
Essential Roles of PPARs in Lipid Metabolism during Mycobacterial Infection

 ,
, 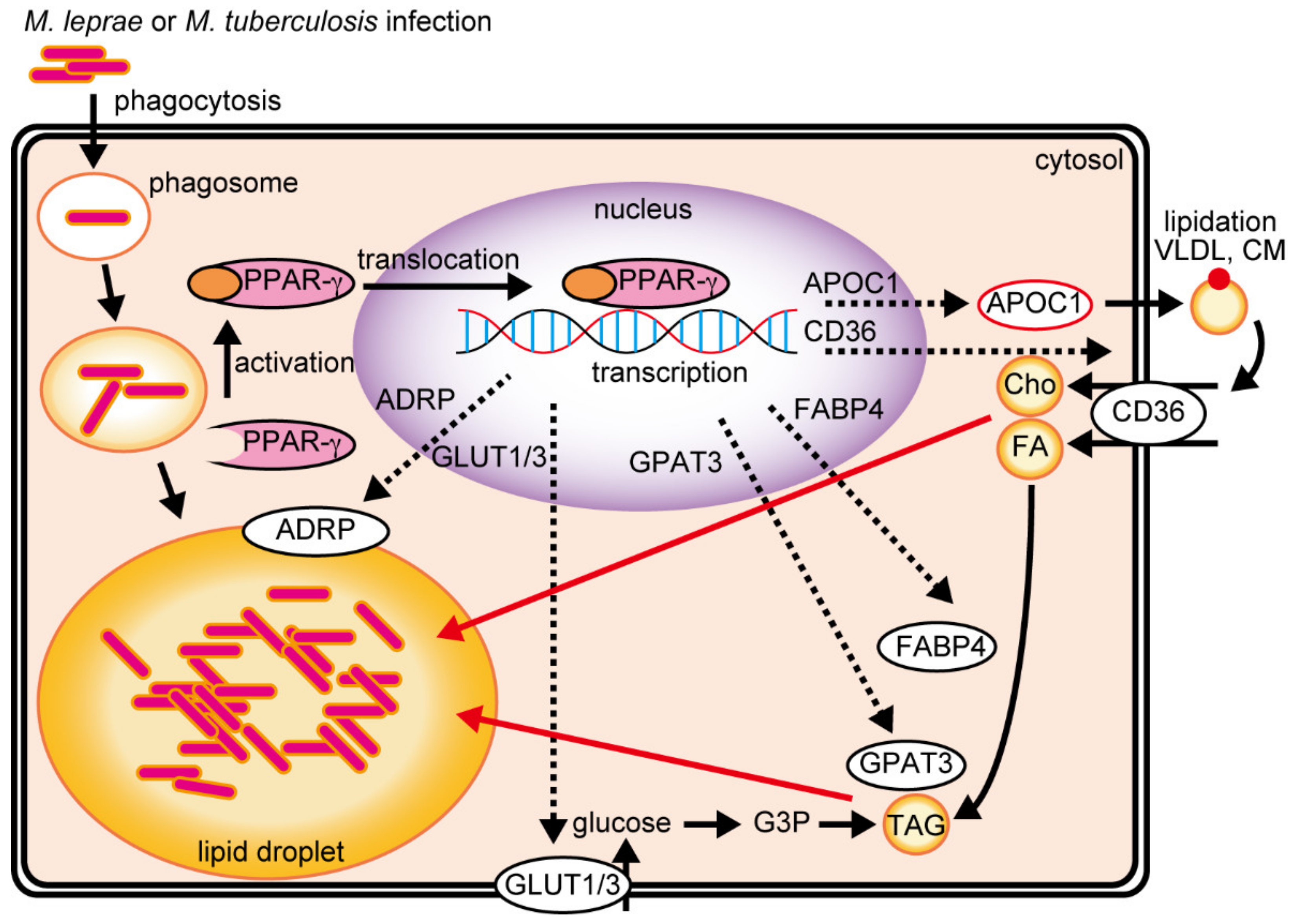{kind=link}
Abstract
:1. Introduction
2. Activation of PPARs by Mycobacteria
3. The Roles of PPARs in Lipid Metabolism
4. Emerging Roles of PPARs in Lipid Metabolism during Mycobacteria Infection
5. Anti-Inflammatory Effects Are Mediated by PPARs during Mycobacterial Infection
6. Mycobacteria-Induced PPAR-Mediated Autophagy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Kawada, T. Lipid metabolism related nuclear receptor—The structure, function, expression and classification of peroxisome proliferation-activated receptor (PPAR). Nihon Rinsho 1998, 56, 1722–1728. [Google Scholar]
- Gottlicher, M.; Widmark, E.; Li, Q.; Gustafsson, J.A. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 4653–4657. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, M.E.; Blumberg, B.; McBride, O.W.; Yi, H.F.; Kronquist, K.; Kwan, K.; Hsieh, L.; Greene, G.; Nimer, S.D. Isolation of the human peroxisome proliferator activated receptor gamma cDNA: Expression in hematopoietic cells and chromosomal mapping. Gene Expr. 1995, 4, 281–299. [Google Scholar]
- Sher, T.; Yi, H.F.; McBride, O.W.; Gonzalez, F.J. cDNA cloning, chromosomal mapping, and functional characterization of the human peroxisome proliferator activated receptor. Biochemistry 1993, 32, 5598–5604. [Google Scholar] [CrossRef]
- Al-Qahtani, A.A.; Lyroni, K.; Aznaourova, M.; Tseliou, M.; Al-Anazi, M.R.; Al-Ahdal, M.N.; Alkahtani, S.; Sourvinos, G.; Tsatsanis, C. Middle east respiratory syndrome corona virus spike glycoprotein suppresses macrophage responses via DPP4-mediated induction of IRAK-M and PPARgamma. Oncotarget 2017, 8, 9053–9066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, S.S.G.; Soares, V.C.; Ferreira, A.C.; Sacramento, C.Q.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Teixeira, L.; Nunes da Silva, M.A.; Barreto, E.; Mattos, M.; et al. Lipid droplets fuel SARS-CoV-2 replication and production of inflammatory mediators. PLoS Pathog. 2020, 16, e1009127. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Bayona, W.; Kallen, C.B.; Harding, H.P.; Ravera, C.P.; McMahon, G.; Brown, M.; Lazar, M.A. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J. Biol. Chem. 1995, 270, 23975–23983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narala, V.R.; Adapala, R.K.; Suresh, M.V.; Brock, T.G.; Peters-Golden, M.; Reddy, R.C. Leukotriene B4 is a physiologically relevant endogenous peroxisome proliferator-activated receptor-alpha agonist. J. Biol. Chem. 2010, 285, 22067–22074. [Google Scholar] [CrossRef] [Green Version]
- O’Flaherty, J.T.; Rogers, L.C.; Paumi, C.M.; Hantgan, R.R.; Thomas, L.R.; Clay, C.E.; High, K.; Chen, Y.Q.; Willingham, M.C.; Smitherman, P.K.; et al. 5-Oxo-ETE analogs and the proliferation of cancer cells. Biochim. Biophys. Acta 2005, 1736, 228–236. [Google Scholar] [CrossRef]
