1. Introduction
More than 10% of patients with cancer develop metastatic brain tumors, and advanced cancers such as lung cancer and breast cancer metastasize to the brain more frequently and rapidly, making surgical resection challenging and chemotherapy refractory in many cases [
1]. In recent years, it has been reported that astrocytes, a type of glial cell, contribute to the malignant transformation of cancer cells [
2,
3]. The blood–brain barrier, comprising vascular endothelial cells, pericytes, and astrocytes, prevents the entry of harmful substances into the brain [
2,
4]. In addition, astrocytes express a variety of transporters that take up and secrete numerous substances to maintain central nervous system (CNS) homeostasis, playing a role in supplying nutrients and protecting neurons [
5,
6].
To clarify the nature of cancer cells of the brain metastases, the interaction between cancer cells and astrocytes has been actively investigated by using their coculture systems. It has been reported that astrocytes contribute to cancer cell survival by suppressing the expression of the tumor suppressor phosphatase and tensin homolog deleted from chromosome 10 (PTEN) [
2] and through endothelin signaling [
7]. There are two possible pathways for the exchange of substances between cells: one is through paracrine factors such as cytokines and exosomes (including small ribonucleic acids (RNAs), such as micro RNAs (miRNAs)), and the other is through intercellular junctions such as gap junctions (GJs). GJs are formed by the aggregation of six connexin (Cx) proteins into a hexamer (hemichannel), which further aggregates between neighboring cells to form a pathway between the cytoplasm of both cells. In particular, connexin 43 (Cx43) is abundantly expressed in astrocytes, and the network constructed by a GJ has been found to affect neural activity in the CNS [
8,
9]. Therefore, if cancer cells infiltrating the brain utilize astrocyte functions for their survival, a GJ may be an intervening molecule in this process [
10].
However, in the early stages of metastasis, astrocytes have also been reported to contribute to cancer suppression by inducing cell death via the secretion of plasminogen activator to produce plasmin, which suppresses brain metastasis by converting membrane-bound astrocytic Fas ligand into a paracrine death signal for cancer cells [
11]. Astrocytes also interfere with the metastasis-promoting signal introduced by the downstream signal of the transient receptor potential ankyrin 1 protein (TRPA1), an ion channel, via miRNA (miRNA-142-3p) secretion [
12]. Taken together, these results indicate that the state of the brain microenvironment, especially that of the astrocytes, comprising most of the brain composition, changes dynamically during sustained contact with cancer cells.
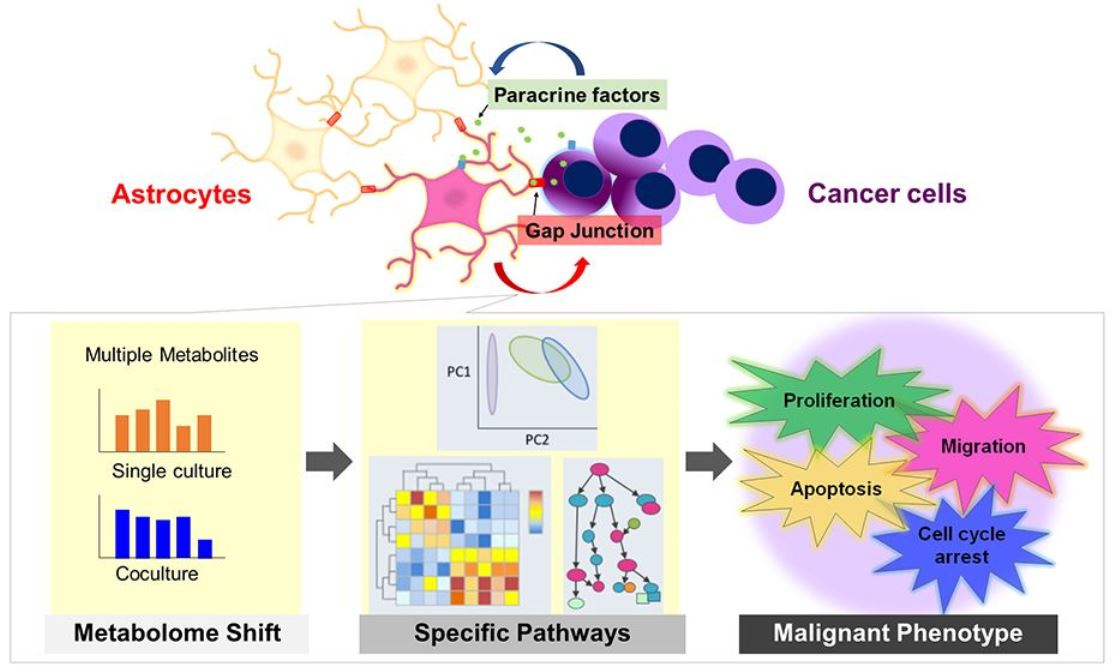
It is generally believed that cancer cells acquire an abnormal proliferative capacity by activating glucose and amino acid metabolism [
13]. On the other hand, astrocytes are regulators of the CNS metabolism, and the metabolites they produce, secrete, and recover affect the survival of the surrounding cells. In other words, a certain relationship between metabolic changes and cellular phenotypes, such as proliferation and motility, is expected, and metabolome shift can be considered as a consequence of early interactions that can occur in the microenvironment. We hypothesized that astrocytes are metabolically altered by cancer cells, resulting in changes in their physiological functions and that direct or indirect contact with altered astrocytes affects the malignant phenotypes of cancer cells. To test this hypothesis, we cocultured astrocytes and cancer cells and aimed to clarify what metabolome shift occurred in both cells, as well as the phenotype transition. In metabolomics, it is possible to capture the metabolic flow of the entire cell by comprehensively measuring small molecules such as sugars, amino acids, and nucleic acids in the cell [
14]. Therefore, we measured 110 compounds using capillary electrophoresis time-of-flight mass spectrometry (CE-TOFMS) and extracted metabolic pathways containing metabolites that showed significant changes to evaluate the transformation of astrocytes and cancer cells, respectively, and to clarify the mechanism of early metastatic brain tumor progression from the viewpoint of cellular metabolism.
