
Liver Bile Acid Changes in Mouse Models of Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
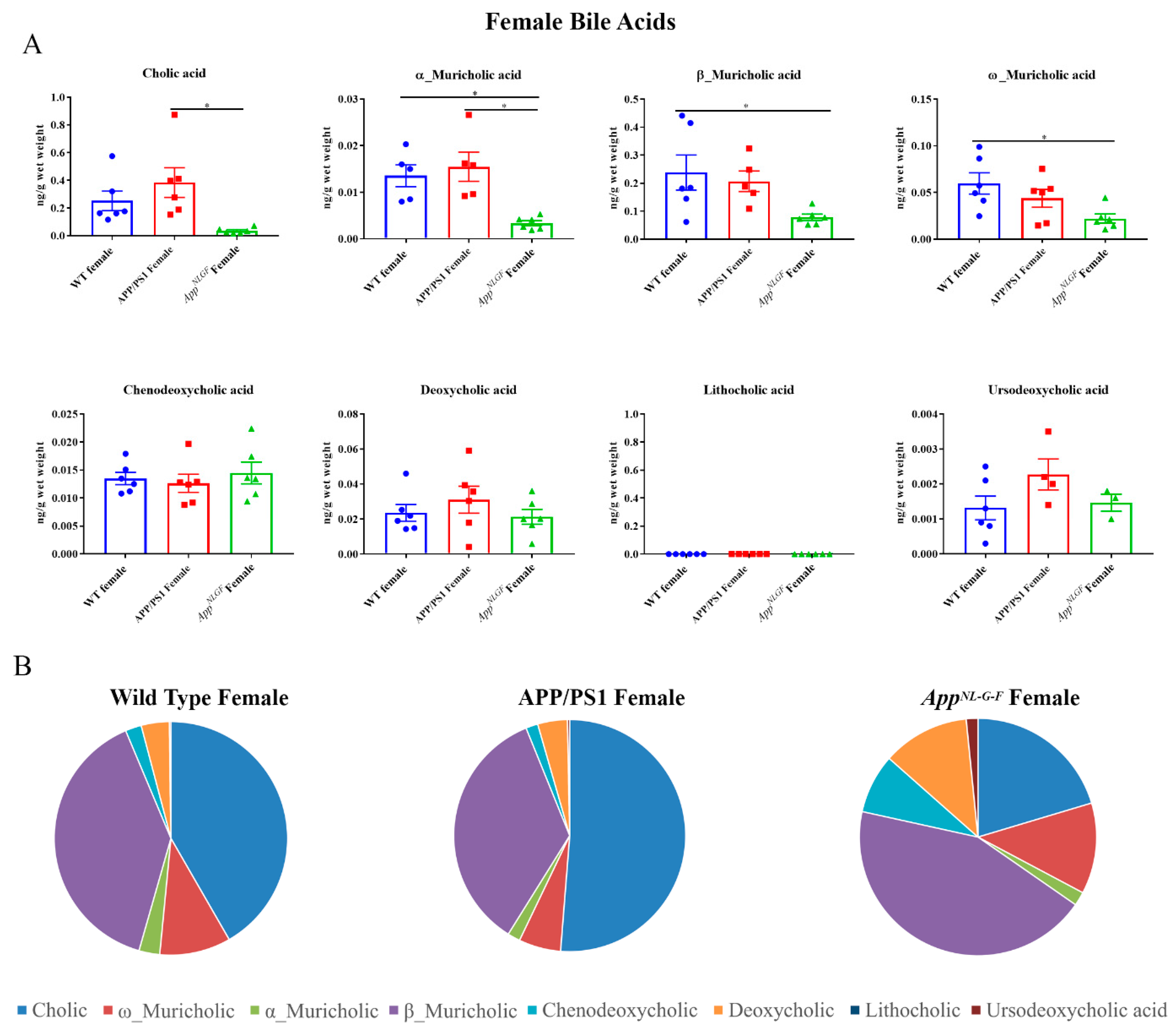
2.1. Primary and Secondary Liver Bile Acid Profile in Female APP/PS1 and AppNL-G-F Mice
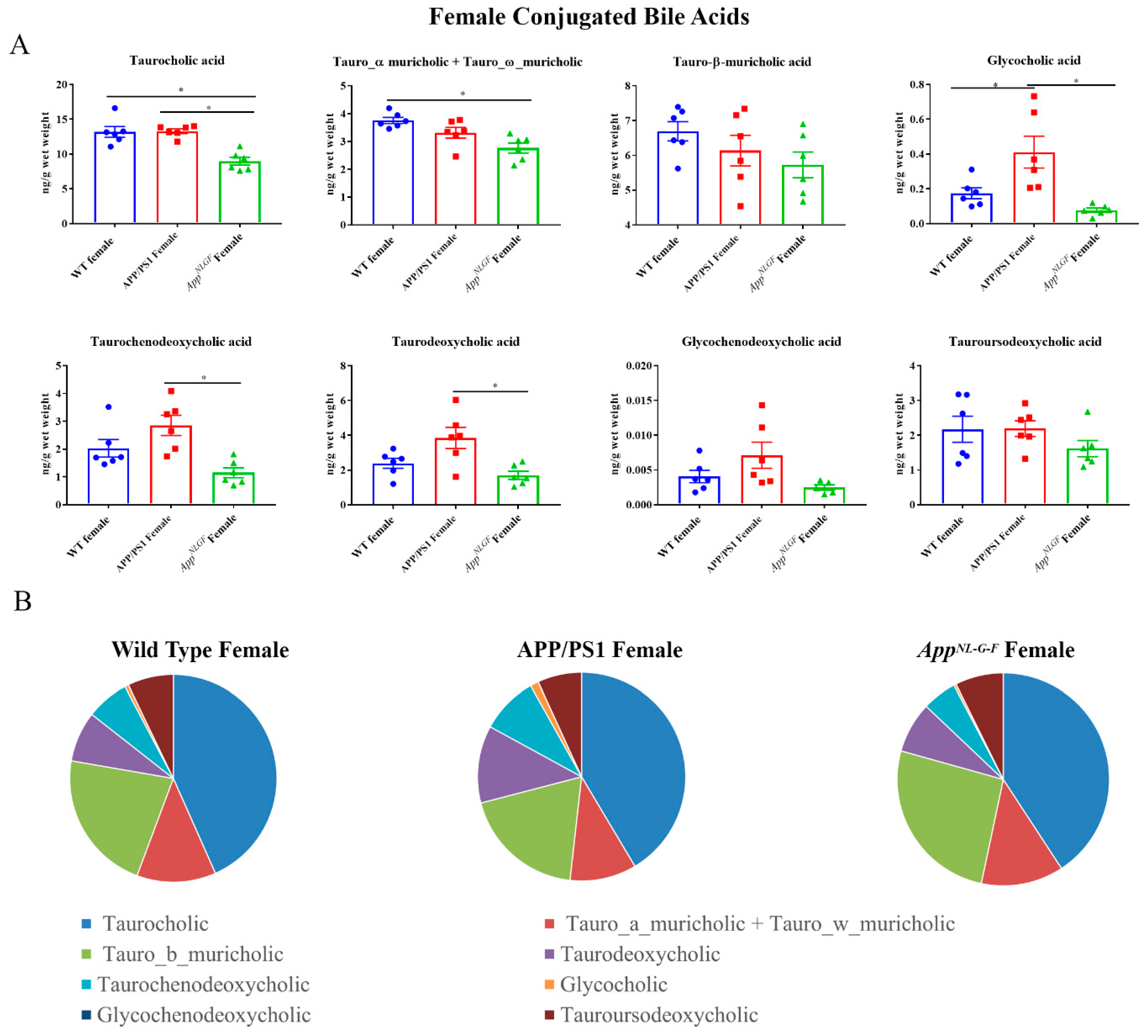
2.2. Conjugated Primary and Secondary Liver Bile Acid Profile in Female APP/PS1 and AppNL-G-F Mice
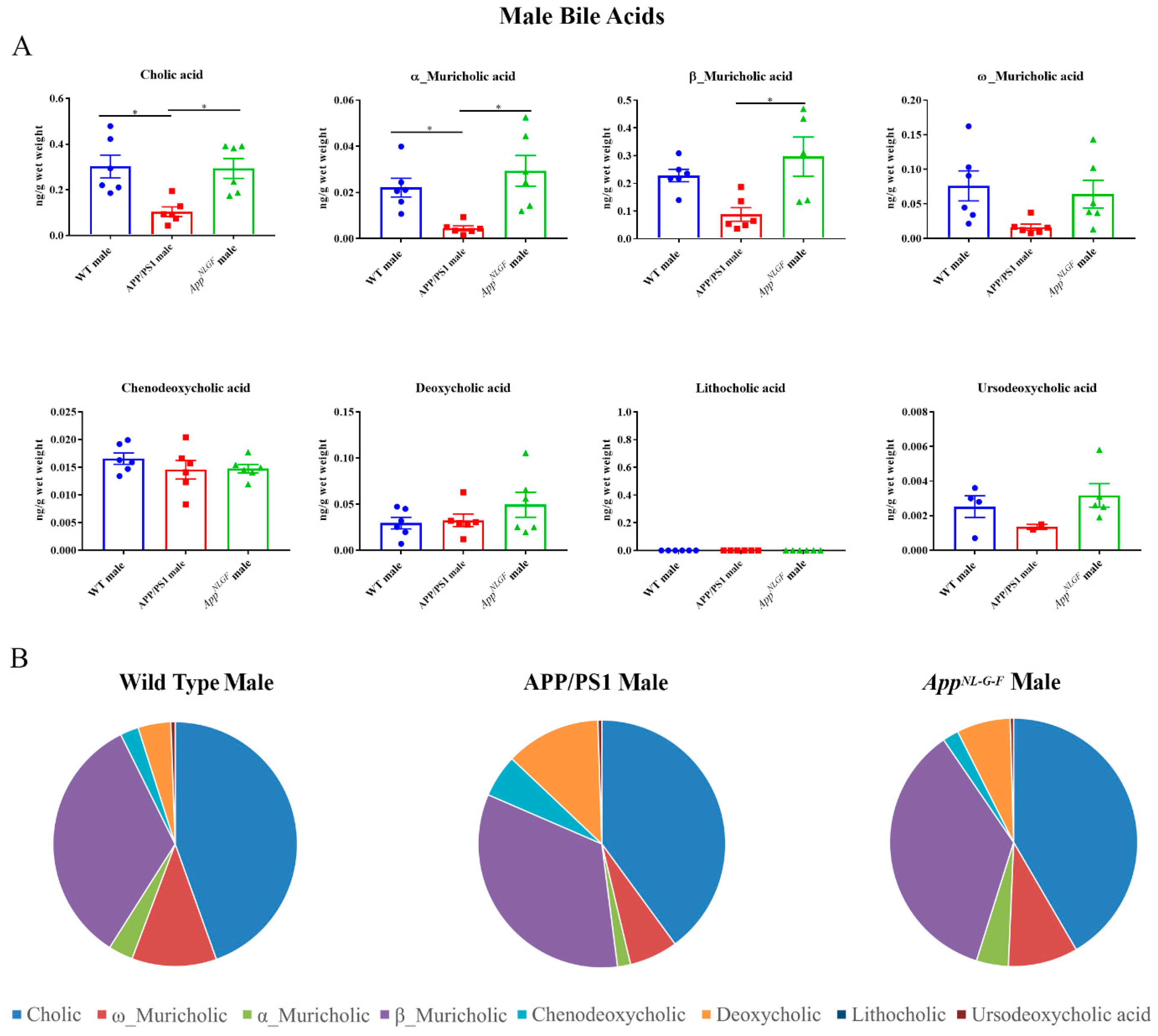
2.3. Primary and Secondary Liver Bile Acid Profile in Male APP/PS1 and AppNL-G-F Mice
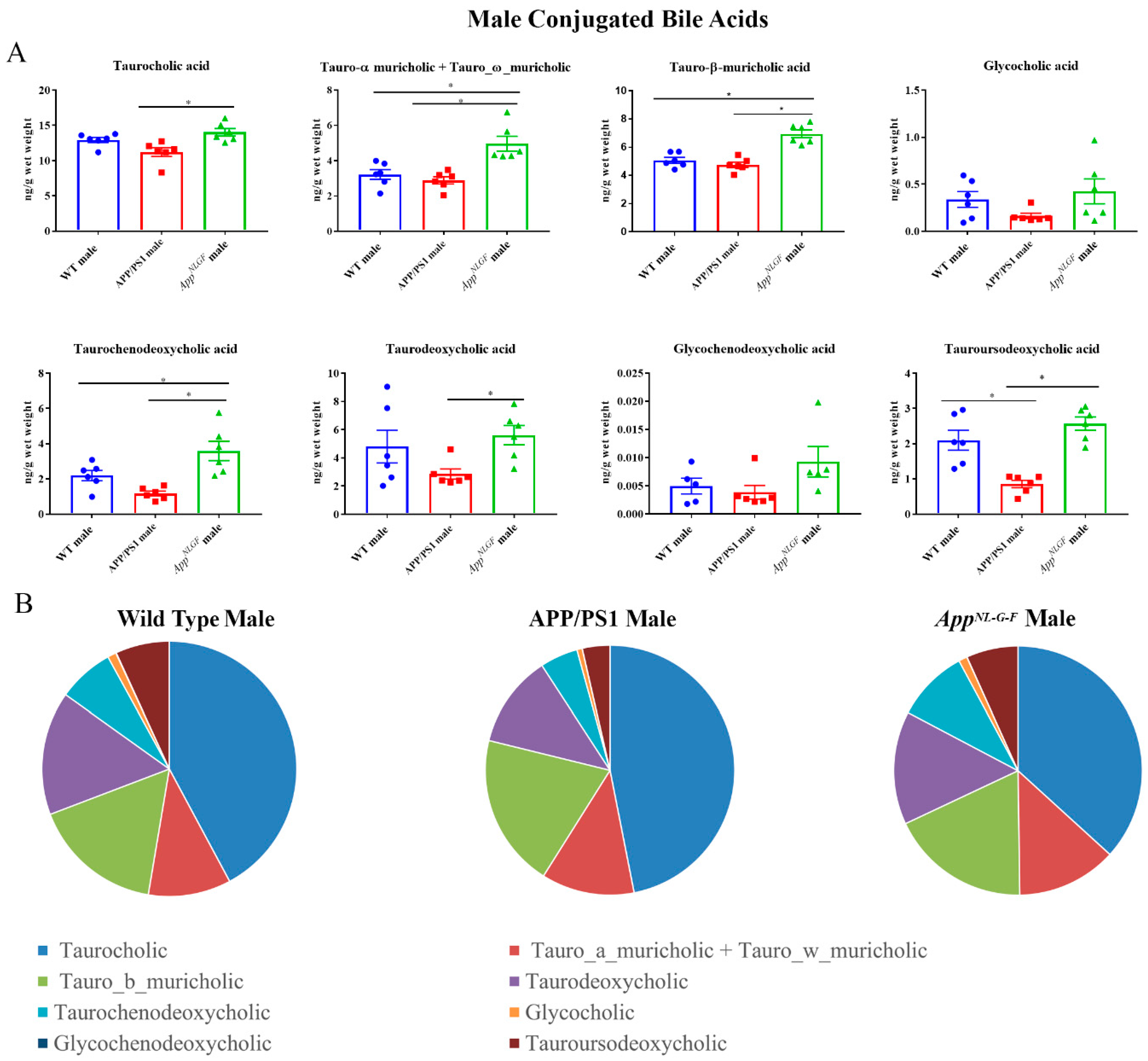
2.4. Conjugated Primary and Secondary Liver Bile Acid Profile in Male APP/PS1 and AppNL-G-F Mice
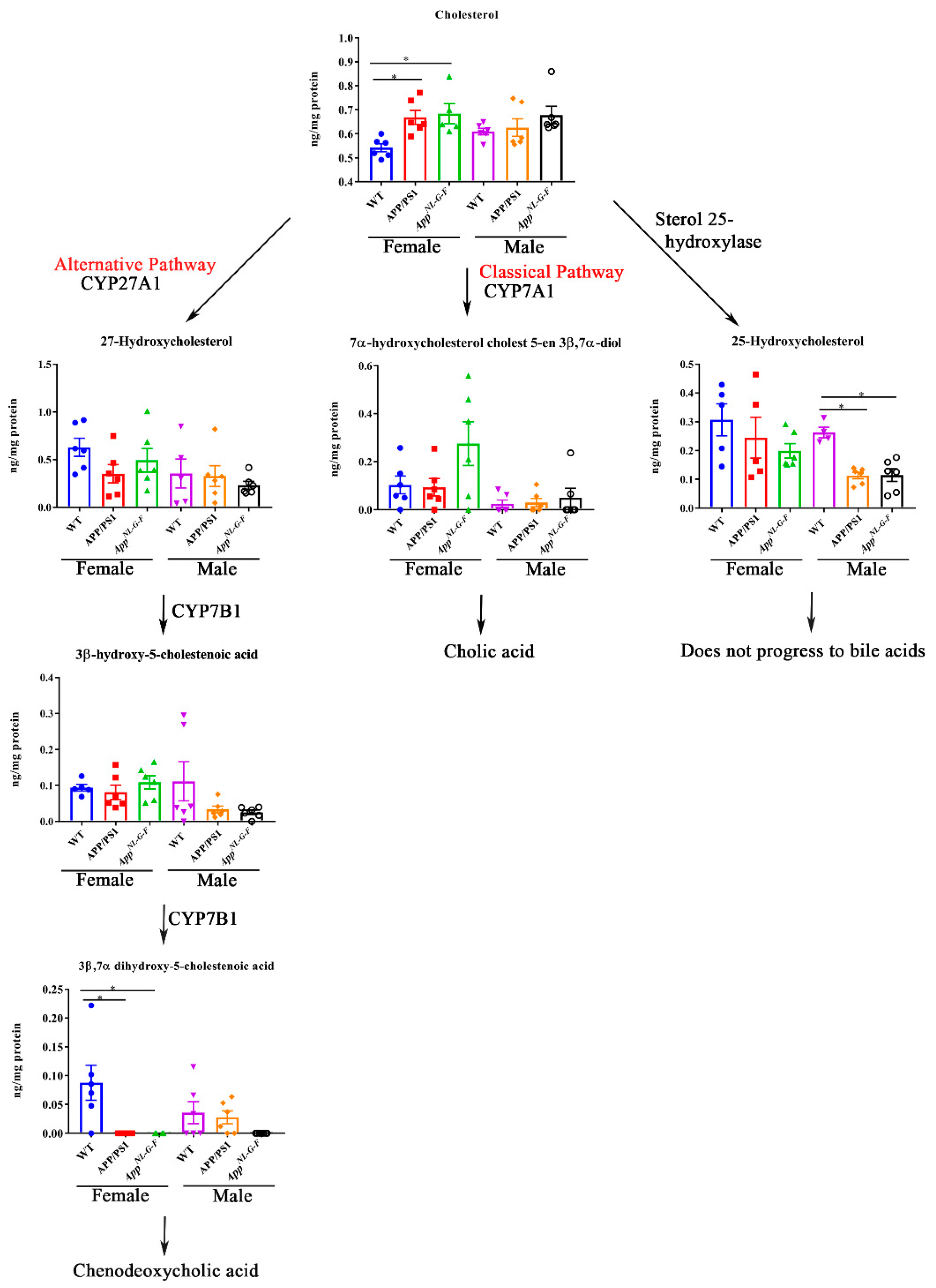
2.5. Cholesterol and Bile Acid Intermediates in Livers of Male and Female APP/PS1 and AppNL-G-F Mice
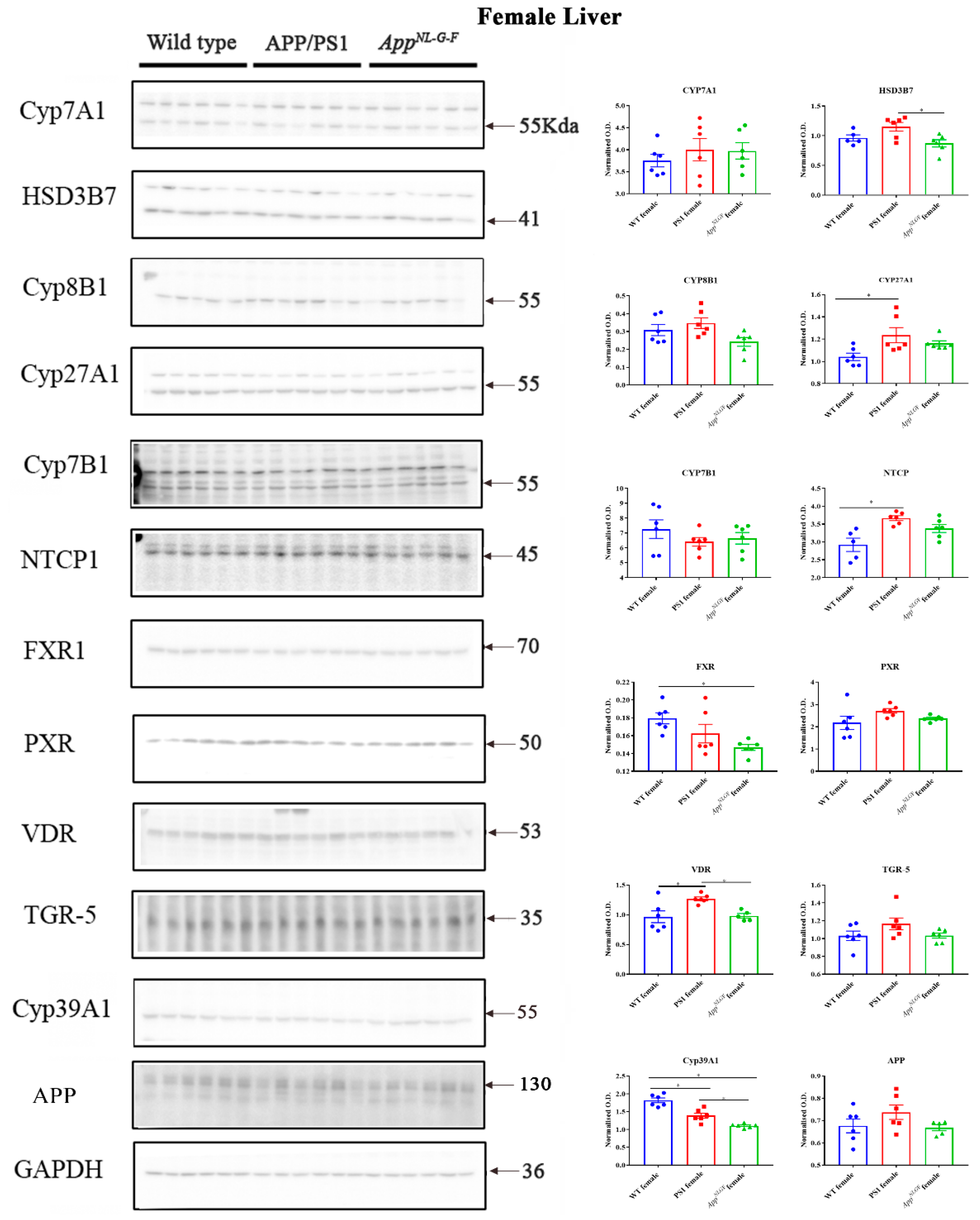
2.6. Changes in Proteins Associated with Bile Acid Synthesis and Signaling in Female Mice
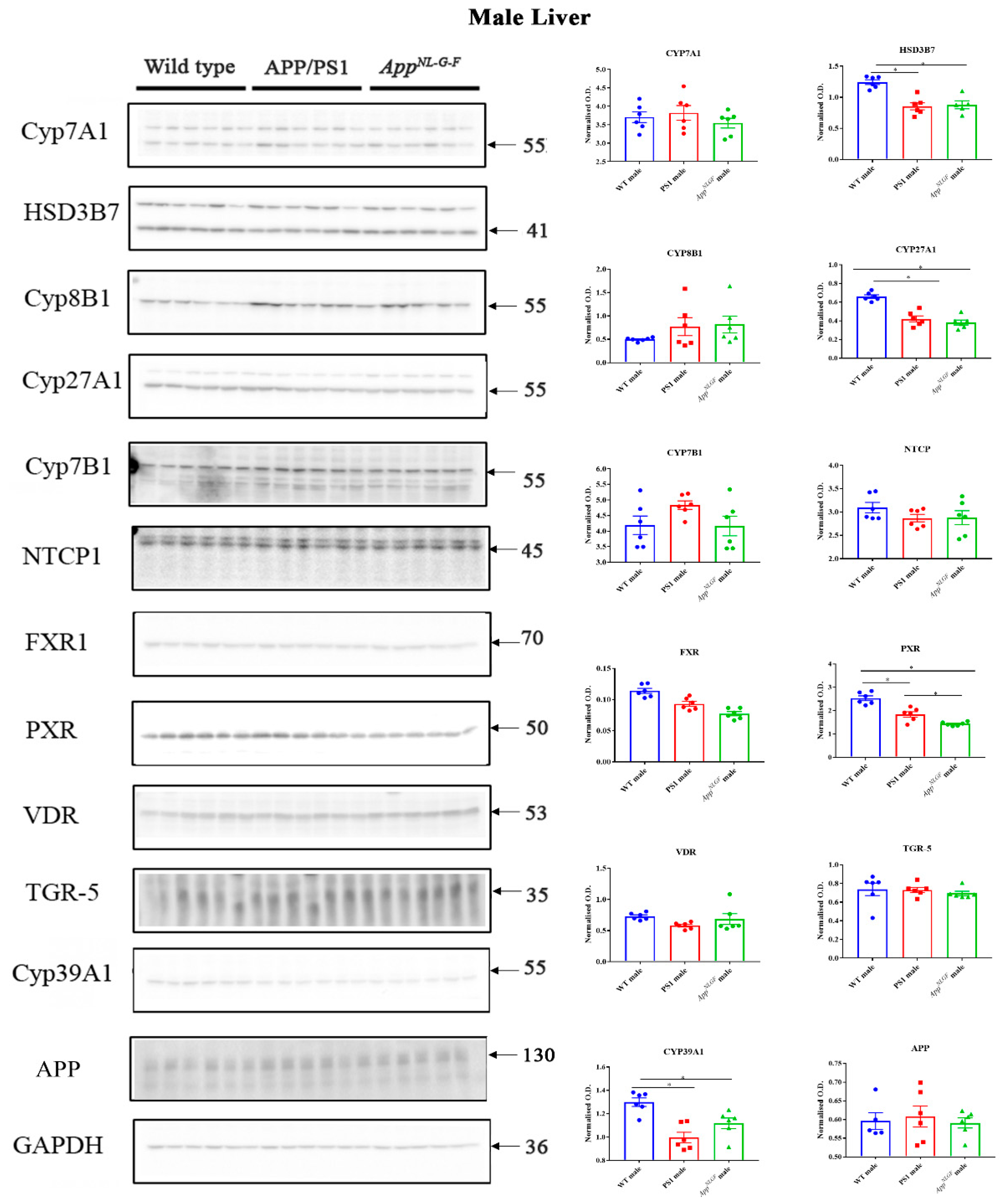
2.7. Changes in Proteins Associated with Bile Acid Synthesis and Signaling in Male Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Animal Use
4.3. Antibodies and Reagents
4.4. Western Blot Analysis
4.5. Cholesterol Metabolites Analysis Using UPLC-MS/MS
4.6. Statistical Analyses
4.7. Ethics Statement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| CYPs | Cytochrome P50s) |
| PS1 | Presenilin |
| CYP7A1 | Cytochrome P450 Family 1 Subfamily A Member 1 |
| HSD3B7 | Hydroxy-Delta-5-Steroid Dehydrogenase, 3 Beta- And Steroid Delta-Isomerase 7 |
| CYP8B1 | Cytochrome P450 Family 8 Subfamily B Member 1 |
| CYP27A1 | Cytochrome P450 Family 27 Subfamily A Member 1 |
| NTCP | Sodium/taurocholate cotransporting polypeptide |
| FXR | Farsenoid X receptor |
| PXR | Pregnane X receptor |
| VDR | Vitamin D receptor |
| TGR-5 | Takeda G-protein coupled receptor 5 |
| CYP46A1 | Cytochrome P450 Family 46 Subfamily A Member 1 |