- Altmann, R.; Hausmann, M.; Spottl, T.; Gruber, M.; Bull, A.W.; Menzel, K.; Vogl, D.; Herfarth, H.; Scholmerich, J.; Falk, W.; et al. 13-Oxo-ODE is an endogenous ligand for PPARgamma in human colonic epithelial cells. Biochem. Pharmacol. 2007, 74, 612–622. [Google Scholar] [CrossRef]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.; Thomazy, V.A.; Evans, R.M. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 1998, 93, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Yki-Jarvinen, H. Thiazolidinediones. N. Engl. J. Med. 2004, 351, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Marion-Letellier, R.; Savoye, G.; Ghosh, S. Fatty acids, eicosanoids and PPAR gamma. Eur. J. Pharmacol. 2016, 785, 44–49. [Google Scholar] [CrossRef]
- Suutari, M.; Laakso, S. Effect of growth temperature on the fatty acid composition of Mycobacterium phlei. Arch. Microbiol. 1993, 159, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Springer, B.; Kirschner, P.; Rost-Meyer, G.; Schroder, K.H.; Kroppenstedt, R.M.; Bottger, E.C. Mycobacterium interjectum, a new species isolated from a patient with chronic lymphadenitis. J. Clin. Microbiol. 1993, 31, 3083–3089. [Google Scholar] [CrossRef] [Green Version]
- Pacifico, C.; Fernandes, P.; de Carvalho, C. Mycobacterial response to organic solvents and possible implications on cross-resistance with antimicrobial agents. Front. Microbiol. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.; Chedore, P.; Kasatiya, S. Use of gas chromatographic fatty acid and mycolic acid cleavage product determination to differentiate among Mycobacterium genavense, Mycobacterium fortuitum, Mycobacterium simiae, and Mycobacterium tuberculosis. J. Clin. Microbiol. 1998, 36, 577–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.G.; Martinelli, R.; Besra, G.S.; Illarionov, P.A.; Szatmari, I.; Brazda, P.; Allen, M.A.; Xu, W.; Wang, X.; Nagy, L.; et al. Identification and characterization of a novel anti-inflammatory lipid isolated from Mycobacterium vaccae, a soil-derived bacterium with immunoregulatory and stress resilience properties. Psychopharmacology 2019, 236, 1653–1670. [Google Scholar] [CrossRef]
- Luo, Y.; Tanigawa, K.; Kawashima, A.; Ishido, Y.; Ishii, N.; Suzuki, K. The function of peroxisome proliferator-activated receptors PPAR-gamma and PPAR-delta in Mycobacterium leprae-induced foam cell formation in host macrophages. PLoS Negl. Trop. Dis. 2020, 14, e0008850. [Google Scholar] [CrossRef]
- Diaz Acosta, C.C.; Dias, A.A.; Rosa, T.; Batista-Silva, L.R.; Rosa, P.S.; Toledo-Pinto, T.G.; Costa, F.; Lara, F.A.; Rodrigues, L.S.; Mattos, K.A.; et al. PGL I expression in live bacteria allows activation of a CD206/PPARgamma cross-talk that may contribute to successful Mycobacterium leprae colonization of peripheral nerves. PLoS Pathog. 2018, 14, e1007151. [Google Scholar] [CrossRef]
- Arnett, E.; Weaver, A.M.; Woodyard, K.C.; Montoya, M.J.; Li, M.; Hoang, K.V.; Hayhurst, A.; Azad, A.K.; Schlesinger, L.S. PPARgamma is critical for Mycobacterium tuberculosis induction of Mcl-1 and limitation of human macrophage apoptosis. PLoS Pathog. 2018, 14, e1007100. [Google Scholar] [CrossRef]
