
Cisplatin Resistance and Redox-Metabolic Vulnerability: A Second Alteration



Abstract
:1. Introduction
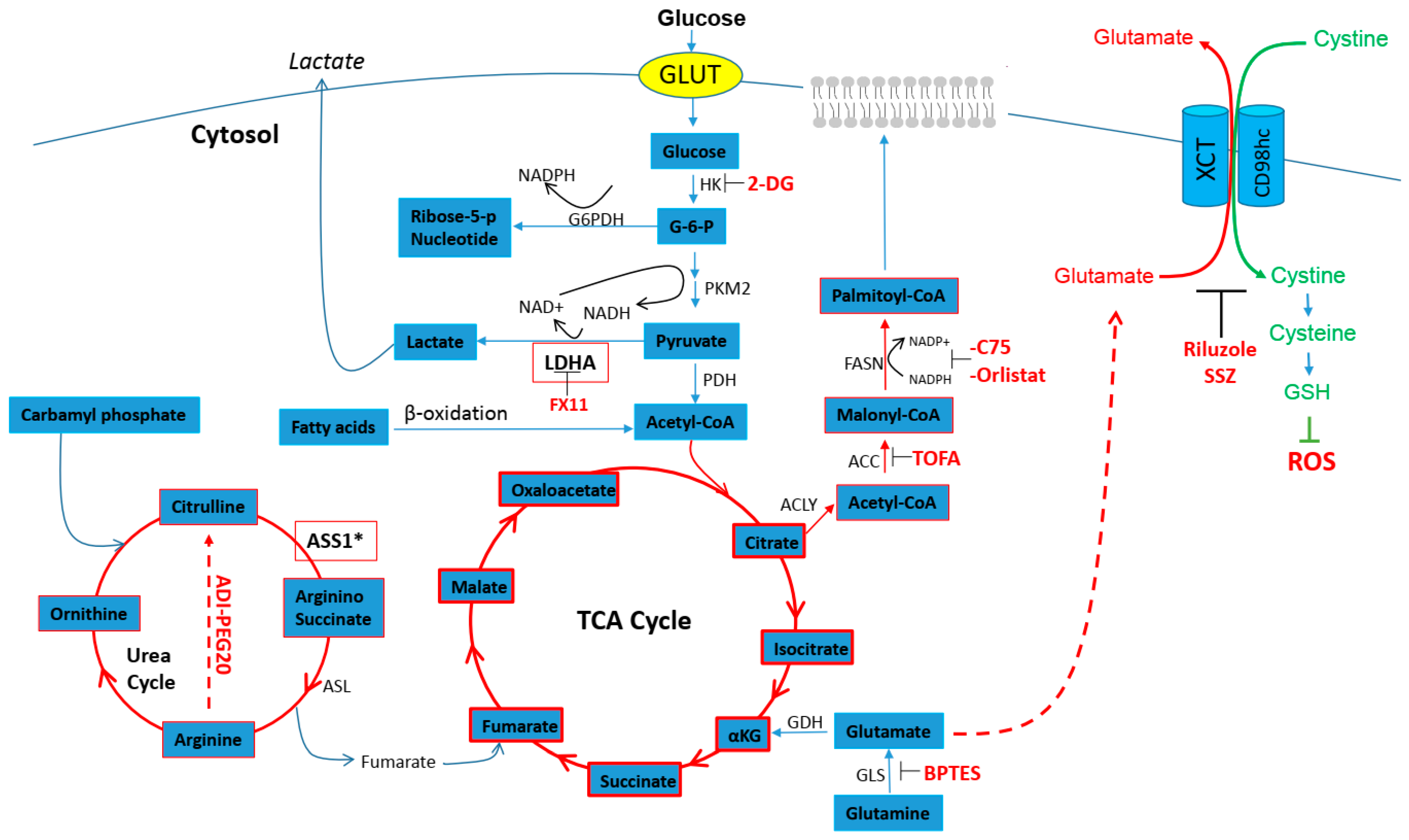
2. Cancer Cell and Metabolic Demand
2.1. Cisplatin Resistance and Glucose Metabolism
2.2. Cisplatin Resistance and Glutamine Anaplerosis
2.3. Cisplatin Resistance and Urea Cycle
2.4. Cisplatin Resistance and Fatty Acids
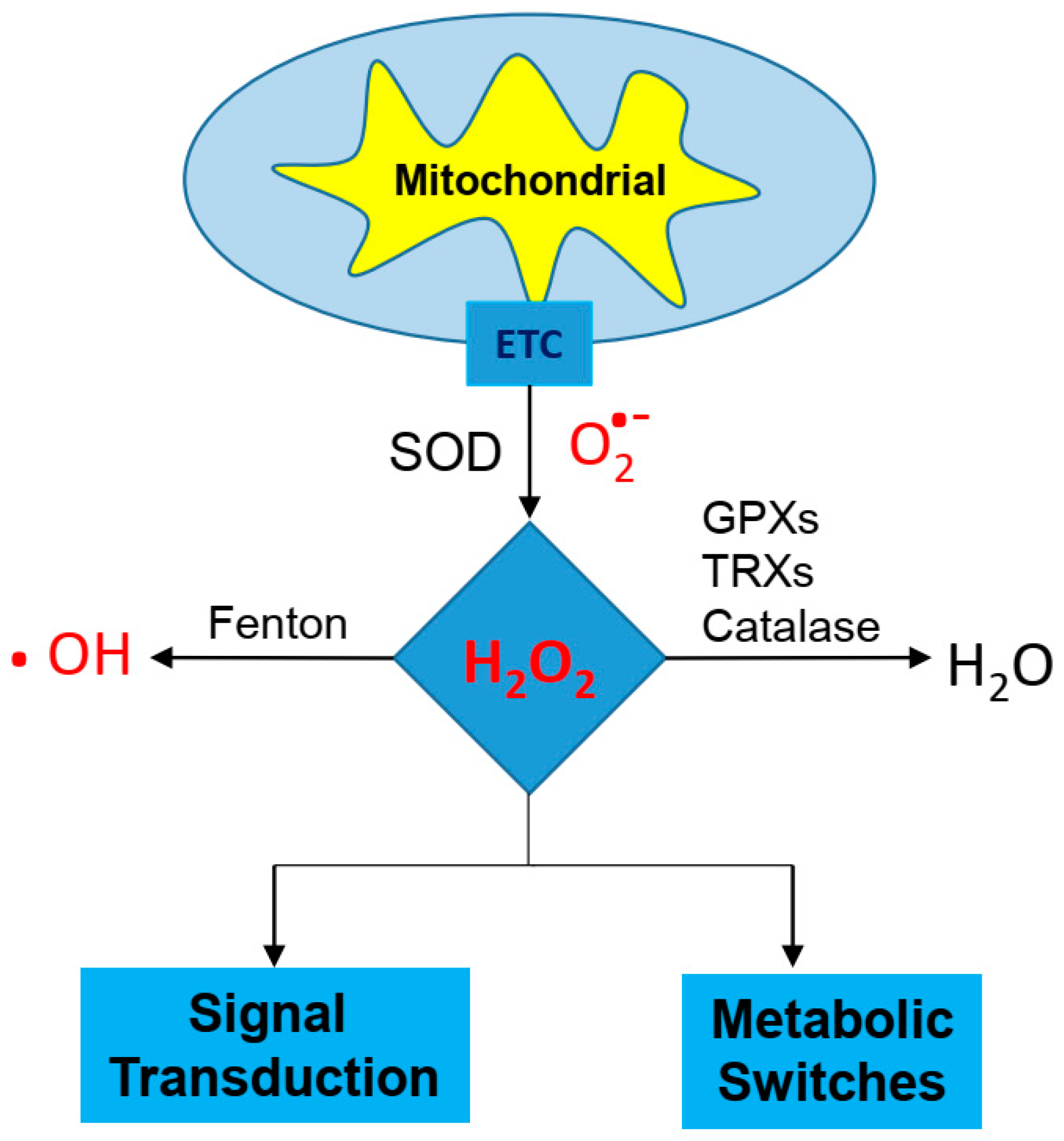
3. Cancer Cell and Redox Balance
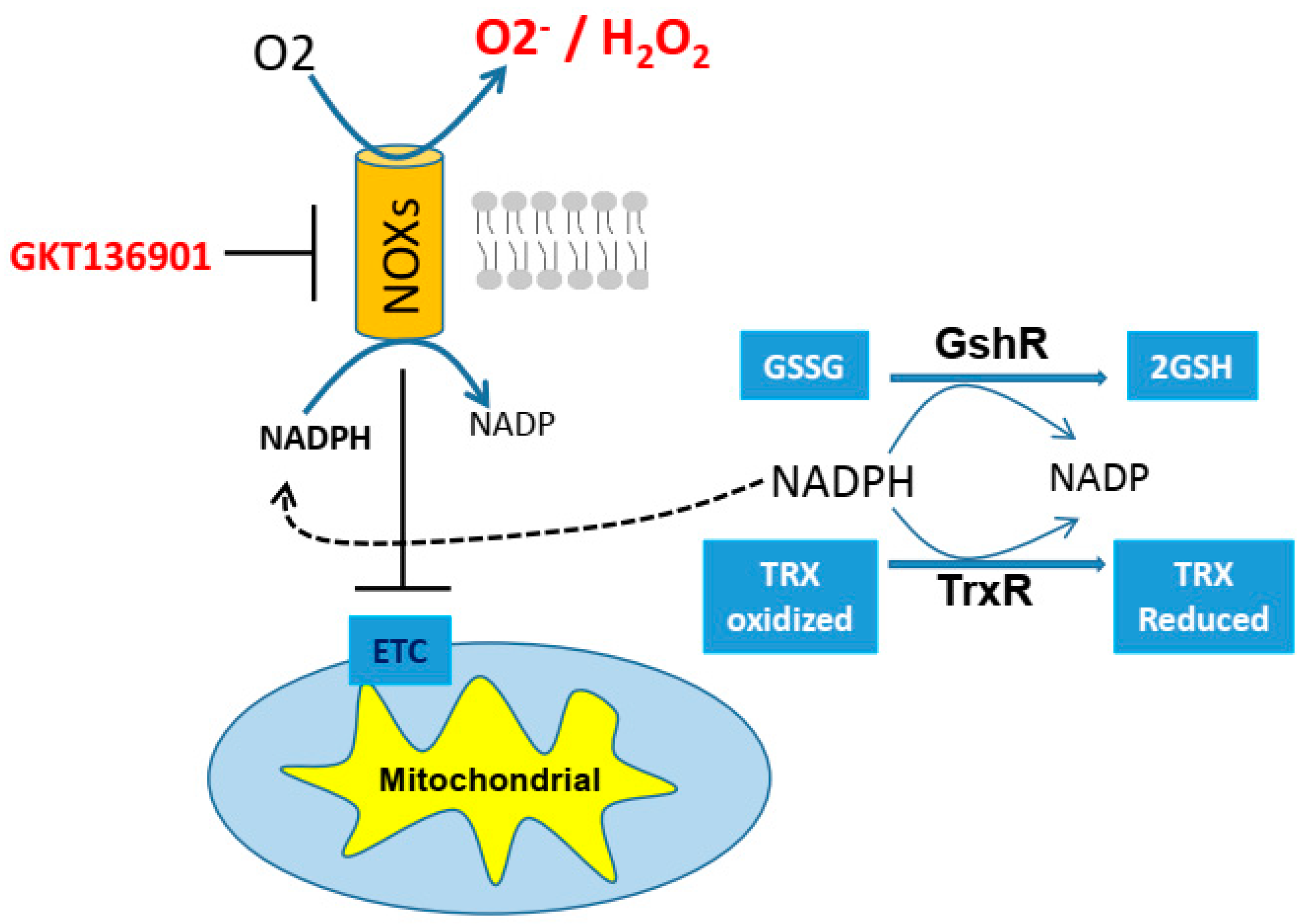
3.1. Cisplatin Resistance and GSH/TRX Antioxidant Systems
3.2. Cisplatin Resistance and NAD+
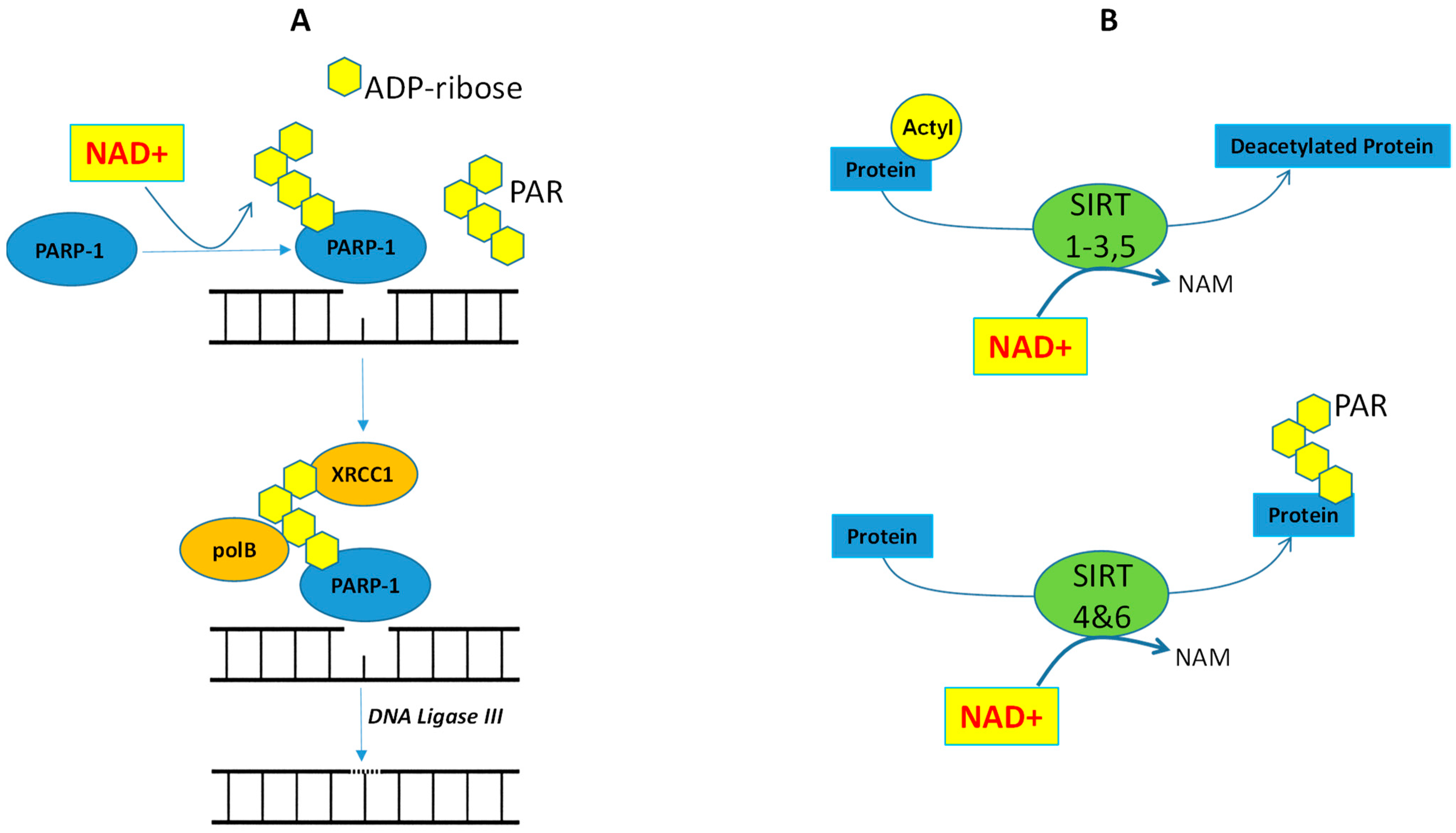
4. Cancer Cells and NAD+/PARP-1/SIRTs Axis
4.1. Cisplatin Resistance and PARP-1
4.2. Cisplatin Resistance and SIRTs
5. Cancer Cell and Immunometabolism
5.1. CR Cells and PD-L1
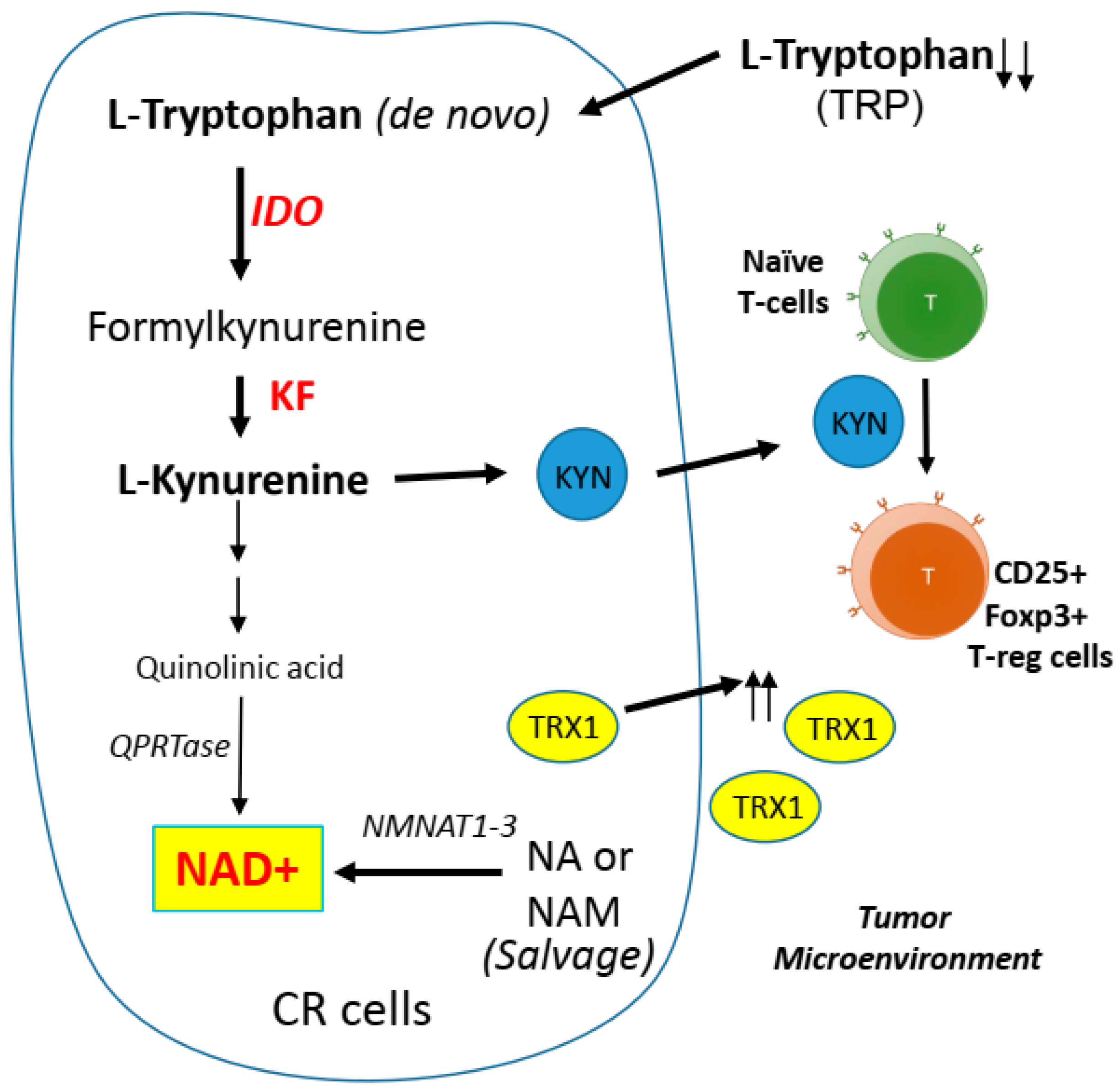
5.2. CR Cells and Kynurenine Pathway
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADI-PEG20 | Pegylated arginine deiminase |
| ACC | Acetyl-CoA Carboxylase |
| ACLY | ATP citrate lyase |
| AMPK | AMP-activated protein kinase |
| AP | Apurinic/apyrimidinic |
| ASS1 | Argininosuccinate synthetase-1 |
| BSO | Buthionine sulfoximine |
| DCA | Dichloroacetate |
| ETC | Electron transport chain |
| FASN | Fatty acid synthase |
| G-6-P | Glucose-6-phosphate |
| G6PDH | Glucose-6-phosphate dehydrogenase |
| GSH | Glutathione |
| GshR | GSH reductase |
| GSSG | Glutathione disulfide |
| GLS | Glutaminase |
| GLUT | Glucose transporter |
| H2O2 | Hydrogen Peroxide |
| HK | Hexokinase |
| IDO | Indoleamine 2,3-dioxygenase |
| ITP3K | Inositol-trisphosphate 3-kinase B |
| KYN | Kynurenine |
| LDHA | Lactate dehydrogenase A |
| mitoKATP | ATP-sensitive potassium channel from the inner mitochondrial membrane |
| MKP-1 | Mitogen-activated protein kinase phosphatase-1 |
| NAD | Nicotinamide adenine dinucleotide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NMNAT1–3 | Nicotinamide mononucleotide adenylyltransferase 1–3 |
| NOX | NADPH oxidase |
| OXMET | Oxidative metabolism |
| PARP-1 | Poly (ADP ribose) polymerase 1 |
| PDH | Pyruvate dehydrogenase |
| PDK | Pyruvate dehydrogenase kinase |
| PKCε | Protein kinase C epsilon type |
| PKM2 | Pyruvate kinase isozymes M2 |
| PPP | Pentose phosphate pathway |
| RECIST | Response evaluation criteria in solid tumors |
| ROS | Reactive oxygen species |
| SIRT | Sirtuin |
| SSZ | Sulfasalazine |
| T reg | Regulatory T cell |
| TCA | Tricarboxylic acid cycle |
| TME | Tumor microenvironment |
| TRP | Tryptophan |
| TRX | Thioredoxin |
| TrxR | TRX reductase |
| xCT | Cystine–glutamate exchange transporter, system xc- |
| XRCC1 | X-ray repair cross-complementing 1 |
References
- Rosenberg, B.; Vancamp, L.; Krigas, T. Inhibition of Cell Division in Escherichia Coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; VanCamp, L. The successful regression of large solid sarcoma 180 tumors by platinum compounds. Cancer Res. 1970, 30, 1799–1802. [Google Scholar]
- Cohen, S.M.; Lippard, S.J. Cisplatin: From DNA damage to cancer chemotherapy. Prog. Nucleic Acid Res. Mol. Biol. 2001, 67, 93–130. [Google Scholar] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangpaichitr, M.; Wu, C.; Li, Y.Y.; Nguyen, D.J.M.; Kandemir, H.; Shah, S.; Chen, S.; Feun, L.G.; Prince, J.S.; Kuo, M.T.; et al. Exploiting ROS and metabolic differences to kill cisplatin resistant lung cancer. Oncotarget 2017, 8, 49275–49292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaal, E.A.; Berkers, C.R. The Influence of Metabolism on Drug Response in Cancer. Front. Oncol. 2018, 8, 500. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Kurtoglu, M.; Maher, J.C.; Lampidis, T.J. Differential toxic mechanisms of 2-deoxy-D-glucose versus 2-fluorodeoxy-D-glucose in hypoxic and normoxic tumor cells. Antioxid. Redox Signal. 2007, 9, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, J.F.; Georgiades, C.S.; Ko, Y.H.; Pedersen, P.L. Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert Rev. Anticancer Ther. 2004, 4, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Sajnani, K.; Islam, F.; Smith, R.A.; Gopalan, V.; Lam, A.K. Genetic alterations in Krebs cycle and its impact on cancer pathogenesis. Biochimie 2017, 135, 164–172. [Google Scholar] [CrossRef]
- Cardaci, S.; Ciriolo, M.R. TCA Cycle Defects and Cancer: When Metabolism Tunes Redox State. Int. J. Cell Biol. 2012, 2012, 161837. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat. Genet. 2018, 50, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.; Lee, D.; Shim, H. Metabolic positron emission tomography imaging in cancer detection and therapy response. Semin. Oncol. 2011, 38, 55–69. [Google Scholar] [CrossRef] [Green Version]
- Song, I.S.; Savaraj, N.; Siddik, Z.H.; Liu, P.; Wei, Y.; Wu, C.J.; Kuo, M.T. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol. Cancer Ther. 2004, 3, 1543–1549. [Google Scholar]
- Kuo, M.T.; Chen, H.H.; Song, I.S.; Savaraj, N.; Ishikawa, T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007, 26, 71–83. [Google Scholar] [CrossRef]