3. Discussion
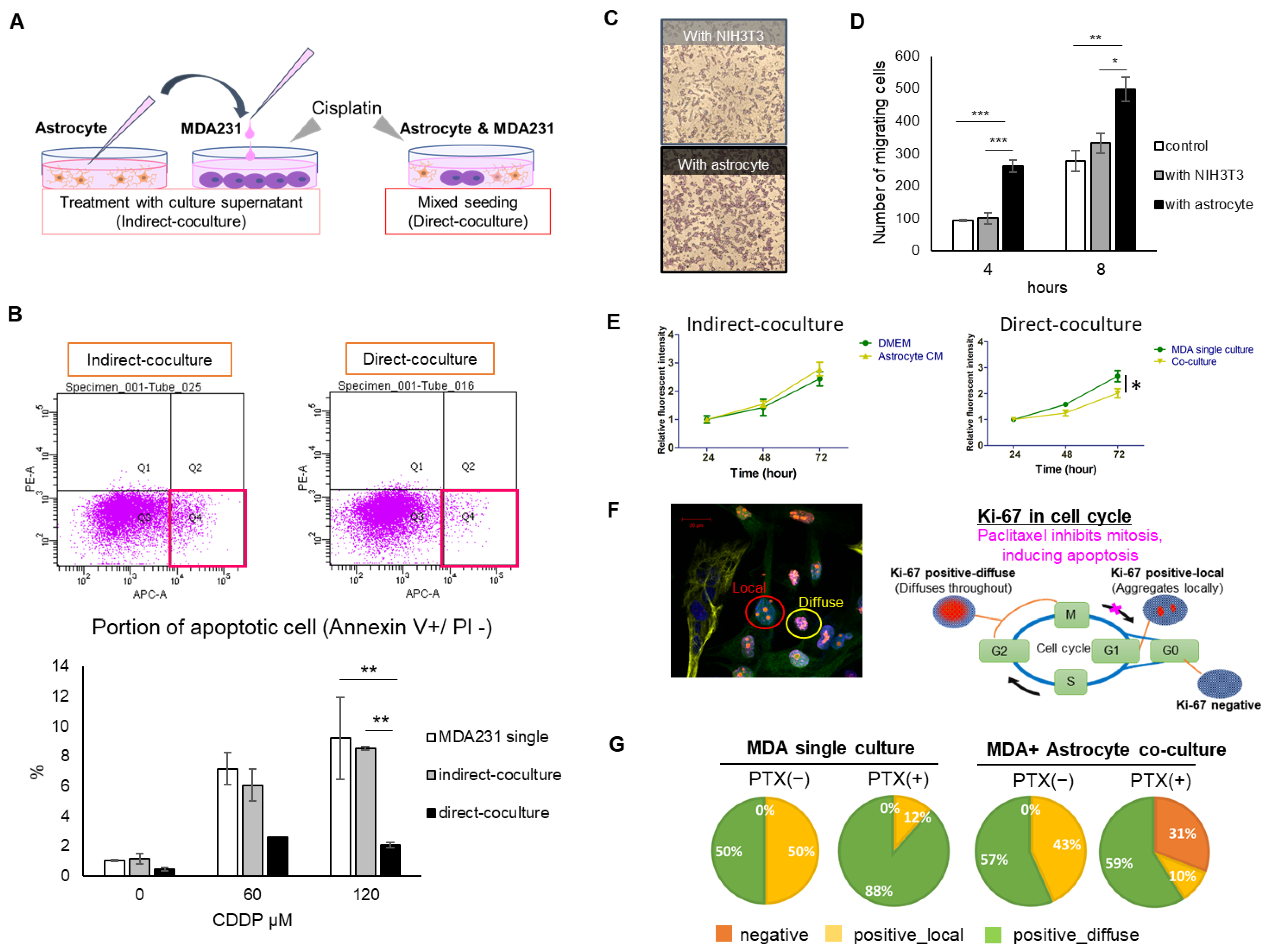
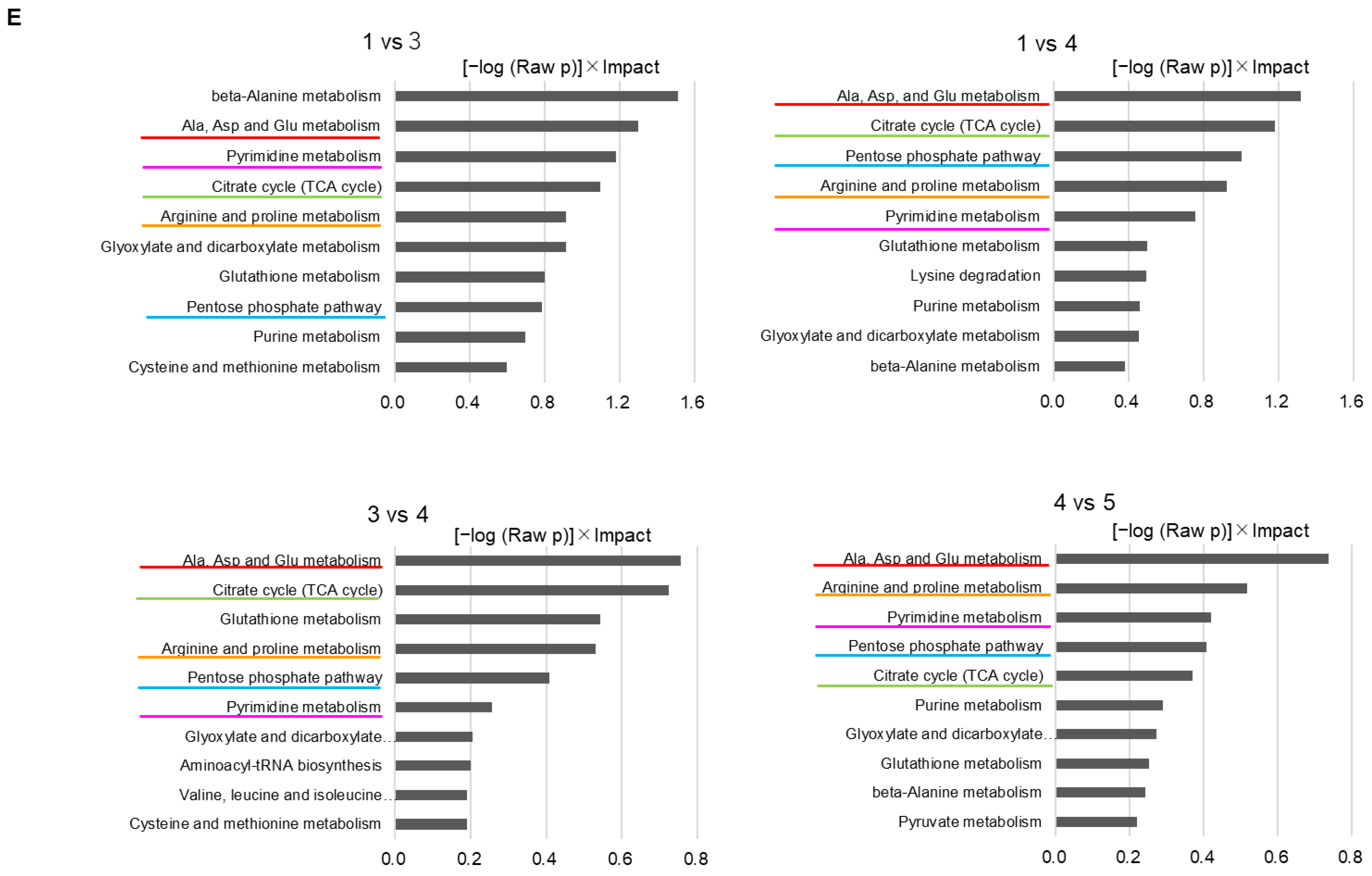
We evaluated two types of culture systems: direct coculture (direct contact between astrocytes and cancer cells) and indirect coculture (contact via liquid factors, i.e., paracrine factors). The important mechanism in direct contact is the existence of gap junction (GJ). However, from the viewpoint of interacting molecules, paracrine factors that are secreted outside the cell and then taken up by the other cell or stimulate the receptor are also evaluated in this direct coculture system. If the contribution of GJ-impermeable paracrine factors to the phenotype under evaluation (drug sensitivity, proliferation, migration, etc.) is not large, no difference can be confirmed by indirect coculture, but direct coculture includes the influence of paracrine factors in addition to GJ-permeable factors. It is possible that the GJ-permeable factor and the paracrine factor are common (i.e., there is a transduction pathway other than GJ). On the other hand, if a difference is found in our indirect coculture, it is clear that the interaction occurred in a GJ-independent transduction pathway.
In the present study, direct coculture showed a decrease in drug sensitivity of MDA231 (
Figure 1B) and a tendency to inhibit growth (
Figure 1E). Indirect coculture using culture supernatant collected at a fixed point did not show a clear change in phenotypes. However, the presence of astrocytes significantly promoted the migration of MDA231 by the indirect coculture (
Figure 1D). It was the only one that could be tested in which both cells remain communication separated by a chamber. Hohensee et al. reported that cancer cells in the transwell insert in the presence of astrocytes under the insert showed increased migration ability [
16], and Kim et al. reported that cancer cells were resistant to various anticancer drugs by direct coculture with astrocyte [
17]. Our results support these reports. It is possible that by prolonging the reaction time or increasing the intensity of exposure, the influence may be seen even with indirect coculture; however, it is thought that direct coculture strengthens the cancer phenotype more aggressively.