- Rajaram, M.V.; Brooks, M.N.; Morris, J.D.; Torrelles, J.B.; Azad, A.K.; Schlesinger, L.S. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J. Immunol. 2010, 185, 929–942. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, M.; Luo, W.; Sui, Y.; Li, Z.; Hua, J. Dietary n-3 PUFA protects mice from con A induced liver injury by modulating regulatory T cells and PPAR-gamma expression. PLoS ONE 2015, 10, e0132741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minnikin, D.E.; Dobson, G.; Goodfellow, M.; Draper, P.; Magnusson, M. Quantitative comparison of the mycolic and fatty acid compositions of Mycobacterium leprae and Mycobacterium gordonae. J. Gen. Microbiol. 1985, 131, 2013–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubins, H.B.; Robins, S.J.; Collins, D.; Fye, C.L.; Anderson, J.W.; Elam, M.B.; Faas, F.H.; Linares, E.; Schaefer, E.J.; Schectman, G.; et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans affairs high-density lipoprotein cholesterol intervention trial study group. N. Engl. J. Med. 1999, 341, 410–418. [Google Scholar] [CrossRef]
- Group, A.S.; Ginsberg, H.N.; Elam, M.B.; Lovato, L.C.; Crouse, J.R., 3rd; Leiter, L.A.; Linz, P.; Friedewald, W.T.; Buse, J.B.; Gerstein, H.C.; et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N. Engl. J. Med. 2010, 362, 1563–1574. [Google Scholar]
- Hossain, M.A.; Tsujita, M.; Gonzalez, F.J.; Yokoyama, S. Effects of fibrate drugs on expression of ABCA1 and HDL biogenesis in hepatocytes. J. Cardiovasc. Pharmacol. 2008, 51, 258–266. [Google Scholar] [CrossRef]
- Manninen, V.; Tenkanen, L.; Koskinen, P.; Huttunen, J.K.; Manttari, M.; Heinonen, O.P.; Frick, M.H. Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study. Implications for treatment. Circulation 1992, 85, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Wang, J.; Hong, F.; Wang, S.; Jin, X.; Xue, T.; Jia, L.; Zhai, Y. Melatonin prevents obesity through modulation of gut microbiota in mice. J. Pineal Res. 2017, 62, e12399. [Google Scholar] [CrossRef]
- Wang, J.; Xu, P.; Xie, X.; Li, J.; Zhang, J.; Wang, J.; Hong, F.; Li, J.; Zhang, Y.; Song, Y.; et al. DBZ (Danshensu Bingpian Zhi), a novel natural compound derivative, attenuates atherosclerosis in apolipoprotein E-deficient mice. J. Am. Heart Assoc. 2017, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bays, H.E.; Schwartz, S.; Littlejohn, T., 3rd; Kerzner, B.; Krauss, R.M.; Karpf, D.B.; Choi, Y.J.; Wang, X.; Naim, S.; Roberts, B.K. MBX-8025, a novel peroxisome proliferator receptor-delta agonist: Lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J. Clin. Endocrinol. Metab. 2011, 96, 2889–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, W.R., Jr.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrins, C.L.; van der Velde, A.E.; van den Oever, K.; Levels, J.H.; Huet, S.; Oude Elferink, R.P.; Kuipers, F.; Groen, A.K. Peroxisome proliferator-activated receptor delta activation leads to increased transintestinal cholesterol efflux. J. Lipid. Res. 2009, 50, 2046–2054. [Google Scholar] [CrossRef] [Green Version]