- Chen, H.H.; Yan, J.J.; Chen, W.C.; Kuo, M.T.; Lai, Y.H.; Lai, W.W.; Liu, H.S.; Su, W.C. Predictive and prognostic value of human copper transporter 1 (hCtr1) in patients with stage III non-small-cell lung cancer receiving first-line platinum-based doublet chemotherapy. Lung Cancer 2011, 75, 228–234. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.J.; Finkel, T.; Shen, D.W.; Yin, J.J.; Aszalos, A.; Gottesman, M.M. SIRT1 contributes in part to cisplatin resistance in cancer cells by altering mitochondrial metabolism. Mol Cancer Res. 2008, 6, 1499–1506. [Google Scholar] [CrossRef] [Green Version]
- Wangpaichitr, M.; Sullivan, E.J.; Theodoropoulos, G.; Wu, C.; You, M.; Feun, L.G.; Lampidis, T.J.; Kuo, M.T.; Savaraj, N. The relationship of thioredoxin-1 and cisplatin resistance: Its impact on ROS and oxidative metabolism in lung cancer cells. Mol. Cancer Ther. 2012, 11, 604–615. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zeigler, M.M.; Lam, G.K.; Hunter, M.G.; Eubank, T.D.; Khramtsov, V.V.; Tridandapani, S.; Sen, C.K.; Marsh, C.B. The role of the NADPH oxidase complex, p38 MAPK, and Akt in regulating human monocyte/macrophage survival. Am. J. Respir Cell Mol. Biol. 2007, 36, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.H.; Ho, C.T. Chemopreventive effects of natural dietary compounds on cancer development. Chem. Soc. Rev. 2008, 37, 2558–2574. [Google Scholar] [CrossRef]
- Sullivan, E.J.; Kurtoglu, M.; Brenneman, R.; Liu, H.; Lampidis, T.J. Targeting cisplatin-resistant human tumor cells with metabolic inhibitors. Cancer Chemother. Pharmacol. 2014, 73, 417–427. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Locasale, J.W.; Swanson, K.D.; Sharfi, H.; Heffron, G.J.; Amador-Noguez, D.; Christofk, H.R.; Wagner, G.; Rabinowitz, J.D.; Asara, J.M.; et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 2010, 329, 1492–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, B.C.; Ku, J.L.; Hong, S.H.; Shin, Y.K.; Park, S.Y.; Kim, H.K.; Park, J.G. Decreased pyruvate kinase M2 activity linked to cisplatin resistance in human gastric carcinoma cell lines. Int. J. Cancer 2004, 108, 532–539. [Google Scholar] [CrossRef]
- Martinez-Balibrea, E.; Plasencia, C.; Gines, A.; Martinez-Cardus, A.; Musulen, E.; Aguilera, R.; Manzano, J.L.; Neamati, N.; Abad, A. A proteomic approach links decreased pyruvate kinase M2 expression to oxaliplatin resistance in patients with colorectal cancer and in human cell lines. Mol. Cancer Ther. 2009, 8, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.K.; Yoo, B.C.; Hong, Y.S.; Chang, H.J.; Jung, K.H.; Jeong, S.Y.; Park, J.G. Upregulation of glycolytic enzymes in proteins secreted from human colon cancer cells with 5-fluorouracil resistance. Electrophoresis 2009, 30, 2182–2192. [Google Scholar] [CrossRef]
- Lu, W.Q.; Hu, Y.Y.; Lin, X.P.; Fan, W. Knockdown of PKM2 and GLS1 expression can significantly reverse oxaliplatin-resistance in colorectal cancer cells. Oncotarget 2017, 8, 44171–44185. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, F.; Wu, X.R. Inhibition of Pyruvate Kinase M2 Markedly Reduces Chemoresistance of Advanced Bladder Cancer to Cisplatin. Sci. Rep. 2017, 7, 45983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.J.; Park, Y.S.; Kang, M.G.; You, Y.M.; Jung, Y.; Koo, H.; Kim, J.A.; Kim, M.J.; Hong, S.M.; Lee, K.B.; et al. Pyruvate kinase isoenzyme M2 is a therapeutic target of gemcitabine-resistant pancreatic cancer cells. Exp. Cell Res. 2015, 336, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, D.; Chen, X.; He, L.; Li, T.; Xu, X.; Li, M. Nuclear PKM2 contributes to gefitinib resistance via upregulation of STAT3 activation in colorectal cancer. Sci. Rep. 2015, 5, 16082. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Zhang, Y.; Chen, T.; Wang, Y.; Xue, J.; Zhang, Y.; Xiao, W.; Mo, X.; Lu, Y. Efficacy of RNAi targeting of pyruvate kinase M2 combined with cisplatin in a lung cancer model. J. Cancer Res. Clin. Oncol. 2011, 137, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.S.; Li, D.; Zhang, J.; Wang, Y.S.; Yang, L.; Zhang, H.L.; Wang, X.H.; Mu, B.; Wang, W.; Ma, Y.; et al. Silencing of pkm2 increases the efficacy of docetaxel in human lung cancer xenografts in mice. Cancer Sci. 2010, 101, 1447–1453. [Google Scholar] [CrossRef]
- Hudson, C.D.; Savadelis, A.; Nagaraj, A.B.; Joseph, P.; Avril, S.; DiFeo, A.; Avril, N. Altered glutamine metabolism in platinum resistant ovarian cancer. Oncotarget 2016, 7, 41637–41649. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [Green Version]
- Lo, M.; Wang, Y.Z.; Gout, P.W. The x(c)- cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef]
- Shukla, K.; Thomas, A.G.; Ferraris, D.V.; Hin, N.; Sattler, R.; Alt, J.; Rojas, C.; Slusher, B.S.; Tsukamoto, T. Inhibition of xc(-) transporter-mediated cystine uptake by sulfasalazine analogs. Bioorg. Med. Chem. Lett. 2011, 21, 6184–6187. [Google Scholar] [CrossRef]