It is also suggested that factors that contribute significantly to cancer cell migration and survival or proliferation are different. The intervening molecules that exert their effects on migration may be those that are released and taken up extracellularly and have multiple pathways for their intracellular and extracellular transport, or those that act on surface receptors to promote downstream signaling. Hohensee et al., who showed that PTEN loss has important implications for the metastatic properties of triple-negative breast cancer (TNBC), found several cytokines released into the supernatant when cocultured with astrocytes, particularly in the context of their close involvement with PTEN, and identified granulocyte-macrophage-colony stimulating factor (GM-CSF) (CSF2) as one of them [
16]. Although they could not distinguish between TNBC and astrocytes as the source of GM-CSF release in their experimental system, there was a high correlation between GM-CSF expression in TNBC tissues and decreased survival [
18]. In addition, the CSF2 receptor appeared on astrocytes only in cocultures with tumor cells [
16], suggesting that GM-CSF is released from TNBCs and astrocytes respond by releasing some chemotactic substances, which may contribute to enhanced migration.
On the other hand, a direct coculture may be influenced by GJs formed by metastatic brain tumors and surrounding astrocytes. The protective effects of astrocytes against drug-induced apoptosis of cancer cells include the sequestration of calcium directly related to cell death from the cytoplasm of tumor cells via GJs, and additional mechanisms have been suggested, including upregulation of survival genes such as BCL2L1, TWIST1, and GSTA5 in tumor cells [
11]. It has also been confirmed that 2′3′ cyclic GMP-AMP (cGAMP) moves through GJs formed between cancer cells and astrocytes, and accelerates the stimulator of interferon genes (STING) pathway in astrocytes, which finally promotes cytokine secretion such as tumor necrosis factor (TNF), which in turn assists cancer cell survival [
3]. Importantly, the loss of GJs by Cx43 knockdown in breast cancer cells did not suppress brain metastasis; however, it suppressed tumor colony growth in the brain. In addition to cGAMP, it has been reported that miR-709, which is involved in lung cancer progression, may permeate GJs in a coculture system of metastatic lung cancer PC14 and astrocytes [