| APP | Amyloid precursor protein |
| UPLC-MS/MS | Ultra performance liquid chromatography—tandem mass spectrometer |
| MS | Mass spectrometer |
| BAAT | Bile acid CoA-amino acid N-acetyltransferase |
References
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 1993, 16, 460–465. [Google Scholar] [CrossRef]
- Morris, J.K.; Honea, R.A.; Vidoni, E.D.; Swerdlow, R.H.; Burns, J.M. Is Alzheimer’s disease a systemic disease? Biochim. Biophys. Acta 2014, 1842, 1340–1349. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Zhao, S.; Zhu, Y.; Fan, Z.; Wang, J.; Zhang, B.; Chen, Y. Microbiota-gut-brain axis and toll-like receptors in Alzheimer’s disease. Comput. Struct. Biotechnol. J. 2019, 17, 1309–1317. [Google Scholar] [CrossRef]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Golovko, S.; Golovko, M.Y.; Singh, S.; Darland, D.C.; Combs, C.K. Effects of Probiotic Supplementation on Short Chain Fatty Acids in the AppNL-G-F Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 76, 1083–1102. [Google Scholar] [CrossRef]
- Duran-Aniotz, C.; Hetz, C. Glucose Metabolism: A Sweet Relief of Alzheimer’s Disease. Curr. Biol. 2016, 26, R806–R809. [Google Scholar] [CrossRef] [Green Version]
- Craft, S.; Asthana, S.; Cook, D.G.; Baker, L.D.; Cherrier, M.; Purganan, K.; Wait, C.; Petrova, A.; Latendresse, S.; Watson, G.S.; et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: Interactions with apolipoprotein E genotype. Psychoneuroendocrinology 2003, 28, 809–822. [Google Scholar] [CrossRef]
- Ferreira, L.S.S.; Fernandes, C.S.; Vieira, M.N.N.; De Felice, F.G. Insulin Resistance in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Velpen, V.; Teav, T.; Gallart-Ayala, H.; Mehl, F.; Konz, I.; Clark, C.; Oikonomidi, A.; Peyratout, G.; Henry, H.; Delorenzi, M.; et al. Systemic and central nervous system metabolic alterations in Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 93. [Google Scholar] [CrossRef] [Green Version]
