
Evidence-Based Second-Line Treatment in RAS Wild-Type/Mutated Metastatic Colorectal Cancer in the Precision Medicine Era

 ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Antiangiogenic Drugs in mCRC Second-Line Therapy
2.1. Bevacizumab
2.2. Aflibercept
2.3. Ramucirumab
3. Anti-EGFR Drugs in mCRC Second-Line Treatment
3.1. Cetuximab
3.2. Panitumumab
4. Braf Inhibitors in mCRC
5. Immune-Checkpoint Inhibitors in mCRC
6. Sequential Second-Line Strategies in Metastatic CRCs
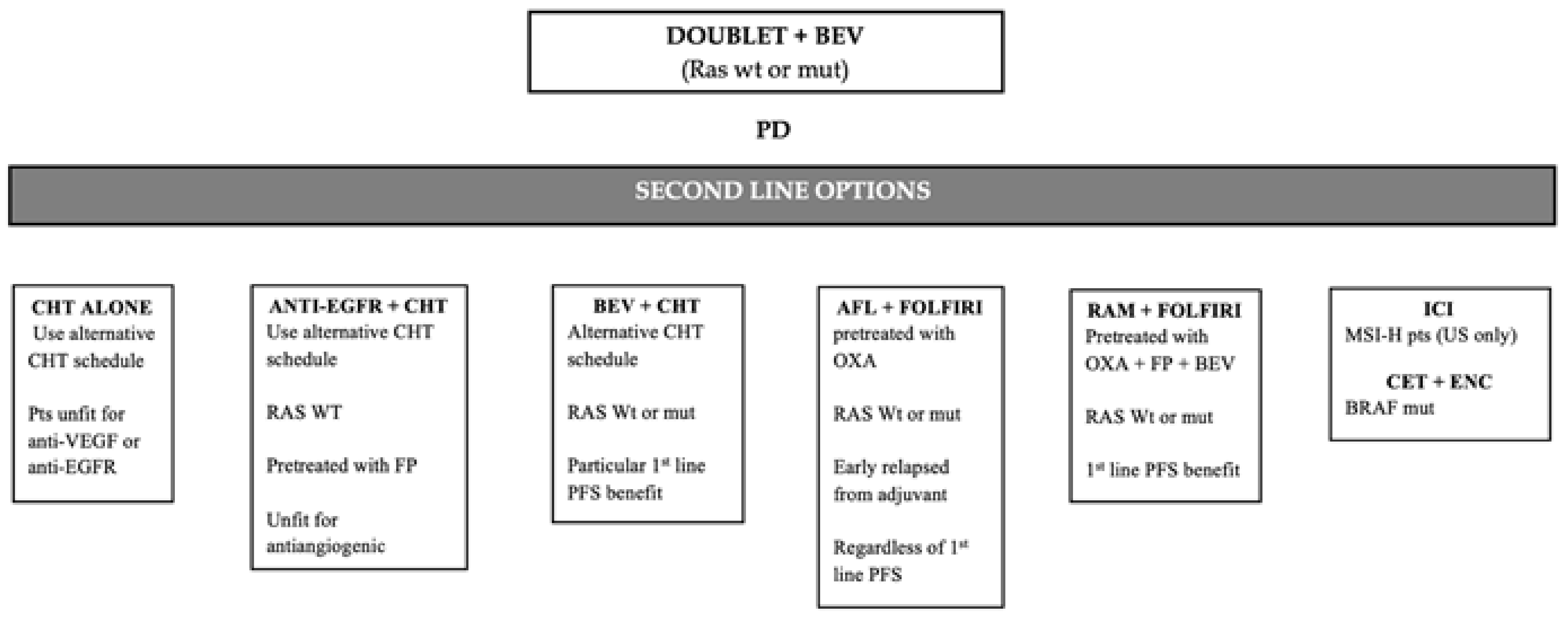
6.1. Second-Line after Progression to Beavacizumab-Based First-Line Treatment
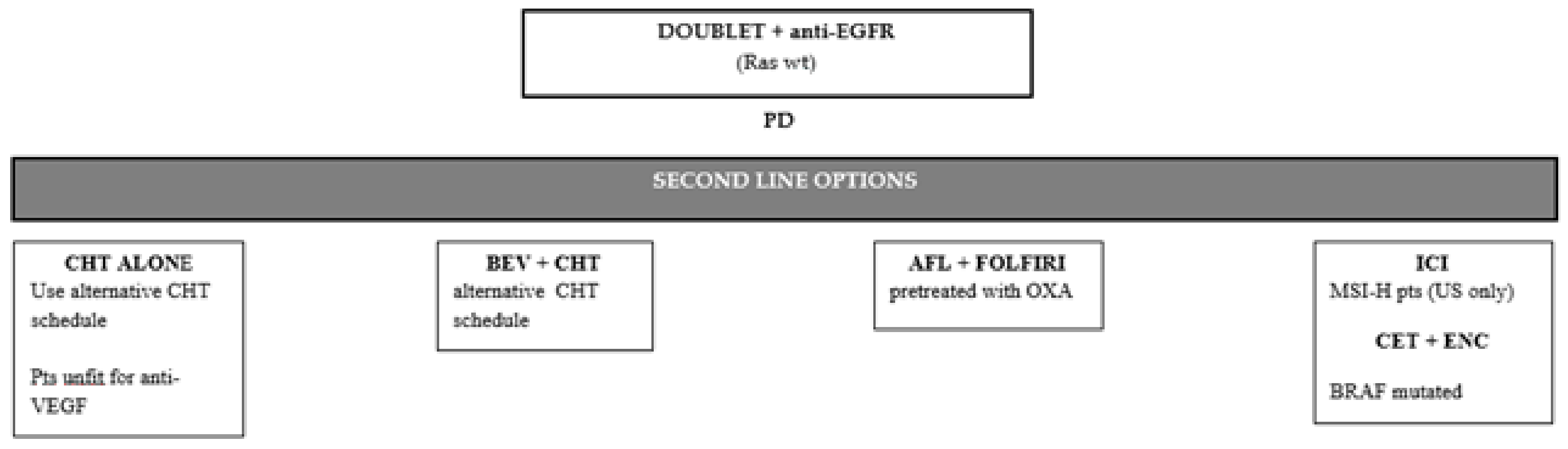
6.2. Second-Line Therapy in Ras Wild-Type Tumors Treated with Anti-Egfr First-Line Therapy
6.3. Second-Line Therapy after First-Line Triplet Chemotherapy Plus Bevacizumab
7. Current Strategies and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer Treatment and Survivorship Statistics. CA Cancer J. Clin. 2016, 66, 271–2892. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [Green Version]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Piawah, S.; Venook, A.P. Targeted therapy for colorectal cancer metastases: A review of current methods of molecularly targeted therapy and the use of tumor biomarkers in the treatment of metastatic colorectal cancer. Cancer 2019, 125, 4139–4147. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaniboni, A.; Formica, V. The Best. First. Anti-EGFR before anti-VEGF, in the first-line treatment of RAS wild-type metastatic colorectal cancer: From bench to bedside. Cancer Chemother. Pharm. 2016, 78, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Köhne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Köhne, C.H.; Láng, I.; Folprecht, G.; Nowacki, M.P.; Cascinu, S.; Shchepotin, I.; Maurel, J.; Cunningham, D.; Tejpar, S.; et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: Updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J. Clin. Oncol. 2011, 29, 2011–2019. [Google Scholar] [CrossRef] [Green Version]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. K-Ras and its inhibitors towards personalized cancer treatment: Pharmacological and structural perspectives. Eur. J. Med. Chem. 2016, 125, 299–314. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausová, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecanimproves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.E.; Portnoy, D.C.; Van Cutsem, E.; Grothey, A.; et al. Ramucirumab versus placebo in combination with second line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): A randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015, 16. [Google Scholar] [CrossRef]