- Peeters, A.; Baes, M. Role of PPARalpha in hepatic carbohydrate metabolism. PPAR Res. 2010, 2010, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finck, B.N.; Bernal-Mizrachi, C.; Han, D.H.; Coleman, T.; Sambandam, N.; LaRiviere, L.L.; Holloszy, J.O.; Semenkovich, C.F.; Kelly, D.P. A potential link between muscle peroxisome proliferator- activated receptor-alpha signaling and obesity-related diabetes. Cell Metab. 2005, 1, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Hatano, B.; Zhao, M.; Yen, C.C.; Kang, K.; Reilly, S.M.; Gangl, M.R.; Gorgun, C.; Balschi, J.A.; Ntambi, J.M.; et al. Role of peroxisome proliferator-activated receptor delta/beta in hepatic metabolic regulation. J. Biol. Chem. 2011, 286, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derosa, G.; Sahebkar, A.; Maffioli, P. The role of various peroxisome proliferator-activated receptors and their ligands in clinical practice. J. Cell Physiol. 2018, 233, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Bojic, L.A.; Telford, D.E.; Fullerton, M.D.; Ford, R.J.; Sutherland, B.G.; Edwards, J.Y.; Sawyez, C.G.; Gros, R.; Kemp, B.E.; Steinberg, G.R.; et al. PPARdelta activation attenuates hepatic steatosis in Ldlr-/- mice by enhanced fat oxidation, reduced lipogenesis, and improved insulin sensitivity. J. Lipid Res. 2014, 55, 1254–1266. [Google Scholar] [CrossRef] [Green Version]
- Corrales, P.; Vidal-Puig, A.; Medina-Gomez, G. PPARs and metabolic disorders associated with challenged adipose tissue plasticity. Int. J. Mol. Sci. 2018, 19, 2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J., Jr.; Liu, X.S.; et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef] [Green Version]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. PPARgamma: A nuclear regulator of metabolism, differentiation, and cell growth. J. Biol. Chem. 2001, 276, 37731–37734. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Xie, Y.; Morrison, R.F.; Bucher, N.L.; Farmer, S.R. PPARgamma induces the insulin-dependent glucose transporter GLUT4 in the absence of C/EBPalpha during the conversion of 3T3 fibroblasts into adipocytes. J. Clin. Investig. 1998, 101, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Gouzy, A.; Larrouy-Maumus, G.; Wu, T.D.; Peixoto, A.; Levillain, F.; Lugo-Villarino, G.; Guerquin-Kern, J.L.; de Carvalho, L.P.; Poquet, Y.; Neyrolles, O. Mycobacterium tuberculosis nitrogen assimilation and host colonization require aspartate. Nat. Chem. Biol. 2013, 9, 674–676. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, C.; Blumenthal, A.; Marrero, J.; Rhee, K.Y.; Schnappinger, D.; Ehrt, S. Triosephosphate isomerase is dispensable in vitro yet essential for Mycobacterium tuberculosis to establish infection. mBio 2014, 5, e00085. [Google Scholar] [CrossRef] [Green Version]
- Daniel, J.; Maamar, H.; Deb, C.; Sirakova, T.D.; Kolattukudy, P.E. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011, 7, e1002093. [Google Scholar] [CrossRef] [Green Version]
- Griffin, J.E.; Gawronski, J.D.; Dejesus, M.A.; Ioerger, T.R.; Akerley, B.J.; Sassetti, C.M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.K.; Sassetti, C.M. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. USA 2008, 105, 4376–4380. [Google Scholar] [CrossRef] [Green Version]