- Razay, G.; Vreugdenhil, A.; Wilcock, G. The metabolic syndrome and Alzheimer disease. Arch. Neurol. 2007, 64, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Aggarwal, N.; Schneider, J.; Wilson, R.S. Dietary fats and the risk of incident Alzheimer disease. Arch. Neurol. 2003, 60, 194–200. [Google Scholar] [CrossRef]
- Wei, S.; Gao, L.; Jiang, Y.; Shang, S.; Chen, C.; Dang, L.; Wang, J.; Huo, K.; Wang, J.; Qu, Q. The Apolipoprotein E epsilon4 Allele-Dependent Relationship Between Serum Lipid Levels and Cognitive Function: A Population-Based Cross-sectional Study. Front. Aging Neurosci. 2020, 12, 44. [Google Scholar] [CrossRef] [Green Version]
- Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch. Neurol. 2011, 68, 1239–1244. [Google Scholar] [CrossRef] [Green Version]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B. Cholesterol and the biology of Alzheimer’s disease. Neuron 2004, 41, 7–10. [Google Scholar] [CrossRef] [Green Version]
- McMillin, M.; DeMorrow, S. Effects of bile acids on neurological function and disease. FASEB J. 2016, 30, 3658–3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.H.; Hong, Y.H.; Sung, J.J.; Kim, S.M.; Lee, J.B.; Lee, K.W. Oral solubilized ursodeoxycholic acid therapy in amyotrophic lateral sclerosis: A randomized cross-over trial. J. Korean Med. Sci. 2012, 27, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.M.; Sola, S.; Nan, Z.; Castro, R.E.; Ribeiro, P.S.; Low, W.C.; Steer, C.J. Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proc. Natl. Acad. Sci. USA 2003, 100, 6087–6092. [Google Scholar] [CrossRef] [Green Version]