- Evonuk, K.S.; Baker, B.J.; Doyle, R.E.; Moseley, C.E.; Sestero, C.M.; Johnston, B.P.; De Sarno, P.; Tang, A.; Gembitsky, I.; Hewett, S.J.; et al. Inhibition of System Xc(-) Transporter Attenuates Autoimmune Inflammatory Demyelination. J. Immunol. 2015, 195, 450–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.Z.; Chen, G.; Wang, P.; Lu, W.H.; Zhu, C.F.; Song, M.; Yang, J.; Wen, S.; Xu, R.H.; Hu, Y.; et al. Xc- inhibitor sulfasalazine sensitizes colorectal cancer to cisplatin by a GSH-dependent mechanism. Cancer Lett. 2015, 368, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Savaraj, N.; Wu, C.; Kuo, M.T.; You, M.; Wangpaichitr, M.; Robles, C.; Spector, S.; Feun, L. The relationship of arginine deprivation, argininosuccinate synthetase and cell death in melanoma. Drug Target Insights 2007, 2, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ensor, C.M.; Holtsberg, F.W.; Bomalaski, J.S.; Clark, M.A. Pegylated arginine deiminase (ADI-SS PEG20,000 mw) inhibits human melanomas and hepatocellular carcinomas in vitro and in vivo. Cancer Res. 2002, 62, 5443–5450. [Google Scholar] [PubMed]
- Dillon, B.J.; Prieto, V.G.; Curley, S.A.; Ensor, C.M.; Holtsberg, F.W.; Bomalaski, J.S.; Clark, M.A. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: A method for identifying cancers sensitive to arginine deprivation. Cancer 2004, 100, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Szlosarek, P.W.; Klabatsa, A.; Pallaska, A.; Sheaff, M.; Smith, P.; Crook, T.; Grimshaw, M.J.; Steele, J.P.; Rudd, R.M.; Balkwill, F.R.; et al. In vivo loss of expression of argininosuccinate synthetase in malignant pleural mesothelioma is a biomarker for susceptibility to arginine depletion. Clin. Cancer Res. 2006, 12, 7126–7131. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Stewart, J.; Wang, H.; Rashid, A.; Zhao, J.; Katz, M.H.; Lee, J.E.; Fleming, J.B.; Maitra, A.; Wolff, R.A.; et al. Reduced expression of argininosuccinate synthetase 1 has a negative prognostic impact in patients with pancreatic ductal adenocarcinoma. PLoS ONE 2017, 12, e0171985. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kobayashi, E.; Kubota, D.; Suehara, Y.; Mukaihara, K.; Akaike, K.; Ito, A.; Kaneko, K.; Chuman, H.; Kawai, A.; et al. Reduced argininosuccinate synthetase expression in refractory sarcomas: Impacts on therapeutic potential and drug resistance. Oncotarget 2016, 7, 70832–70844. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.H.; Chen, C.M.; Strathdee, G.; Harnsomburana, J.; Shyu, C.R.; Rahmatpanah, F.; Shi, H.; Ng, S.W.; Yan, P.S.; Nephew, K.P.; et al. Methylation microarray analysis of late-stage ovarian carcinomas distinguishes progression-free survival in patients and identifies candidate epigenetic markers. Clin. Cancer Res. 2002, 8, 2246–2252. [Google Scholar]
- Long, Y.; Tsai, W.B.; Chang, J.T.; Estecio, M.; Wangpaichitr, M.; Savaraj, N.; Feun, L.G.; Chen, H.H.; Kuo, M.T. Cisplatin-induced synthetic lethality to arginine-starvation therapy by transcriptional suppression of ASS1 is regulated by DEC1, HIF-1alpha, and c-Myc transcription network and is independent of ASS1 promoter DNA methylation. Oncotarget 2016, 7, 82658–82670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, L.J.; Smith, P.R.; Hiller, L.; Szlosarek, P.W.; Kimberley, C.; Sehouli, J.; Koensgen, D.; Mustea, A.; Schmid, P.; Crook, T. Epigenetic silencing of argininosuccinate synthetase confers resistance to platinum-induced cell death but collateral sensitivity to arginine auxotrophy in ovarian cancer. Int. J. Cancer 2009, 125, 1454–1463. [Google Scholar] [CrossRef]
- Tsai, W.B.; Aiba, I.; Lee, S.Y.; Feun, L.; Savaraj, N.; Kuo, M.T. Resistance to arginine deiminase treatment in melanoma cells is associated with induced argininosuccinate synthetase expression involving c-Myc/HIF-1alpha/Sp4. Mol. Cancer Ther. 2009, 8, 3223–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, M.T.; Savaraj, N.; Feun, L.G. Targeted cellular metabolism for cancer chemotherapy with recombinant arginine-degrading enzymes. Oncotarget 2010, 1, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Feun, L.; Savaraj, N. Pegylated arginine deiminase: A novel anticancer enzyme agent. Expert Opin. Investig. Drugs 2006, 15, 815–822. [Google Scholar] [CrossRef]
- Yau, T.; Cheng, P.N.; Chan, P.; Chen, L.; Yuen, J.; Pang, R.; Fan, S.T.; Wheatley, D.N.; Poon, R.T. Preliminary efficacy, safety, pharmacokinetics, pharmacodynamics and quality of life study of pegylated recombinant human arginase 1 in patients with advanced hepatocellular carcinoma. Invest. New Drugs 2015, 33, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Feun, L.G.; Kuo, M.T.; Savaraj, N. Arginine deprivation in cancer therapy. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 78–82. [Google Scholar] [CrossRef]
- Duan, J.; Sun, L.; Huang, H.; Wu, Z.; Wang, L.; Liao, W. Overexpression of fatty acid synthase predicts a poor prognosis for human gastric cancer. Mol. Med. Rep. 2016, 13, 3027–3035. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Wang, J.; Zhang, L.; Wu, D.; Yu, D.; Tian, X.; Liu, J.; Jiang, X.; Shen, Y.; Zhang, L.; et al. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med. Oncol. 2015, 32, 391. [Google Scholar] [CrossRef] [Green Version]
- Takahiro, T.; Shinichi, K.; Toshimitsu, S. Expression of fatty acid synthase as a prognostic indicator in soft tissue sarcomas. Clin. Cancer Res. 2003, 9, 2204–2212. [Google Scholar]