- Griffin, J.E.; Pandey, A.K.; Gilmore, S.A.; Mizrahi, V.; McKinney, J.D.; Bertozzi, C.R.; Sassetti, C.M. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem. Biol. 2012, 19, 218–227. [Google Scholar] [CrossRef] [Green Version]
- VanderVen, B.C.; Fahey, R.J.; Lee, W.; Liu, Y.; Abramovitch, R.B.; Memmott, C.; Crowe, A.M.; Eltis, L.D.; Perola, E.; Deininger, D.D.; et al. Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog. 2015, 11, e1004679. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Wainwright, H.C.; Locketz, M.; Bekker, L.G.; Walther, G.B.; Dittrich, C.; Visser, A.; Wang, W.; Hsu, F.F.; Wiehart, U.; et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2010, 2, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.A.; Berredo-Pinho, M.; Rosa, T.L.; Pujari, V.; Lemes, R.M.; Lery, L.M.; Silva, C.A.; Guimaraes, A.C.; Atella, G.C.; Wheat, W.H.; et al. The essential role of cholesterol metabolism in the intracellular survival of Mycobacterium leprae is not coupled to central carbon metabolism and energy production. J. Bacteriol. 2015, 197, 3698–3707. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, K.R.; Das Gupta, N.N.; De, M.L. Electron microscopic observations on the morphology of Mycobacterium leprae. Exp. Cell Res. 1959, 18, 521–527. [Google Scholar] [CrossRef]
- Mattos, K.A.; Lara, F.A.; Oliveira, V.G.; Rodrigues, L.S.; D’Avila, H.; Melo, R.C.; Manso, P.P.; Sarno, E.N.; Bozza, P.T.; Pessolani, M.C. Modulation of lipid droplets by Mycobacterium leprae in Schwann cells: A putative mechanism for host lipid acquisition and bacterial survival in phagosomes. Cell Microbiol. 2011, 13, 259–273. [Google Scholar] [CrossRef]
- Mattos, K.A.; Oliveira, V.C.; Berredo-Pinho, M.; Amaral, J.J.; Antunes, L.C.; Melo, R.C.; Acosta, C.C.; Moura, D.F.; Olmo, R.; Han, J.; et al. Mycobacterium leprae intracellular survival relies on cholesterol accumulation in infected macrophages: A potential target for new drugs for leprosy treatment. Cell Microbiol. 2014, 16, 797–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanigawa, K.; Hayashi, Y.; Hama, K.; Yamashita, A.; Yokoyama, K.; Luo, Y.; Kawashima, A.; Maeda, Y.; Nakamura, Y.; Harada, A.; et al. Mycobacterium leprae promotes triacylglycerol de novo synthesis through induction of GPAT3 expression in human premonocytic THP-1 cells. PLoS ONE 2021, 16, e0249184. [Google Scholar] [CrossRef]
- Medeiros, R.C.; Girardi, K.D.; Cardoso, F.K.; Mietto, B.S.; Pinto, T.G.; Gomez, L.S.; Rodrigues, L.S.; Gandini, M.; Amaral, J.J.; Antunes, S.L.; et al. Subversion of schwann cell glucose metabolism by Mycobacterium leprae. J. Biol. Chem. 2016, 291, 21375–21387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanigawa, K.; Degang, Y.; Kawashima, A.; Akama, T.; Yoshihara, A.; Ishido, Y.; Makino, M.; Ishii, N.; Suzuki, K. Essential role of hormone-sensitive lipase (HSL) in the maintenance of lipid storage in Mycobacterium leprae-infected macrophages. Microb. Pathog. 