- Dehkordi, S.M.; Arnold, M.; Nho, K.; Ahmad, S.; Jia, W.; Xie, G.; Louie, G.; Kueider-Paisley, A.; Moseley, M.A.; Thompson, J.W.; et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease-An emerging role for gut microbiome. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2019, 15, 76–92. [Google Scholar]
- Kaur, H.; Nagamoto-Combs, K.; Golovko, S.; Golovko, M.Y.; Klug, M.G.; Combs, C.K. Probiotics ameliorate intestinal pathophysiology in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2020, 92, 114–134. [Google Scholar] [CrossRef]
- Manocha, G.D.; Floden, A.M.; Miller, N.M.; Smith, A.J.; Nagamoto-Combs, K.; Saito, T.; Saido, T.C.; Combs, C.K. Temporal progression of Alzheimer’s disease in brains and intestines of transgenic mice. Neurobiol. Aging 2019, 81, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Einarsson, K.; Ericsson, S.; Ewerth, S.; Reihner, E.; Rudling, M.; Stahlberg, D.; Angelin, B. Bile acid sequestrants: Mechanisms of action on bile acid and cholesterol metabolism. Eur. J. Clin. Pharmacol. 1991, 40 (Suppl. 1), S53–S58. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F. The continuing importance of bile acids in liver and intestinal disease. Arch. Intern. Med. 1999, 159, 2647–2658. [Google Scholar] [CrossRef]
- Xie, G.; Wang, X.; Huang, F.; Zhao, A.; Chen, W.; Yan, J.; Zhang, Y.; Lei, S.; Ge, K.; Zheng, X.; et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int. J. Cancer 2016, 139, 1764–1775. [Google Scholar] [CrossRef]
- Shao, Y.; Ouyang, Y.; Li, T.; Liu, X.; Xu, X.; Li, S.; Xu, G.; Le, W. Alteration of Metabolic Profile and Potential Biomarkers in the Plasma of Alzheimer’s Disease. Aging Dis. 2020, 11, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.M.; DeMorrow, S. Bile Acid Signaling in Neurodegenerative and Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 5982. [Google Scholar] [CrossRef]