- Uddin, S.; Jehan, Z.; Ahmed, M.; Alyan, A.; Al-Dayel, F.; Hussain, A.; Bavi, P.; Al-Kuraya, K.S. Overexpression of fatty acid synthase in Middle Eastern epithelial ovarian carcinoma activates AKT and Its inhibition potentiates cisplatin-induced apoptosis. Mol. Med. 2011, 17, 635–645. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Colomer, R.; Brunet, J.; Menendez, J.A. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int. J. Oncol. 2007, 31, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Vellon, L.; Lupu, R. The antiobesity drug Orlistat induces cytotoxic effects, suppresses Her-2/neu (erbB-2) oncogene overexpression, and synergistically interacts with trastuzumab (Herceptin) in chemoresistant ovarian cancer cells. Int. J. Gynecol. Cancer 2006, 16, 219–221. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Y.; Zhang, J.T. A new mechanism of drug resistance in breast cancer cells: Fatty acid synthase overexpression-mediated palmitate overproduction. Mol. Cancer Ther. 2008, 7, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, H.; Li, Z.; Zhao, Z.; Yip-Schneider, M.; Fan, Q.; Schmidt, C.M.; Chiorean, E.G.; Xie, J.; Cheng, L.; et al. Role of fatty acid synthase in gemcitabine and radiation resistance of pancreatic cancers. Int. J. Biochem. Mol. Biol. 2011, 2, 89–98. [Google Scholar] [PubMed]
- Warmoes, M.; Jaspers, J.E.; Xu, G.; Sampadi, B.K.; Pham, T.V.; Knol, J.C.; Piersma, S.R.; Boven, E.; Jonkers, J.; Rottenberg, S.; et al. Proteomics of genetically engineered mouse mammary tumors identifies fatty acid metabolism members as potential predictive markers for cisplatin resistance. Mol. Cell Proteom. 2013, 12, 1319–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kant, S.; Kumar, A.; Singh, S.M. Tumor growth retardation and chemosensitizing action of fatty acid synthase inhibitor orlistat on T cell lymphoma: Implication of reconstituted tumor microenvironment and multidrug resistance phenotype. Biochim. Biophys. Acta 2014, 1840, 294–302. [Google Scholar] [CrossRef]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. The multidrug resistance protein family. Biochim. Biophys. Acta 1999, 1461, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Savaraj, N.; Wu, C.; Wangpaichitr, M.; Kuo, M.T.; Lampidis, T.; Robles, C.; Furst, A.J.; Feun, L. Overexpression of mutated MRP4 in cisplatin resistant small cell lung cancer cell line: Collateral sensitivity to azidothymidine. Int. J. Oncol. 2003, 23, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Preston, T.J.; Muller, W.J.; Singh, G. Scavenging of extracellular H2O2 by catalase inhibits the proliferation of HER-2/Neu-transformed rat-1 fibroblasts through the induction of a stress response. J. Biol. Chem. 2001, 276, 9558–9564. [Google Scholar] [CrossRef] [Green Version]
- Lillig, C.H.; Holmgren, A. Thioredoxin and related molecules--from biology to health and disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Forman, H.J. Glutathione synthesis and its role in redox signaling. Semin. Cell Dev. Biol. 2012, 23, 722–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, J.C.; Beutler, E. Electrophoretic study of glutathione reductase in human erythrocytes and leucocytes. Nature 1968, 217, 256–258. [Google Scholar] [CrossRef]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Kirkman, H.N.; Gaetani, G.F. Catalase: A tetrameric enzyme with four tightly bound molecules of NADPH. Proc. Natl. Acad. Sci. USA 1984, 81, 4343–4347. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Tanaka, M.; Miura, Y.; Numanami, H.; Karnan, S.; Ota, A.; Konishi, H.; Hosokawa, Y.; Hanyuda, M. Inhibition of NADPH oxidase 4 induces apoptosis in malignant mesothelioma: Role of reactive oxygen species. Oncol. Rep. 2015, 34, 1726–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushio-Fukai, M. Localizing NADPH oxidase-derived ROS. Sci. STKE 2006, 2006, re8. [Google Scholar] [CrossRef]
- Chang, G.; Chen, L.; Lin, H.M.; Lin, Y.; Maranchie, J.K. Nox4 inhibition enhances the cytotoxicity of cisplatin in human renal cancer cells. J. Exp. Ther. Oncol. 2012, 10, 9–18. [Google Scholar]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. Embo J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Rigas, B. The thioredoxin system mediates redox-induced cell death in human colon cancer cells: Implications for the mechanism of action of anticancer agents. Cancer Res. 2008, 68, 8269–8277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, A.B.; Anestal, K.; Jerremalm, E.; Ehrsson, H.; Arner, E.S. Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic. Biol. Med. 2005, 39, 696–703. [Google Scholar] [CrossRef]
- Mostert, V.; Hill, K.E.; Burk, R.F. Loss of activity of the selenoenzyme thioredoxin reductase causes induction of hepatic heme oxygenase-1. FEBS Lett. 2003, 541, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Cederbaum, A. The mode of cisplatin-induced cell death in CYP2E1-overexpressing HepG2 cells: Modulation by ERK, ROS, glutathione, and thioredoxin. Free Radic. Biol. Med. 2007, 43, 1061–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangpaichitr, M.; Wu, C.; You, M.; Maher, J.C.; Dinh, V.; Feun, L.G.; Savaraj, N. N1,N3-Dimethyl-N1,N3-bis(phenylcarbonothioyl) Propanedihydrazide (Elesclomol) Selectively Kills Cisplatin Resistant Lung Cancer Cells through Reactive Oxygen Species (ROS). Cancers 2009, 1, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met. Based Drugs 2010, 2010. [Google Scholar] [CrossRef]
- Haendeler, J.; Popp, R.; Goy, C.; Tischler, V.; Zeiher, A.M.; Dimmeler, S. Cathepsin D and H2O2 stimulate degradation of thioredoxin-1: Implication for endothelial cell apoptosis. J. Biol. Chem. 2005, 280, 42945–42951. [Google Scholar] [CrossRef] [Green Version]