19]. Substances that permeate GJs may directly affect cell survival and subsequent growth. In this study, we did not directly identify any changes in the expression of antiapoptotic or proapoptotic factors that might explain the interaction, which needs to be pursued in the future.
Taken together, the present study clearly shows that it is likely that GJ-dependent mechanisms are strongly involved in drug sensitivity and cell proliferation, while GJ-independent transduction pathways are involved in the interaction for migration. However, since GJ-permeable substances and paracrine factors may be common, it is difficult at present to narrow down the interacting molecules that affect each phenotype or to estimate their influence. Furthermore, another challenge in the mixed seeding conditions is the need to carefully compensate for the effect of metabolite loss due to cell separating manipulation by cell sorting on metabolome evaluation [
20]. Considering the technical challenges, in proving our hypothesis that the interaction between cancer cells and astrocytes would first appear in each other’s metabolome, we applied the evaluation system of indirect coculture to metabolome analysis, expecting the contribution of paracrine factors not limited to GJ permeabilization.
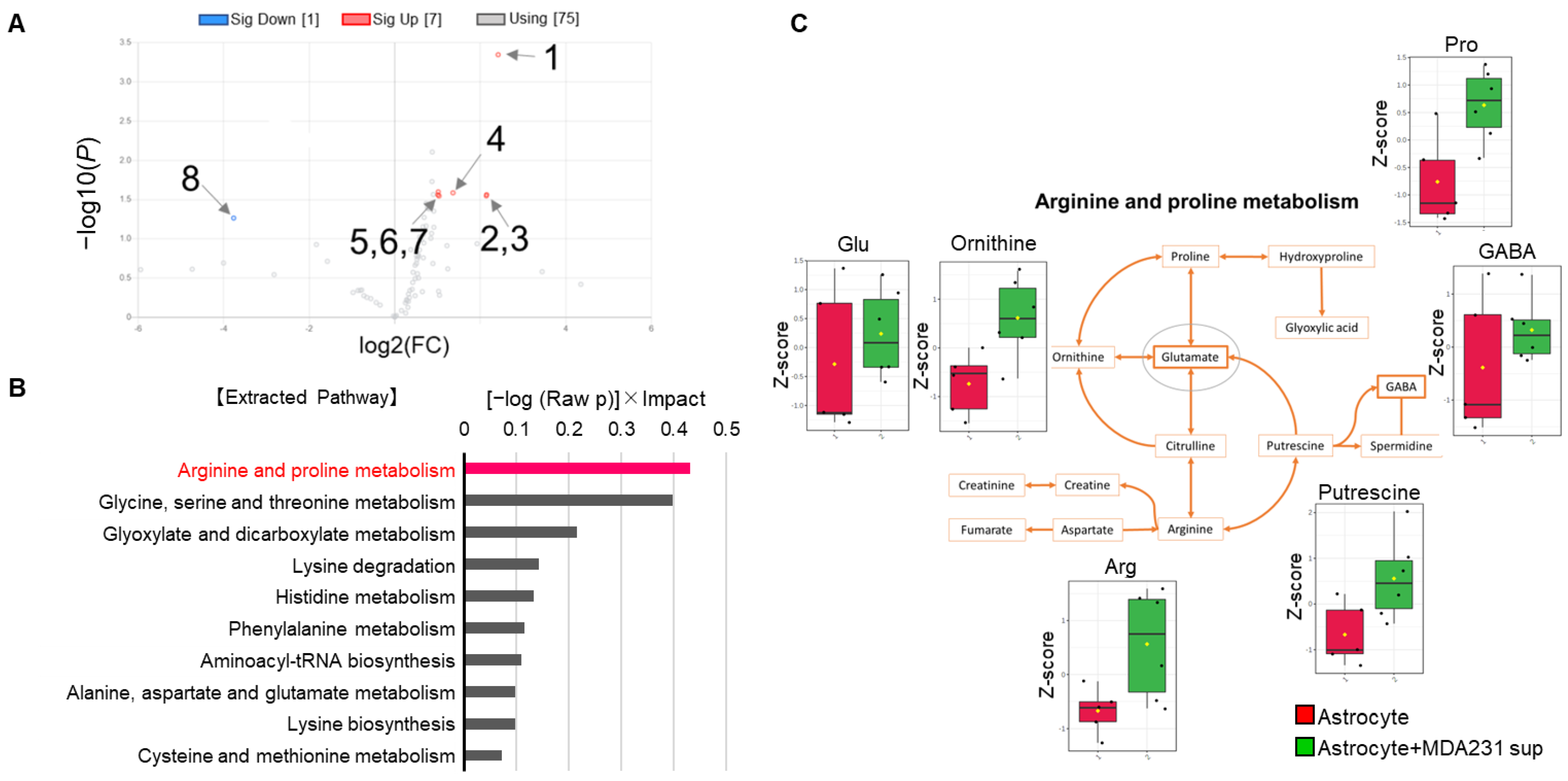
First, the changes in astrocytes induced by MDA231 were particularly pronounced in amino acid metabolism. Astrocytes are known to take up Glu produced by neural activity to suppress the cytotoxicity caused by excess Glu [
8]. Considering that the metabolic pathways of Glu-related substances such as arginine and proline were extracted from pathway analysis when cancer cell supernatants were added to astrocytes (
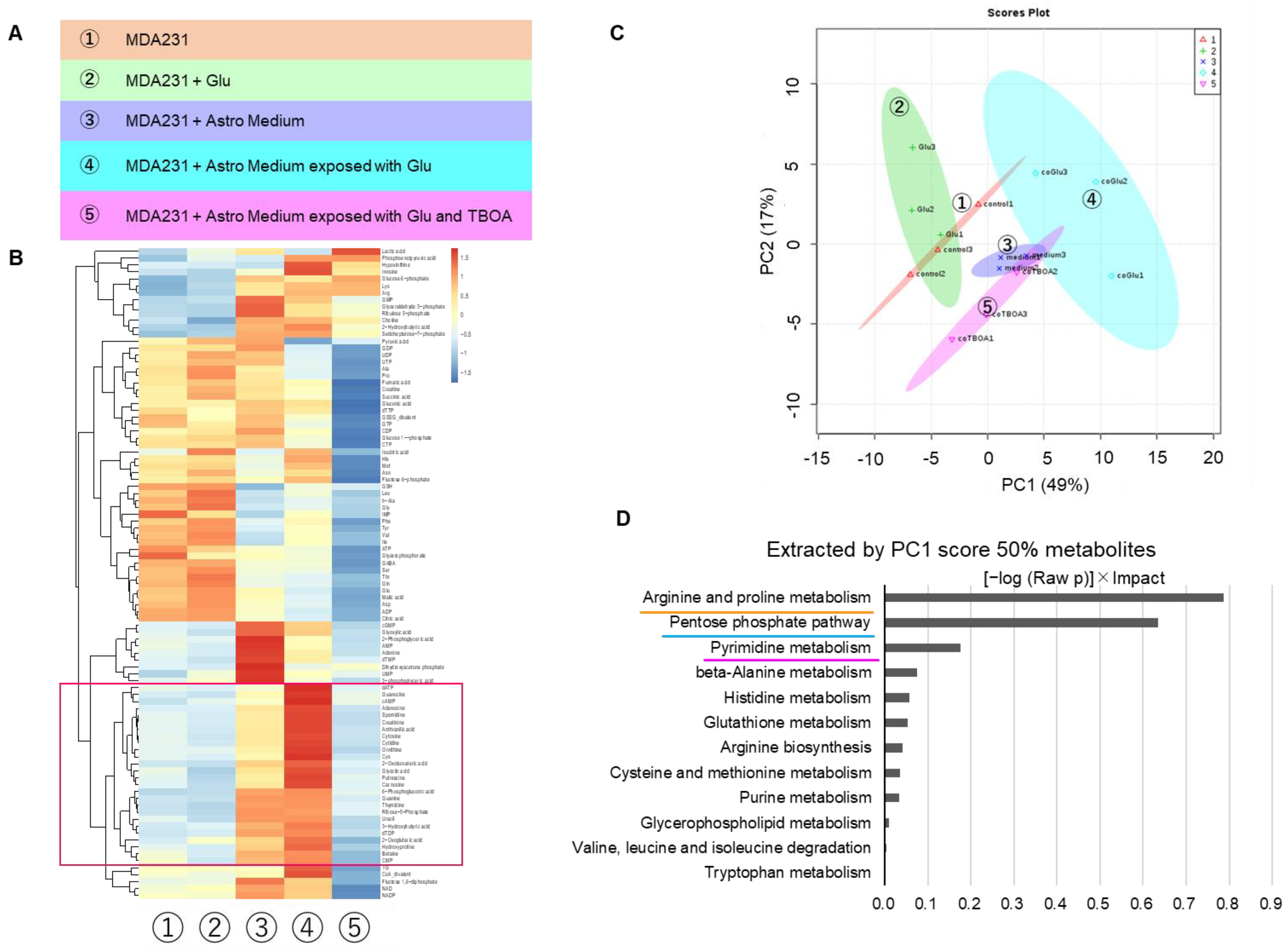
Figure 2B,C), the interaction between cancer cells and astrocytes is likely to depend on the major physiological function of astrocytic Glu uptake and associated Glu-peripheral metabolism. Based on this hypothesis, we attempted to culture MDA231 cells with astrocyte culture supernatants that were exposed to Glu, to evaluate the effect on the phenotype and metabolism of cancer cells.