- Schubring, S.R.; Fleischer, W.; Lin, J.S.; Haas, H.L.; Sergeeva, O.A. The bile steroid chenodeoxycholate is a potent antagonist at NMDA and GABA(A) receptors. Neurosci. Lett. 2012, 506, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, X.; Cong, Y.; Deng, Y.; Xu, Y.; Chen, A.; Yin, Y. Effects of bile acids and the bile acid receptor FXR agonist on the respiratory rhythm in the in vitro brainstem medulla slice of neonatal Sprague-Dawley rats. PLoS ONE 2014, 9, e112212. [Google Scholar] [CrossRef]
- Theofilopoulos, S.; Wang, Y.; Kitambi, S.S.; Sacchetti, P.; Sousa, K.M.; Bodin, K.; Kirk, J.; Salto, C.; Gustafsson, M.; Toledo, E.M.; et al. Brain endogenous liver X receptor ligands selectively promote midbrain neurogenesis. Nat. Chem. Biol. 2013, 9, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Baloni, P.; Funk, C.C.; Yan, J.; Yurkovich, J.T.; Kueider-Paisley, A.; Nho, K.; Heinken, A.; Jia, W.; Mahmoudiandehkordi, S.; Louie, G.; et al. Metabolic Network Analysis Reveals Altered Bile Acid Synthesis and Metabolism in Alzheimer’s Disease. Cell Rep. Med. 2020, 1, 100138. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [PubMed] [Green Version]
- Li, X.; Feng, Y.; Wu, W.; Zhao, J.; Fu, C.; Li, Y.; Ding, Y.; Wu, B.; Gong, Y.; Yang, G.; et al. Sex differences between APPswePS1dE9 mice in A-beta accumulation and pancreatic islet function during the development of Alzheimer’s disease. Lab. Anim. 2016, 50, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Fu, B.; Lei, H.; Tang, H.; Wang, Y. Gender differences of peripheral plasma and liver metabolic profiling in APP/PS1 transgenic AD mice. Neuroscience 2016, 332, 160–169. [Google Scholar] [CrossRef]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Abeta Plaques. Cell Rep. 2019, 27, 1293–1306.e1296. [Google Scholar] [CrossRef] [Green Version]