- Rubartelli, A.; Bajetto, A.; Allavena, G.; Wollman, E.; Sitia, R. Secretion of thioredoxin by normal and neoplastic cells through a leaderless secretory pathway. J. Biol. Chem. 1992, 267, 24161–24164. [Google Scholar] [CrossRef]
- Rubartelli, A.; Bajetto, A.; Bonifaci, N.; Di Blas, E.; Solito, E.; Sitia, R. A novel way to get out of the cell. Cytotechnology 1993, 11 (Suppl. S1), S37–S40. [Google Scholar] [CrossRef]
- Rubartelli, A.; Bonifaci, N.; Sitia, R. High rates of thioredoxin secretion correlate with growth arrest in hepatoma cells. Cancer Res. 1995, 55, 675–680. [Google Scholar]
- Miyamoto, S.; Kawano, H.; Sakamoto, T.; Soejima, H.; Kajiwara, I.; Hokamaki, J.; Hirai, N.; Sugiyama, S.; Yoshimura, M.; Yasue, H.; et al. Increased plasma levels of thioredoxin in patients with coronary spastic angina. Antioxid. Redox Signal. 2004, 6, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; De Rosa, S.; Roederer, M.; Anderson, M.T.; Dubs, J.G.; Yodoi, J.; Holmgren, A.; Herzenberg, L.A.; Herzenberg, L.A. Elevation of plasma thioredoxin levels in HIV-infected individuals. Int. Immunol. 1996, 8, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Vaage, J.; Valen, G.; Padilla, C.A.; Bjornstedt, M.; Holmgren, A. Measurements of plasma glutaredoxin and thioredoxin in healthy volunteers and during open-heart surgery. Free Radic. Biol. Med. 1998, 24, 1176–1186. [Google Scholar] [CrossRef]
- Sasada, T.; Nakamura, H.; Ueda, S.; Iwata, S.; Ueno, M.; Takabayashi, A.; Yodoi, J. Secretion of thioredoxin enhances cellular resistance to cis-diamminedichloroplatinum (II). Antioxid. Redox Signal. 2000, 2, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Sasada, T.; Nakamura, H.; Ueda, S.; Sato, N.; Kitaoka, Y.; Gon, Y.; Takabayashi, A.; Spyrou, G.; Holmgren, A.; Yodoi, J. Possible involvement of thioredoxin reductase as well as thioredoxin in cellular sensitivity to cis-diamminedichloroplatinum (II). Free Radic. Biol. Med. 1999, 27, 504–514. [Google Scholar] [CrossRef]
- Baker, A.F.; Dragovich, T.; Tate, W.R.; Ramanathan, R.K.; Roe, D.; Hsu, C.H.; Kirkpatrick, D.L.; Powis, G. The antitumor thioredoxin-1 inhibitor PX-12 (1-methylpropyl 2-imidazolyl disulfide) decreases thioredoxin-1 and VEGF levels in cancer patient plasma. J. Lab. Clin. Med. 2006, 147, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, C.R.; Garcia, C.C.; Vieira, D.B.; Quinet, A.; de Andrade-Lima, L.C.; Munford, V.; Belizario, J.E.; Menck, C.F. Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1505. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Watson, J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013, 3, 120144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Outten, C.E.; Culotta, V.C. A novel NADH kinase is the mitochondrial source of NADPH in Saccharomyces cerevisiae. EMBO J. 2003, 22, 2015–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, G.S.; Kim, H.J.; Choi, J.H.; Shen, A.; Choe, S.K.; Karna, A.; Lee, S.H.; Jo, H.J.; Yang, S.H.; Kwak, T.H.; et al. Pharmacological activation of NQO1 increases NAD(+) levels and attenuates cisplatin-mediated acute kidney injury in mice. Kidney Int. 2014, 85, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Chen, Y.; Dubrulle, J.; Stossi, F.; Putluri, V.; Sreekumar, A.; Putluri, N.; Baluya, D.; Lai, S.Y.; Sandulache, V.C. Cisplatin generates oxidative stress which is accompanied by rapid shifts in central carbon metabolism. Sci. Rep. 2018, 8, 4306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dworakowski, R.; Anilkumar, N.; Zhang, M.; Shah, A.M. Redox signalling involving NADPH oxidase-derived reactive oxygen species. Biochem. Soc. Trans. 2006, 34, 960–964. [Google Scholar] [CrossRef]
- Infanger, D.W.; Sharma, R.V.; Davisson, R.L. NADPH oxidases of the brain: Distribution, regulation, and function. Antioxid. Redox Signal. 2006, 8, 1583–1596. [Google Scholar] [CrossRef]
- Spencer, N.Y.; Yan, Z.; Boudreau, R.L.; Zhang, Y.; Luo, M.; Li, Q.; Tian, X.; Shah, A.M.; Davisson, R.L.; Davidson, B.; et al. Control of hepatic nuclear superoxide production by glucose 6-phosphate dehydrogenase and NADPH oxidase-4. J. Biol. Chem. 2011, 286, 8977–8987. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Jin, L.; Wang, X.; Li, Y.; Chun, J.; Boese, A.C.; Li, D.; Kang, H.B.; Zhang, G.; Zhou, L.; et al. Inositol-triphosphate 3-kinase B confers cisplatin resistance by regulating NOX4-dependent redox balance. J. Clin. Invest. 2019, 130. [Google Scholar] [CrossRef] [PubMed]
- Virag, L.; Szabo, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef]
- Ying, W. NAD+ and NADH in cellular functions and cell death. Front Biosci. 2006, 11, 3129–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strom, C.E.; Johansson, F.; Uhlen, M.; Szigyarto, C.A.; Erixon, K.; Helleday, T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39, 3166–3175. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Canto, C.; Brunyanszki, A.; Huber, A.; Szanto, M.; Cen, Y.; Yamamoto, H.; Houten, S.M.; Kiss, B.; Oudart, H.; et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 2011, 13, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolthur-Seetharam, U.; Dantzer, F.; McBurney, M.W.; de Murcia, G.; Sassone-Corsi, P. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle 2006, 5, 873–877. [Google Scholar] [CrossRef] [Green Version]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef] [Green Version]