The metabolites with high contribution of PC1 in PCA showed an increasing trend with astrocyte culture supernatant (group 3) compared to control (group 1). It also occurred both when only astrocyte culture supernatant was added (group 3) and when astrocyte supernatant cultured under Glu-exposed conditions was added (group 4) (
Figure 4C,E). This supports the hypothesis that the metabolic changes, which occurred in cancer cells when astrocyte supernatants were added, depended on “Glu uptake and associated peri-Glu metabolism by astrocytes.” Since the addition of Glu alone did not cause significant changes in the MDA231 metabolome (
Figure 4B,C), Glu itself was not directly responsible for the metabolic changes.
Pathway analysis showed that the arginine–proline pathway, which promotes polyamine synthesis and is involved in RNA translation and cell proliferation, contributes to changes in both MDA231 and astrocytes. On the other hand, it was observed that mixed coculture with astrocytes tended to decrease the proliferation rate of MDA231 (
Figure 1E) as described above. In the early stage of brain metastasis, when the survival of cancer cells is unstable, it has been reported that cancer cells slow down cell cycle progression and enhance apoptosis-resistant survival signals. This switching is thought to involve a mechanism in which a component secreted by peripheral astrocytes suppresses DNMT1, a methyltransferase of cancer cells [
21]. It is suggested that there is a mechanism by which the responsiveness of peripheral astrocytes is dynamically altered upon some stimuli or oxidative stress loading and that such changes are first manifested in metabolomic shifts. We have previously shown that the histone deacetylase inhibitor, trichostatin A, attenuates drug resistance by metabolic shift in renal cell carcinoma [
22], and epigenetic regulation may consequently induce a metabolomic shift [
23,
24]. Considering the suppressive effect against proliferation in the coculture group, the arginine–proline pathway in this context may involve the promotion of certain gene expression rather than cell growth.
The pentose phosphate pathway (PPP) is a major source of nicotinamide adenine dinucleotide phosphate (NADPH) and enhances metabolic flux [
25], which may lead to cell growth. The TCA cycle is generally incomplete and slows down in cancer cells [
26]; however, mitochondrial oxidative phosphorylation (OXPHOS) is often maintained [
27,
28], allowing large amounts of ATP production, which is effective for energy production throughout the entire cell. However, the activation of OXPHOS also promotes the production of reactive oxygen species (ROS) from oxidative stress sources, and the capture of ROS by NADPH is important for cell survival. In addition, the TCA cycle produces NADH and FADH
2, which serve as important electron carriers for OXPHOS. Therefore, activation of both the PPP and TCA cycle may be beneficial for cancer cell survival. Although cancer stem cells (CSCs) are known for their glycolytic-dependent metabolism and high glucose uptake capacity, certain quiescent or low-metabolism-turnover tumor-initiating CSCs, including breast cancer and glioblastoma, are more dependent on OXPHOS than differentiated progenitor cancer cells [
28,
29,
30]. Conversely, when OXPHOS is inhibited, there is a shift to glycolytic-dependent metabolism [
30], and trends in metabolomic changes suggest that the cancer microenvironment, where astrocytes are the majority, contributes to this early CSC flexibility.
Although we were unable to fully examine the metabolome under the direct coculture conditions, it was preliminarily observed that direct coculture of MDA231 and astrocytes resulted in a greater degree of change compared with the indirect coculture performed in this study (data not shown). While GJs have been identified between astrocytes and cancer cells by mixed seeding [
3,
19,
31], the causal relationship between this presence and metabolomic changes in both cells is unknown. However, changes in cellular phenotype due to GJ-mediated exchange of molecules may allow for rapid response independent of gene expression regulation [
32], which may be an advantage for cancer cells with survival at stake. For dynamic astrocyte reactivity in response to circumstances, molecules transferred from cancer cells in contact via GJs would be a direct trigger for a metabolomic shift. It is also possible that the molecule is common with a paracrine factor that is secreted out of the cell and taken up into the other cell in sustained cultures of both cells. In this study, we proposed the hypothesis that there are intracellular metabolomic changes prior to the appearance of cancer malignant phenotype on the surface, and actually identified metabolic pathways that were altered in conjunction with contact. The significance of these enhanced metabolic pathways found in the present study needs to be explored by assessing the impact of these changes on the malignant phenotype of cancer under experimental conditions in which cell–cell interactions are maintained.