- Masuda, A.; Kobayashi, Y.; Kogo, N.; Saito, T.; Saido, T.C.; Itohara, S. Cognitive deficits in single App knock-in mouse models. Neurobiol. Learn. Mem. 2016, 135, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Phelps, T.; Snyder, E.; Rodriguez, E.; Child, H.; Harvey, P. The influence of biological sex and sex hormones on bile acid synthesis and cholesterol homeostasis. Biol. Sex Differ. 2019, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xue, R.; Yang, C.; Gu, J.; Chen, S.; Zhang, S. Cholestasis-induced bile acid elevates estrogen level via farnesoid X receptor-mediated suppression of the estrogen sulfotransferase SULT1E1. J. Biol. Chem. 2018, 293, 12759–12769. [Google Scholar] [CrossRef] [Green Version]
- Swann, J.R.; Want, E.J.; Geier, F.M.; Spagou, K.; Wilson, I.D.; Sidaway, J.E.; Nicholson, J.K.; Holmes, E. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4523–4530. [Google Scholar] [CrossRef] [Green Version]
- Claudel, T.; Zollner, G.; Wagner, M.; Trauner, M. Role of nuclear receptors for bile acid metabolism, bile secretion, cholestasis, and gallstone disease. Biochim. Biophys. Acta 2011, 1812, 867–878. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, G.; Renga, B.; Mencarelli, A.; Pellicciari, R.; Fiorucci, S. Role of FXR in regulating bile acid homeostasis and relevance for human diseases. Curr. Drug Targets Immune. Endocr. Metab. Disord. 2005, 5, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G554–G573. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.R.; Holmstrom, S.R.; Fon Tacer, K.; Bookout, A.L.; Kliewer, S.A.; Mangelsdorf, D.J. Regulation of bile acid synthesis by fat-soluble vitamins A and D. J. Biol. Chem. 2010, 285, 14486–14494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barchetta, I.; Carotti, S.; Labbadia, G.; Gentilucci, U.V.; Muda, A.O.; Angelico, F.; Silecchia, G.; Leonetti, F.; Fraioli, A.; Picardi, A.; et al. Liver vitamin D receptor, CYP2R1, and CYP27A1 expression: Relationship with