- Michels, J.; Vitale, I.; Galluzzi, L.; Adam, J.; Olaussen, K.A.; Kepp, O.; Senovilla, L.; Talhaoui, I.; Guegan, J.; Enot, D.P.; et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res. 2013, 73, 2271–2280. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Kho, D.H.; Zhou, J.Y.; Davis, R.J.; Wu, G.S. MKP-1 suppresses PARP-1 degradation to mediate cisplatin resistance. Oncogene 2017, 36, 5939–5947. [Google Scholar] [CrossRef] [Green Version]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.I.; Guarente, L. It takes two to tango: NAD(+) and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [Green Version]
- Mvunta, D.H.; Miyamoto, T.; Asaka, R.; Yamada, Y.; Ando, H.; Higuchi, S.; Ida, K.; Kashima, H.; Shiozawa, T. SIRT1 Regulates the Chemoresistance and Invasiveness of Ovarian Carcinoma Cells. Transl. Oncol. 2017, 10, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Asaka, R.; Miyamoto, T.; Yamada, Y.; Ando, H.; Mvunta, D.H.; Kobara, H.; Shiozawa, T. Sirtuin 1 promotes the growth and cisplatin resistance of endometrial carcinoma cells: A novel therapeutic target. Lab. Invest. 2015, 95, 1363–1373. [Google Scholar] [CrossRef]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.L.; Xiong, Y.; Lei, Q.Y. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Xie, Q.R.; Chen, Z.; Lu, S.; Xia, W. Regulation of SIRT2 levels for human non-small cell lung cancer therapy. Lung Cancer 2013, 82, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Schlicker, C.; Gertz, M.; Papatheodorou, P.; Kachholz, B.; Becker, C.F.; Steegborn, C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J. Mol. Biol. 2008, 382, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Invest. 2015, 125, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Schwer, B.; Carobbio, S.; Waltregny, D.; North, B.J.; Castronovo, V.; Maechler, P.; Verdin, E. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 2007, 282, 33583–33592. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.M.; Hwang, S.; Seong, R.H. SIRT4 regulates cancer cell survival and growth after stress. Biochem. Biophys. Res. Commun. 2016, 470, 251–256. [Google Scholar] [CrossRef]
- Mathias, R.A.; Greco, T.M.; Oberstein, A.; Budayeva, H.G.; Chakrabarti, R.; Rowland, E.A.; Kang, Y.; Shenk, T.; Cristea, I.M. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 2014, 159, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Zuo, Y.; Feng, Y.; Zhang, M. SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer. Tumour. Biol. 2014, 35, 10699–10705. [Google Scholar] [CrossRef]
- Mao, Z.; Hine, C.; Tian, X.; Van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar] [CrossRef] [Green Version]
- Dominy, J.E., Jr.; Lee, Y.; Jedrychowski, M.P.; Chim, H.; Jurczak, M.J.; Camporez, J.P.; Ruan, H.B.; Feldman, J.; Pierce, K.; Mostoslavsky, R.; et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol. Cell 2012, 48, 900–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Miyasato, Y.; Yoshizawa, T.; Sato, Y.; Nakagawa, T.; Miyasato, Y.; Kakizoe, Y.; Kuwabara, T.; Adachi, M.; Ianni, A.; Braun, T.; et al. Sirtuin 7 Deficiency Ameliorates Cisplatin-induced Acute Kidney Injury Through Regulation of the Inflammatory Response. Sci. Rep. 2018, 8, 5927. [Google Scholar] [CrossRef] [PubMed]
- Kouidhi, S.; Ben Ayed, F.; Benammar Elgaaied, A. Targeting Tumor Metabolism: A New Challenge to Improve Immunotherapy. Front. Immunol. 2018, 9, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Revving the engine: Signal transduction fuels T cell activation. Immunity 2007, 27, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Triplett, T.A.; Garrison, K.C.; Marshall, N.; Donkor, M.; Blazeck, J.; Lamb, C.; Qerqez, A.; Dekker, J.D.; Tanno, Y.; Lu, W.C.; et al. Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat. Biotechnol. 2018, 36, 758–764. [Google Scholar] [CrossRef]
- Ben-Shoshan, J.; Maysel-Auslender, S.; Mor, A.; Keren, G.; George, J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur. J. Immunol. 2008, 38, 2412–2418. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Mu, C.Y.; Huang, J.A.; Chen, Y.; Chen, C.; Zhang, X.G. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med. Oncol. 2011, 28, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Fournel, L.; Wu, Z.; Stadler, N.; Damotte, D.; Lococo, F.; Boulle, G.; Segal-Bendirdjian, E.; Bobbio, A.; Icard, P.; Tredaniel, J.; et al. Cisplatin increases PD-L1 expression and optimizes immune check-point blockade in non-small cell lung cancer. Cancer Lett. 2019, 464, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Zerdes, I.; Wallerius, M.; Sifakis, E.G.; Wallmann, T.; Betts, S.; Bartish, M.; Tsesmetzis, N.; Tobin, N.P.; Coucoravas, C.; Bergh, J.; et al. STAT3 Activity Promotes Programmed-Death Ligand 1 Expression and Suppresses Immune Responses in Breast Cancer. Cancers 2019, 11, 1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Wang, H.Y.; Woetmann, A.; Raghunath, P.N.; Odum, N.; Wasik, M.A. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood 2006, 108, 1058–1064. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Pang, J.; Peng, Y.; Molina, J.R.; Yang, P.; Liu, S. Elevated Cellular PD1/PD-L1 Expression Confers Acquired Resistance to Cisplatin in Small Cell Lung Cancer Cells. PLoS ONE 2016, 11, e0162925. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Brew, B.J. Implications of the kynurenine pathway and quinolinic acid in Alzheimer’s disease. Redox Rep. 2002, 7, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Henderson, L.M.; Gross, C.J. Metabolism of niacin and niacinamide in perfused rat intestine. J. Nutr. 1979, 109, 654–662. [Google Scholar] [CrossRef]
- Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate; Other B Vitamins; Choline. Dietary Reference Intakes for Thiamin, Riboflovin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline; National Academies Press (US): Washington, DC, USA, 1998. [Google Scholar]
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Hashimoto, T.; Tsuchiya, M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J. Biol. Chem. 2007, 282, 24574–24582. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.C. Tryptophan nutrition and metabolism: An overview. Adv. Exp. Med. Biol. 1991, 294, 345–358. [Google Scholar]
- Davis, I.; Liu, A. What is the tryptophan kynurenine pathway and why is it important to neurotherapeutics? Expert Rev. Neurother. 2015, 15, 719–721. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Dong, H.; Li, Q.; Li, Y.; Hong, A. Thioredoxin induces Tregs to generate an immunotolerant tumor microenvironment in metastatic melanoma. Oncoimmunology 2015, 4, e1027471. [Google Scholar] [CrossRef] [Green Version]
- de Araujo, E.F.; Feriotti, C.; Galdino, N.A.L.; Preite, N.W.; Calich, V.L.G.; Loures, F.V. The IDO-AhR Axis Controls Th17/Treg Immunity in a Pulmonary Model of Fungal Infection. Front. Immunol. 2017, 8, 880. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; von Knebel Doeberitz, N.; Oezen, I.; Wick, W.; Ochs, K. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front. Immunol. 2014, 5, 673. [Google Scholar] [CrossRef]
- Campia, I.; Buondonno, I.; Castella, B.; Rolando, B.; Kopecka, J.; Gazzano, E.; Ghigo, D.; Riganti, C. An Autocrine Cytokine/JAK/STAT-Signaling Induces Kynurenine Synthesis in Multidrug Resistant Human Cancer Cells. PLoS ONE 2015, 10, e0126159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O.; et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiya, T.; Huang, C.H. Updates in the Clinical Development of Epacadostat and Other Indoleamine 2,3-Dioxygenase 1 Inhibitors (IDO1) for Human Cancers. Front. Oncol. 2018, 8, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullard, A. IDO takes a blow. Nat. Rev. Drug Discov. 2018, 17, 307. [Google Scholar] [CrossRef] [PubMed]
- Simons, A.L.; Ahmad, I.M.; Mattson, D.M.; Dornfeld, K.J.; Spitz, D.R. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007, 67, 3364–3370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangpaichitr, M.; Savaraj, N.; Maher, J.; Kurtoglu, M.; Lampidis, T.J. Intrinsically lower AKT, mammalian target of rapamycin, and hypoxia-inducible factor activity correlates with increased sensitivity to 2-deoxy-D-glucose under hypoxia in lung cancer cell lines. Mol. Cancer Ther. 2008, 7, 1506–1513. [Google Scholar] [CrossRef] [Green Version]
- Manerba, M.; Di Ianni, L.; Fiume, L.; Roberti, M.; Recanatini, M.; Di Stefano, G. Lactate dehydrogenase inhibitors sensitize lymphoma cells to cisplatin without enhancing the drug effects on immortalized normal lymphocytes. Eur. J. Pharm. Sci. 2015, 74, 95–102. [Google Scholar] [CrossRef]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxidative Med. Cell. Longev. 2019, 2019, 8201079. [Google Scholar] [CrossRef] [PubMed]
- Pillozzi, S.; D’Amico, M.; Bartoli, G.; Gasparoli, L.; Petroni, G.; Crociani, O.; Marzo, T.; Guerriero, A.; Messori, L.; Severi, M.; et al. The combined activation of KCa3.1 and inhibition of Kv11.1/hERG1 currents contribute to overcome Cisplatin resistance in colorectal cancer cells. Br. J. Cancer 2018, 118, 200–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Cui, H.; Fang, J.; Deng, H.; Kuang, P.; Guo, H.; Wang, X.; Zhao, L. Glutamine deprivation plus BPTES alters etoposide- and cisplatin-induced apoptosis in triple negative breast cancer cells. Oncotarget 2016, 7, 54691–54701. [Google Scholar] [CrossRef] [Green Version]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Wang, S.; Zaal, E.A.; Wang, C.; Wu, H.; Bosma, A.; Jochems, F.; Isima, N.; Jin, G.; Lieftink, C.; et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. eLife 2020, 9. [Google Scholar] [CrossRef]
- Mazumder, M.E.; Beale, P.; Chan, C.; Yu, J.Q.; Huq, F. Epigallocatechin gallate acts synergistically in combination with cisplatin and designed trans-palladiums in ovarian cancer cells. AntiCancer Res. 2012, 32, 4851–4860. [Google Scholar]
- Hu, F.; Wei, F.; Wang, Y.; Wu, B.; Fang, Y.; Xiong, B. EGCG synergizes the therapeutic effect of cisplatin and oxaliplatin through autophagic pathway in human colorectal cancer cells. J. Pharmacol. Sci. 2015, 128, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Martin, A.; Colomer, R.; Brunet, J.; Lupu, R.; Menendez, J.A. Overexpression of fatty acid synthase gene activates HER1/HER2 tyrosine kinase receptors in human breast epithelial cells. Cell Prolif. 2008, 41, 59–85. [Google Scholar] [CrossRef]
