0-C1P↑, C24:1-C1P↑ | In-MS/nCNS and In-MS/Ac-MS | Plaque | [128] |
| Total LacCer↑ | MS/WM of MS | Plaque | [25] |
| C18:0-Cer↑ | Ac-MS/nCNS | Plaque | [30] |
| Total Cer↓, Sph↑, S1P↓, C16:0/C24:0-Cer ratio↑, C18:0/C24:0-Cer ratio↑ | MS/nCNS, WM of AD | Plaque | [32] |
| C26:1-C1P↑, C18:0/C15:0-DG↑, C18:2/C19:0-DG↑, C10:0-lysoPC↑, C17:0-lysoPC↓, C22:2-lysoPE↑, C18:1/C18:0-PA↑, C18:1/C21:0-PA↓, C20:4/C20:0-PA↑, C21:2/C24:0-PA↓, C22:6/C18:1-PA↓, C20:5/C18:2-PC↑, C18:0/C18:1-PC-P↓, C18:1/C22:1-PE↓, C18:2/C16:0-PE↓, C18:2/C21:0-PE↓, C18:2/C22:1-PE↓, C20:2/C18:2-PE↓, C20:3/C20:2-PE↓, C20:3/C22:0-PE↓, C20:4/C18:0-PE↓, C20:4/C20:0-PE↓, C16:0/C18:0-PE-P↓, C18:0/C13:0-PE-P↓, C18:0/C17:0-PE-P↓, C18:1/C22:1-PE-P↓, C18:1/C22:1-PE-P↓, C18:1/C22:1-PE-P↓, C18:0/C20:4-PE-P↑, C18:0/C20:5-PE-P↑, C18:0/C22:1-PE-P↓, 20:0/22:6-PE-P↓, C18:0/C16:0-PG↑, C18:1/C18:0-PG↑, C22:6/C20:1-PG↓, C18:2/C18:1-PI↓, C22:6/C16:0-PI↓, C18:0/C21:0-PS↓, C18:1/C20:3-PS↑, C18:1/C22:0-PS↓, C18:1/C24:1-PS↓, 18:2/22:1-PS↓, C20:1/C18:0-PS↓, C20:3/C21:0-PS↓, C22:6/C17:2-PS↑, C22:6/C18:2-PS↓, C18:0/C12:0-SQDG↑ | SPMS/PPMS | NAWM | [8] |
| C20:4/C22:2-DG↓, C18:2/C17:0-PA↓, C20:5/C18:1-PA↓, C18:2/C20:0-PE↑, C20:4/C20:0-PE↑, C20:4/C20:1-PE↓, C22:6/C22:0-PE↓, C18:0/C16:0-PI↓, C18:2/C19:0-PS↓, C20:0-sulfatide↓ | PPMS/nCNS and SPMS/nCNS | NAWM | [8] |
| Total Cer↓, Sph↑, S1P↓, C16:0/C24:0-Cer ratio↑, C18:0/C24:0-Cer ratio↑ | MS/nCNS and WM of AD | NAWM | [32] |
| C18:0-Cer↓, C20:0-Cer↓, C20:0-SM↓, C22:0-SM↓ | Ac-MS/nCNS | NAGM | [27] |
| C20:0-Cer↓, C22:0-Cer↓, C16:0-SM↓ | Ac-MS and In-MS/nCNS | NAGM | [27] |
| C18:0-Cer↓, C20:0-Cer↓, C22:0-SM↓, C24:0-SM↓ | Ac-MS and In-MS/nCNS | NAWM | [27] |
| C18:1(9Z)-lysoPC↑, C18:0-lysoPC↑, C16:0-lysoPI↑, PA↓, PC↑, PI↑ | RRMS/ONDs | CSF | [135] |
| C52:3-TG↓, C58:3-TG↓, C57:4-TG↓, C52:3-TG↓, C61:10-TG↓, C37:2-TG↓, C55:5-TG↓, C57:7-TG↓, C61:8-TG↓, C60:10-TG↓, C62:8-TG↓, C50:1-TG↓, C44:5-TG↓, C59:6-TG↓, C44:4-TG↓, C58:1-TG↓, C56:6-TG↓, C64:10-TG↑, C63:8-TG↑, C59:2-TG↑, C56:4-TG↑, C57:6-TG↑, C32:1-DG↓, C38:7-DG↑, C38:6-DG↑, C32:2-DG↑, C18:3-DG↑, C39:2-DG↑, C42:5-DG↑, C36:6-DG↑, 5beta-cholestane-3alpha↑, 7alpha-diol, 5beta-dihydrotestosterone↑, 22:0 cholesteryl ester↓, cholest-5-en-3alpha-ol↓, 12-methyl-10-oxo- tridecanoic acid↑, N-oleoylethanolamine↑, PE-NMe(O,O-28:0)↑, C21:0-PE↑, C27:1-PC-P↓, C40:3-PS↓, C42:6-PC↑, C25:2-PC↑, C42:0-GluCer↑, C20:0-sulfatide↓, C42:2-C1P↓ | RRMS/ONDs | CSF | [136] |