liver histology and vitamin D3 levels in patients with nonalcoholic steatohepatitis or hepatitis C virus. Hepatology 2012, 56, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Liddle, C.; Downes, M. FXR and PXR: Potential therapeutic targets in cholestasis. J. Steroid Biochem. Mol. Biol. 2012, 130, 147–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, K.; Cowie, D.E.; Konstantinou, D.K.; Hill, S.J.; Tjelle, T.E.; Axon, A.; Koruth, M.; White, S.A.; Carlsen, H.; Mann, D.A.; et al. The PXR is a drug target for chronic inflammatory liver disease. J. Steroid Biochem. Mol. Biol. 2010, 120, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.C.; Callaerts-Vegh, Z.; Nunes, A.F.; Rodrigues, C.M.; D’Hooge, R. Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol. Dis. 2013, 50, 21–29. [Google Scholar] [CrossRef]
- Vang, S.; Longley, K.; Steer, C.J.; Low, W.C. The Unexpected Uses of Urso-and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases. Glob. Adv. Health Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Nunes, A.F.; Amaral, J.D.; Lo, A.C.; Fonseca, M.B.; Viana, R.J.; Callaerts-Vegh, Z.; D’Hooge, R.; Rodrigues, C.M. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-beta deposition in APP/PS1 mice. Mol. Neurobiol. 2012, 45, 440–454. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid regulation of gene expression: Roles of nuclear hormone receptors. Endocr. Rev. 2002, 23, 443–463. [Google Scholar] [CrossRef] [Green Version]
- Cohen, D.E. Balancing cholesterol synthesis and absorption in the gastrointestinal tract. J. Clin. Lipidol. 2008, 2, S1–S3. [Google Scholar] [CrossRef] [Green Version]