- Puig, T.; Turrado, C.; Benhamu, B.; Aguilar, H.; Relat, J.; Ortega-Gutierrez, S.; Casals, G.; Marrero, P.F.; Urruticoechea, A.; Haro, D.; et al. Novel Inhibitors of Fatty Acid Synthase with Anticancer Activity. Clin. Cancer Res. 2009, 15, 7608–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaevangelou, E.; Almeida, G.S.; Box, C.; deSouza, N.M.; Chung, Y.L. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int. J. Cancer 2018, 143, 992–1002. [Google Scholar] [CrossRef]
- Imai, H.; Kaira, K.; Oriuchi, N.; Shimizu, K.; Tominaga, H.; Yanagitani, N.; Sunaga, N.; Ishizuka, T.; Nagamori, S.; Promchan, K.; et al. Inhibition of L-type amino acid transporter 1 has antitumor activity in non-small cell lung cancer. AntiCancer Res. 2010, 30, 4819–4828. [Google Scholar]
- Huttunen, K.M.; Gynther, M.; Huttunen, J.; Puris, E.; Spicer, J.A.; Denny, W.A. A Selective and Slowly Reversible Inhibitor of l-Type Amino Acid Transporter 1 (LAT1) Potentiates Antiproliferative Drug Efficacy in Cancer Cells. J. Med. Chem. 2016, 59, 5740–5751. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Liu, X.; Li, F. Esophageal Angiolipoma: A Tongue-Like Mucosal Mass. Clin. Gastroenterol. Hepatol. 2021, 19, e49–e50. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.J.M.; Theodoropoulos, G.; Li, Y.Y.; Wu, C.; Sha, W.; Feun, L.G.; Lampidis, T.J.; Savaraj, N.; Wangpaichitr, M. Targeting the Kynurenine Pathway for the Treatment of Cisplatin-Resistant Lung Cancer. Mol. Cancer Res. 2020, 18, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Zhang, W.; Liu, H.; Peng, L.; Yang, Z.; Lou, J. Inhibition of glutathione synthesis reverses Kruppel-like factor 4-mediated cisplatin resistance. Cancer Chemother. Pharmacol. 2012, 69, 377–385. [Google Scholar] [CrossRef]
- Haas, B.; Schutte, L.; Wos-Maganga, M.; Weickhardt, S.; Timmer, M.; Eckstein, N. Thioredoxin Confers Intrinsic Resistance to Cytostatic Drugs in Human Glioma Cells. Int. J. Mol. Sci. 2018, 19, 2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirshner, J.R.; He, S.; Balasubramanyam, V.; Kepros, J.; Yang, C.Y.; Zhang, M.; Du, Z.; Barsoum, J.; Bertin, J. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol. Cancer Ther. 2008, 7, 2319–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogihara, K.; Kikuchi, E.; Okazaki, S.; Hagiwara, M.; Takeda, T.; Matsumoto, K.; Kosaka, T.; Mikami, S.; Saya, H.; Oya, M. Sulfasalazine could modulate the CD44v9-xCT system and enhance cisplatin-induced cytotoxic effects in metastatic bladder cancer. Cancer Sci. 2019, 110, 1431–1441. [Google Scholar] [CrossRef]
- Stanford, E.A.; Ramirez-Cardenas, A.; Wang, Z.; Novikov, O.; Alamoud, K.; Koutrakis, P.; Mizgerd, J.P.; Genco, C.A.; Kukuruzinska, M.; Monti, S.; et al. Role for the Aryl Hydrocarbon Receptor and Diverse Ligands in Oral Squamous Cell Carcinoma Migration and Tumorigenesis. Mol. Cancer Res. 2016, 14, 696–706. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Target (Propose Target) | References |
|---|---|---|
| Glycolysis pathway | ||
| 2-DG | HK | [160,161] |
| FX-11 | LDHA | [6] |
| Oxamate | LDHA | [162] |
| DCA | PDK | [163] |
| Riluzole | LDHA via NAD+ | [6,164] |
| Glutaminolysis pathway | ||
| BPTES | GLS | [165] |
| CB-839 | GLS | [166,167] |
| EGCG | GDH | [168,169] |
| Fatty acid synthesis pathway | ||
| C75 | FASN | [22,170,171] |
| Cerulenin | FASN | [171] |
| Orlistat | FASN | [171,172] |
| TOFA | ACC | [22,171] |
| Arginine synthesis pathway | ||
| ADI-PEG20 | arginine degradation | [54,173,174] |
| Arginase1 | arginine degradation | [51,175] |
| Redox pathway | ||
| BSO | GSH | [100,176,177] |
| PX-12 | TRX | [178] |
| Elesclomol | ETC (Cu++) | [22,88,179] |
| Riluzole | xCT | [6,164] |
| SSZ | xCT | [43,180] |
| EX527 | SIRT1 | [122] |
| Kynurenine pathway | ||
| BCH | LAT1 | [173,174] |
| CH-223191 | AHR | [181] |
| Epacadostat | IDO | [176] |
| Indoximod | IDO | [176] |
| Navoximod | IDO | [176] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wangpaichitr, M.; Theodoropoulos, G.; Nguyen, D.J.M.; Wu, C.; Spector, S.A.; Feun, L.G.; Savaraj, N. Cisplatin Resistance and Redox-Metabolic Vulnerability: A Second Alteration. Int. J. Mol. Sci. 2021, 22, 7379. https://doi.org/10.3390/ijms22147379
Wangpaichitr M, Theodoropoulos G, Nguyen DJM, Wu C, Spector SA, Feun LG, Savaraj N. Cisplatin Resistance and Redox-Metabolic Vulnerability: A Second Alteration. International Journal of Molecular Sciences. 2021; 22(14):7379. https://doi.org/10.3390/ijms22147379
Chicago/Turabian StyleWangpaichitr, Medhi, George Theodoropoulos, Dan J. M. Nguyen, Chunjing Wu, Sydney A. Spector, Lynn G. Feun, and Niramol Savaraj. 2021. "Cisplatin Resistance and Redox-Metabolic Vulnerability: A Second Alteration" International Journal of Molecular Sciences 22, no. 14: 7379. https://doi.org/10.3390/ijms22147379
APA StyleWangpaichitr, M., Theodoropoulos, G., Nguyen, D. J. M., Wu, C., Spector, S. A., Feun, L. G., & Savaraj, N. (2021). Cisplatin Resistance and Redox-Metabolic Vulnerability: A Second Alteration. International Journal of Molecular Sciences, 22(14), 7379. https://doi.org/10.3390/ijms22147379