2012, 52, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, K.; Suzuki, K.; Nakamura, K.; Akama, T.; Kawashima, A.; Wu, H.; Hayashi, M.; Takahashi, S.; Ikuyama, S.; Ito, T.; et al. Expression of adipose differentiation-related protein (ADRP) and perilipin in macrophages infected with Mycobacterium leprae. FEMS Microbiol. Lett. 2008, 289, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Karasawa, K.; Tanigawa, K.; Harada, A.; Yamashita, A. Transcriptional Regulation of Acyl-CoA:Glycerol-sn-3-Phosphate Acyltransferases. Int. J. Mol. Sci. 2019, 20, 964. [Google Scholar] [CrossRef] [Green Version]
- Degang, Y.; Akama, T.; Hara, T.; Tanigawa, K.; Ishido, Y.; Gidoh, M.; Makino, M.; Ishii, N.; Suzuki, K. Clofazimine modulates the expression of lipid metabolism proteins in Mycobacterium leprae-infected macrophages. PLoS Negl. Trop. Dis. 2012, 6, e1936. [Google Scholar] [CrossRef] [Green Version]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffe, M.; Emile, J.F.; Marchou, B.; Cardona, P.J.; et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef]
- Singh, V.; Jamwal, S.; Jain, R.; Verma, P.; Gokhale, R.; Rao, K.V. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 2012, 12, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Almeida, P.E.; Silva, A.R.; Maya-Monteiro, C.M.; Torocsik, D.; D’Avila, H.; Dezso, B.; Magalhaes, K.G.; Castro-Faria-Neto, H.C.; Nagy, L.; Bozza, P.T. Mycobacterium bovis bacillus Calmette-Guerin infection induces TLR2-dependent peroxisome proliferator-activated receptor gamma expression and activation: Functions in inflammation, lipid metabolism, and pathogenesis. J. Immunol. 2009, 183, 1337–1345. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, S.; Dkhar, H.K.; Chandra, V.; Dave, S.; Nanduri, R.; Janmeja, A.K.; Agrewala, J.N.; Gupta, P. Mycobacterium tuberculosis modulates macrophage lipid-sensing nuclear receptors PPARgamma and TR4 for survival. J. Immunol. 2012, 188, 5593–5603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Avila, H.; Melo, R.C.; Parreira, G.G.; Werneck-Barroso, E.; Castro-Faria-Neto, H.C.; Bozza, P.T. Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: Intracellular domains for eicosanoid synthesis in vivo. J. Immunol. 2006, 176, 3087–3097. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Li, J.L.; Li, D.; Tobin, J.F.; Gimeno, R.E. Molecular identification of microsomal acyl-CoA:glycerol-3-phosphate acyltransferase, a key enzyme in de novo triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19695–19700. [Google Scholar] [CrossRef] [Green Version]
- Gorga, A.; Rindone, G.M.; Regueira, M.; Pellizzari, E.H.; Camberos, M.C.; Cigorraga, S.B.; Riera, M.F.; Galardo, M.N.; Meroni, S.B. PPARgamma activation regulates lipid droplet formation and lactate production in rat Sertoli cells. Cell Tissue Res. 2017, 369, 611–624. [Google Scholar] [CrossRef]