4. Materials and Methods
4.1. Reagents
All cultures and reagents were procured from Sigma Chemical Company (St. Louis, MO, USA) unless otherwise indicated. Cellmatrix Type IC (Nitta Gelatin Inc., Osaka, Japan) was diluted with HCl (pH 3.0) to make 0.1 mg/mL type-I collagen solution. CDDP (Wako Pure Chemicals, Osaka, Japan) was prepared in dimethyl sulfoxide (DMSO) (Wako Pure Chemicals, Osaka, Japan) at a concentration of 100 mM just before use, and the final concentration of DMSO in the cell culture was <0.1%. DL-TBOA (R&D Systems, Minneapolis, MN, USA) was used as an inhibitor of the glutamate uptake transporter (EAAT2). Precrystalline deoxyribonuclease (DNase) I (Wako Pure Chemicals), from bovine pancreas, was dissolved in distilled water to make a 25 mM stock solution.
4.2. Cell Culture
Astrocyte cultures were prepared from neonatal Wistar rats as described previously by Kajitani et al. [
33]. Briefly, the isolated cerebral cortices and hippocampi were minced and incubated with trypsin and DNase. Dissociated cells were plated in 75 cm
2 tissue culture flasks (8–15 × 10
6 cells/flask) precoated with 0.1 mg/mL type-I collagen solution. After 8–12 days, the cells were purified to remove fewer adherent neurons and microglia with a rotary shaker at 100 rpm for 15 h. Adherent cells were trypsinized (0.25%) and plated into 75 cm
2 flasks. After the cells reached confluence (10 days), the confluent cells were shaken by hand for 10 min. Adherent cells were trypsinized (0.25%) and plated on new dishes. Using this method, more than 90% of the cells expressed glial fibrillary acidic protein (GFAP), a marker of astrocytes. Animal experiments were performed in accordance with the protocols approved by the Animal Research Committee of Chiba University.
The human breast cancer cell line MDA231 was obtained from the American Type Culture Collection (Manassas, VA, USA). MDA231 cells were transfected with the enhanced green fluorescent protein (eGFP) using the pEGFP-N1 plasmid with Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. To obtain stable transfected cells, cells were further subjected to cell sorting using BD FACS AriaTM II (BD Biosciences, San Jose, CA, USA). The pEGFP-N1 plasmid was gifted by Dr. Aoki, S. to Chiba University.
The murine fibroblast cell line NIH3T3 was used as a control for astrocytes. NIH3T3 was gifted by Dr. Nishimura, K. to International University of Health and Welfare.
All cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Wako Pure Chemicals) supplemented with 10% fetal bovine serum (FBS) (Biowest, Riverside, MO, USA), penicillin (0.5 U/mL), and streptomycin (1 µg/mL), and were maintained at 37 °C in a fully humidified atmosphere of 5% CO2.
4.3. Coculture of Astrocytes and MDA231
Two types of coculture methods were used: (1) indirect coculture: addition of culture supernatant (generally used a 3-day culture medium of astrocytes or MDA231 diluted to 50% with DMEM was used), and (2) direct coculture: mixed seeding (astrocyte cell number:MDA231 cell number = 1:1), except for the migration assay, where cells were incubated with the sharing medium during the coculture period as MDA231 in a cell-culture insert with 8.0 µm pores and astrocytes in a well of 24-well plate under the insert.
4.4. Migration Assay
The migration assay was performed as previously described [
34]. Briefly, MDA231 was treated with FBS-free DMEM for 24 h before the day of the experiment. As an attractant of migration, a total of 500 µL DMEM with 10% FBS containing 5 × 10
4 astrocytes or NIH3T3 cells was seeded in a well of a 24-well plate, and then MDA231 cells were trypsinized and suspended in an FBS-free medium. A total of 200 µL of FBS-free medium containing 5 × 10
4 cells was placed into the top of the migration chambers with 8 µm filters (24-well plate format; Becton Dickinson Labware, Franklin Lakes, NJ, USA), which were placed in wells containing astrocytes or NIH3T3 cells. The cells were incubated at 37 °C and 5% CO
2 for 4 h or 8 h. Following this, the chambers were then removed from the wells and coded for analysis by a blinded observer. Cells that had migrated to the bottom of filters were fixed with methanol, rinsed thoroughly with phosphate-buffered saline (PBS), and stained with Giemsa’s solution. Four visual fields were randomly selected for each chamber, and the total number of cells at the bottom of each field was counted under a microscope and then averaged between the four.
4.5. Cell Proliferation Assay
MDA231 was cocultured with type (1) or (2) astrocytes as described in
Section 4.3. The eGFP fluorescence intensity of MDA231 was measured at 488 nm excitation and 515 nm emission using SpectraMax
TM i3 (Molecular Devices, Sunnyvale, CA, USA), 24, 48, and 72 h after the start of coculture. It was confirmed that eGFP emission is proportional to cell number in advance (
Supplementary Figure S1).