| C16:0-Cer↑, C24:0-Cer↑, C16:0-HexCer↑ | MS/ONDs | CSF | [130] |
| C16:0-HexCer↑ | MS/ONDs | CSF | [155] |
| 24(S)-OHC↑, 24(S)-OHC /27-OHC↑ | MS/nCNS | CSF | [48] |
| 24(S)-OHC↓, 27-OHC↓ | NZ treated RRMS/untreated RRMS | CSF | [156] |
| 24(S)-OHC↓ | MS/ONDs | Serum | [157] |
| C18:1-lysoPE↓, C18:2-lysoPE↓, C22:4-lysoPE↓, C16:0-lysoPC-P↓, C18:0-lysoPC-P↓, C18:1-lysoPC-P↓, C44:12-PC↓, C20:1-lysoPC↓, C20:0-lysoPC↓, C36:5-PE↓, C35:5-PC↓, C18:1/C18:1-PC↓, C18:0/C18:3-PC↓, tiglylcarnitine↓, 2(R)-HOT↓, GPC(14:0)↓, gamma-linolenic acid↓ | PPMS/HS | Plasma | [134] |
| C18:2-lysoPE↓, C20:0-lysoPC↓, tiglylcarnitine↑ | PPMS/RRMS | Plasma | [134] |
| Gamma-linolenic acid↓, C20:0-lysoPC↓ | PPMS/RRMS and PD | Plasma | [134] |
| C16:0-Cer↑, C24:1-Cer↑, C16:0-GluCer↑, C24:1-GluCer↑, C16:0-LacCer↓ | MS/HS | Plasma | [31] |
| C20:0-HexCer↑, C14:0-SM↓ | PPMS/HS | Plasma | [137] |
| C18:2-LPA↓ | PPMS with rapid progression/PPMS with mild progression and SPMS with rapid progression/SPMS with mild progression | Plasma | [137] |
| LDL-C↓, HDL-C↑ | RRMS and progressive MS/HS | Plasma | [158] |
| C24:0-Cer↓, C16:0-LacCer↓ | MS/HS | White blood cells | [31] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Podbielska, M.; O’Keeffe, J.; Pokryszko-Dragan, A. New Insights into Multiple Sclerosis Mechanisms: Lipids on the Track to Control Inflammation and Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 7319. https://doi.org/10.3390/ijms22147319
Podbielska M, O’Keeffe J, Pokryszko-Dragan A. New Insights into Multiple Sclerosis Mechanisms: Lipids on the Track to Control Inflammation and Neurodegeneration. International Journal of Molecular Sciences. 2021; 22(14):7319. https://doi.org/10.3390/ijms22147319
Chicago/Turabian StylePodbielska, Maria, Joan O’Keeffe, and Anna Pokryszko-Dragan. 2021. "New Insights into Multiple Sclerosis Mechanisms: Lipids on the Track to Control Inflammation and Neurodegeneration" International Journal of Molecular Sciences 22, no. 14: 7319. https://doi.org/10.3390/ijms22147319
APA StylePodbielska, M., O’Keeffe, J., & Pokryszko-Dragan, A. (2021). New Insights into Multiple Sclerosis Mechanisms: Lipids on the Track to Control Inflammation and Neurodegeneration. International Journal of Molecular Sciences, 22(14), 7319. https://doi.org/10.3390/ijms22147319