- Li-Hawkins, J.; Gafvels, M.; Olin, M.; Lund, E.G.; Andersson, U.; Schuster, G.; Bjorkhem, I.; Russell, D.W.; Eggertsen, G. Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J. Clin. Investig. 2002, 110, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Slunt, H.H.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. APP processing and amyloid deposition in mice haplo-insufficient for presenilin 1. Neurobiol. Aging 2004, 25, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Golovko, S.A.; Golovko, M.Y. Plasma Unesterified Fatty-Acid Profile Is Dramatically and Acutely Changed under Ischemic Stroke in the Mouse Model. Lipids 2018, 53, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Brose, S.A.; Golovko, S.A.; Golovko, M.Y. Brain 2-Arachidonoylglycerol Levels Are Dramatically and Rapidly Increased Under Acute Ischemia-Injury Which Is Prevented by Microwave Irradiation. Lipids 2016, 51, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Seeger, D.R.; Golovko, S.A.; Golovko, M.Y. Blood-Brain Barrier Is the Major Site for a Rapid and Dramatic Prostanoid Increase upon Brain Global Ischemia. Lipids 2020, 55, 79–85. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, H.; Seeger, D.; Golovko, S.; Golovko, M.; Combs, C.K. Liver Bile Acid Changes in Mouse Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7451. https://doi.org/10.3390/ijms22147451
Kaur H, Seeger D, Golovko S, Golovko M, Combs CK. Liver Bile Acid Changes in Mouse Models of Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(14):7451. https://doi.org/10.3390/ijms22147451
Chicago/Turabian StyleKaur, Harpreet, Drew Seeger, Svetlana Golovko, Mikhail Golovko, and Colin Kelly Combs. 2021. "Liver Bile Acid Changes in Mouse Models of Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 14: 7451. https://doi.org/10.3390/ijms22147451
APA StyleKaur, H., Seeger, D., Golovko, S., Golovko, M., & Combs, C. K. (2021). Liver Bile Acid Changes in Mouse Models of Alzheimer’s Disease. International Journal of Molecular Sciences, 22(14), 7451. https://doi.org/10.3390/ijms22147451