4.6. Apoptosis Assay
Cells were arranged at a density of 2.0 × 105 in a 60 mm dish for one tube of flow cytometry. MDA231 single culture or MDA231 cells with astrocyte culture supernatant (50% conditioned medium diluted by DMEM) were seeded at 2.0 × 105 cells/well in a 6-well plate. In the mixed seeding of MDA231 and the astrocyte group, cells were mixed in a 1:1 ratio to obtain a total of 4 × 105 cells/well. After 48 h of culture, the medium was removed and replaced with 120 µM CDDP medium. After an additional 48 h, the cells were trypsinized and washed with PBS. Cells were resuspended in Annexin V binding buffer (BioLegend, San Diego, CA, USA) to 1 × 106 cells/mL. Cell fluid (100 µL) was taken and 5 µL of allophycocyanin (APC) -Annexin V (BioLegend) and 10 µL of propidium iodide (PI) (Invitrogen, Carlsbad, CA, USA) were added, mixed gently, and incubated for another 15 min in the dark. Finally, 400 µL of Annexin V binding buffer was added. The cell fluid was passed through a mesh and transferred to a fluorescence-activated cell sorting tube for flow cytometry using BD FACS Canto IITM (BD Biosciences). For the early apoptotic marker, the Annexin V-positive population was detected, while the late apoptotic marker, PI-positive population, was detected. Finally, Annexin V-positive and PI-negative populations in each culture group were compared.
4.7. Immunofluorescence Staining
A total of 4 × 105 cells were seeded on coverslips, which were placed in a 12-well plate for 24 h. The cells were treated with 20 nM paclitaxel or DMSO (control) for 24 h and then fixed in 4% paraformaldehyde PBS (Wako Pure Chemicals, Tokyo, Japan) for 20 min at 37 °C. After blocking with PBS containing 0.1% saponin (MP Biomedicals, Santa Ana, CA, USA) and 3% bovine serum albumin (BSA) for 30 min, the cells were incubated with primary antibodies for 1 h, followed by incubation with secondary donkey anti-rabbit IgG H&L, Alexa Fluor 405 (Abcam, Cambridge, UK, ab175651) or goat anti-mouse IgG (H+L), Alexa Fluor 647 (Thermo Fisher Scientific, Boston, MA, USA)-conjugated antibodies (diluted in PBS containing 0.1% saponin, 1:500) for 1 h; all reactions were carried out at room temperature. Coverslips were picked and mounted with DAPI-Fluormount-G® (SouthernBiotech, Birmingham, AL, USA). Finally, the cells were observed under a Zeiss LSM 780 confocal microscope (Carl Zeiss MicroImaging GmbH, Jena, Germany). The dilutions used for the primary antibodies were as follows: GFAP (1:300; Bioss, Woburn, MA, USA, bs 0199R) and Ki67 (1:200) (Proteintech, Rosemont, IL, USA, 27309-1-AP). All primary antibodies were diluted in PBS containing 0.1% saponin and 3% BSA. The obtained images were analyzed using the ImageJ2 software (National Institute of Health, Bethesda, MD, USA).
4.8. Metabolite Extraction and Metabolome Analysis Using CE-TOFMS
Extractions were performed according to a previous report by Hatakeyama et al. [
35]. Briefly, 1.0 × 10
6 astrocytes or MDA231 were collected in one tube. Cells were detached by trypsin-EDTA treatment and washed twice with 5% mannitol. Cell pellets were suspended in 1 mL of methanol, sonicated for 30 s, and vortexed with 1 mL of chloroform and 0.4 mL of ultra-pure water for 30 s. After centrifugation at 2300×
g at 4 °C for 5 min, the aqueous layers were filtered through a Nanosep/3K (3-kDa cut-off) filter (Nihon Pall Ltd., Tokyo, Japan) at 9100×
g at 4 °C for 2.5 h to remove proteins and phospholipids. The resultant filtrates were used as ionic metabolites; they were lyophilized and dissolved in 25 μL ultra-pure water prior to CE-TOFMS analysis. CE-TOFMS was carried out using an Agilent 7100 CE system equipped with an Agilent 6230 TOF-MS system (Agilent Technologies, Santa Clara, CA, USA). Raw data were processed using the Mass Hunter software (Qualitative and Quantitative Analysis, Agilent Technologies, Santa Clara, CA, USA) for metabolite quantification.
4.9. Statistical Analysis
Each experiment was performed independently at least three times and data are presented as mean ± standard deviation (SD) or standard error of the mean (SEM), except for metabolome analysis. Statistical analysis was performed using the Tukey–Kramer test for comparing multiple groups, or t-test for comparing two groups.
p-values < 0.05 were considered statistically significant. Regarding metabolome analysis, for clustering analysis (heat map), each square indicates a normalized value derived from three samples in each group. Analysis was performed by statistical analysis and pathway analysis using MetaboAnalyst 4.0 (
http://www.metaboanalyst.ca/ (accessed on 10 May 2021)). In statistical analysis, the number of metabolites was converted into a Z score and analyzed. The fold changes and
p-values for the metabolites in the astrocyte and MDA231 coculture cells as compared with astrocyte or MDA231 single culture cells were calculated. For the volcano plot, the
p-values were transformed by −log10 so that the more significant features (
p-values < 0.1 as statistically significant) could be expressed higher on the graph.
4.10. Pathway Analysis
This study elucidated aspects of both the metabolic enrichment of pathways using metabolite set enrichment analysis and the biological importance of the metabolic pathway detected by the centrality theory via topology analysis. The differentially changed metabolites extracted from the volcano plot metabolome or heat map were subjected to pathway analysis with the MetaboAnalyst 4.0 platform (
http://www.metaboanalyst.ca/ (accessed on 10 May 2021)), according to previous reports [
22,
36,
37].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