- Lee, M.J.; Jash, S.; Jones, J.E.C.; Puri, V.; Fried, S.K. Rosiglitazone remodels the lipid droplet and britens human visceral and subcutaneous adipocytes ex vivo. J. Lipid Res. 2019, 60, 856–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, C.E.; Pyle, C.J.; Glowinski, R.; Rajaram, M.V.; Schlesinger, L.S. CD36-Mediated Uptake of Surfactant Lipids by Human Macrophages Promotes Intracellular Growth of Mycobacterium tuberculosis. J. Immunol. 2016, 197, 4727–4735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, P.E.; Roque, N.R.; Magalhaes, K.G.; Mattos, K.A.; Teixeira, L.; Maya-Monteiro, C.; Almeida, C.J.; Castro-Faria-Neto, H.C.; Ryffel, B.; Quesniaux, V.F.; et al. Differential TLR2 downstream signaling regulates lipid metabolism and cytokine production triggered by Mycobacterium bovis BCG infection. Biochim. Biophys. Acta 2014, 1841, 97–107. [Google Scholar] [CrossRef]
- Dasgupta, S.; Rai, R.C. PPAR-gamma and Akt regulate GLUT1 and GLUT3 surface localization during Mycobacterium tuberculosis infection. Mol. Cell Biochem. 2018, 440, 127–138. [Google Scholar] [CrossRef]
- Brzostek, A.; Pawelczyk, J.; Rumijowska-Galewicz, A.; Dziadek, B.; Dziadek, J. Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J. Bacteriol. 2009, 191, 6584–6591. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Lee, H.M.; Kim, J.K.; Yang, C.S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-alpha Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Kim, J.K.; Hanh, B.T.B.; Kim, S.Y.; Kim, H.J.; Kim, Y.J.; Jeon, S.M.; Park, C.R.; Oh, G.T.; Park, J.W.; et al. The peroxisome proliferator-activated receptor alpha- agonist gemfibrozil promotes defense against Mycobacterium abscessus infections. Cells 2020, 9, 648. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef]
- Guirado, E.; Rajaram, M.V.; Chawla, A.; Daigle, J.; La Perle, K.M.; Arnett, E.; Turner, J.; Schlesinger, L.S. Deletion of PPARgamma in lung macrophages provides an immunoprotective response against M. tuberculosis infection in mice. Tuberculosis 2018, 111, 170–177. [Google Scholar] [CrossRef]
- Pan, Q.; Wang, Q.; Sun, X.; Xia, X.; Wu, S.; Luo, F.; Zhang, X.L. Aptamer against mannose-capped lipoarabinomannan inhibits virulent Mycobacterium tuberculosis infection in mice and rhesus monkeys. Mol. Ther. 2014, 22, 940–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; Yan, J.; Liu, Q.; Yuan, C.; Zhang, X.L. A single-stranded DNA aptamer against mannose-capped lipoarabinomannan enhances anti-tuberculosis activity of macrophages through downregulation of lipid-sensing nuclear receptor peroxisome proliferator-activated receptor gamma expression. Microbiol. Immunol. 2017, 61, 92–102. [Google Scholar] [CrossRef]
- Jiao, M.; Ren, F.; Zhou, L.; Zhang, X.; Zhang, L.; Wen, T.; Wei, L.; Wang, X.; Shi, H.; Bai, L.; et al. Peroxisome proliferator-activated receptor alpha activation attenuates the inflammatory response to protect the liver from acute failure by promoting the autophagy pathway. Cell Death Dis. 2014, 5, e1397. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, X.; Qu, N.; Zhang, B.; Xia, C. Chondroprotection of PPARalpha activation by WY14643 via autophagy involving Akt and ERK in LPS-treated mouse chondrocytes and osteoarthritis model. J. Cell Mol. Med. 2019, 23, 2782–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.S.; Jin, Y.B.; Kim, Y.S.; Kim, S.; Kim, J.K.; Lee, H.M.; Suh, H.W.; Choe, J.H.; Kim, Y.J.; Koo, B.S.; et al. SIRT3 promotes antimycobacterial defenses by coordinating mitochondrial and autophagic functions. Autophagy 2019, 15, 1356–1375. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Jana, M.; Modi, K.; Gonzalez, F.J.; Sims, K.B.; Berry-Kravis, E.; Pahan, K. Activation of peroxisome proliferator-activated receptor alpha induces lysosomal biogenesis in brain cells: Implications for lysosomal storage disorders. J. Biol. Chem 2015, 290, 10309–10324. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, O.A.; Martina, J.A.; Puertollano, R. Emerging roles for TFEB in the immune response and inflammation. Autophagy 2018, 14, 181–189. [Google Scholar] [CrossRef]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Halder, P.; Kumar, R.; Jana, K.; Chakraborty, S.; Ghosh, Z.; Kundu, M.; Basu, J. Gene expression profiling of Mycobacterium tuberculosis lipoarabinomannan-treated macrophages: A role of the Bcl-2 family member A1 in inhibition of apoptosis in mycobacteria-infected macrophages. IUBMB Life 2015, 67, 726–736. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Kim, J.J.; Lee, H.M.; Jin, H.S.; Lee, S.H.; Park, J.H.; Kim, S.J.; Kim, J.M.; Han, Y.M.; Lee, M.S.; et al. The AMPK-PPARGC1A pathway is required for antimicrobial host defense through activation of autophagy. Autophagy 2014, 10, 785–802. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Yang, C.S.; Lee, H.M.; Kim, J.K.; Kim, Y.S.; Kim, Y.R.; Kim, J.S.; Kim, T.S.; Yuk, J.M.; Dufour, C.R.; et al. ESRRA (estrogen-related receptor alpha) is a key coordinator of transcriptional and post-translational activation of autophagy to promote innate host defense. Autophagy 2018, 14, 152–168. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Gutierrez, N.M.; Marzuki, M.B.; Lu, X.; Foreman, T.W.; Paleja, B.; Lee, B.; Balachander, A.; Chen, J.; Tsenova, L.; et al. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci. Immunol. 2017, 2, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanigawa, K.; Luo, Y.; Kawashima, A.; Kiriya, M.; Nakamura, Y.; Karasawa, K.; Suzuki, K. Essential Roles of PPARs in Lipid Metabolism during Mycobacterial Infection. Int. J. Mol. Sci. 2021, 22, 7597. https://doi.org/10.3390/ijms22147597
Tanigawa K, Luo Y, Kawashima A, Kiriya M, Nakamura Y, Karasawa K, Suzuki K. Essential Roles of PPARs in Lipid Metabolism during Mycobacterial Infection. International Journal of Molecular Sciences. 2021; 22(14):7597. https://doi.org/10.3390/ijms22147597
Chicago/Turabian StyleTanigawa, Kazunari, Yuqian Luo, Akira Kawashima, Mitsuo Kiriya, Yasuhiro Nakamura, Ken Karasawa, and Koichi Suzuki. 2021. "Essential Roles of PPARs in Lipid Metabolism during Mycobacterial Infection" International Journal of Molecular Sciences 22, no. 14: 7597. https://doi.org/10.3390/ijms22147597
APA StyleTanigawa, K., Luo, Y., Kawashima, A., Kiriya, M., Nakamura, Y., Karasawa, K., & Suzuki, K. (2021). Essential Roles of PPARs in Lipid Metabolism during Mycobacterial Infection. International Journal of Molecular Sciences, 22(14), 7597. https://doi.org/10.3390/ijms22147597