- Grothey, A.; Sargent, D.; Goldberg, R.M.; Schmoll, H.J. Survival of patients with advanced colorectal cancer improves with the availability of fluorouracil-leucovorin, irinotecan, and oxaliplatin in the course of treatment. J. Clin. Oncol. 2004, 22, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- NCCN guidelines-colonrectalcancer/version 2.2021.
- Wu, C.C.; Wang, J.H.; Lin, P.C.; Liang, C.A.; Huang, C.Y.; Lien, H.C.; Chen, C.Y.; Chou, K.J.; Su, Y.C. Tumor sidedness and efficacy of first-line therapy in patients with RAS/BRAF wild-type metastatic colorectal cancer: A network meta-analysis. Crit. Rev. Oncol. Hematol. 2020, 145, 102823. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Fucà, G.; Rossini, D.; Schmoll, H.J.; Bendell, J.C.; Morano, F.; Antoniotti, C.; Corallo, S.; Borelli, B.; Raimondi, A.; et al. FOLFOXIRI-Bevacizumab or FOLFOX-Panitumumab in Patients with Left-Sided RAS/BRAF Wild-Type Metastatic Colorectal Cancer: A Propensity Score-Based Analysis. Oncologist 2020. [Google Scholar] [CrossRef]
- Modest, D.P.; Stintzing, S.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Kahl, C.; Seipelt, G.; et al. Exploring the effect of primary tumor sidedness on therapeutic efficacy across treatment lines in patients with metastatic colorectal cancer: Analysis of FIRE-3 (AIOKRK0306). Oncotarget 2017, 8, 105749–105760. [Google Scholar] [CrossRef]
- Huang, D.; Sun, W.; Zhou, Y.; Li, P.; Chen, F.; Chen, H.; Xia, D.; Xu, E.; Lai, M.; Wu, Y.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef]
- Cohen, R.; Cervera, P.; Svrcek, M.; Pellat, A.; Dreyer, C.; de Gramont, A.; André, T. BRAF-Mutated Colorectal Cancer: What Is the Optimal Strategy for Treatment? Curr. Treat. Opt. Oncol. 2017, 18, 9. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Salazar, R.; Tabernero, J. Molecular Subtypes and the Evolution of Treatment Decisions in Metastatic Colorectal Cancer. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 231–238. [Google Scholar] [CrossRef]
- Lee, M.K.C.; Loree, J.M. Current and emerging biomarkers in metastatic colorectal cancer. Curr. Oncol. 2019, 26 (Suppl. S1), S7–S15. [Google Scholar] [CrossRef] [Green Version]
- Stintzing, S.; Miller-Phillips, L.; Modest, D.P.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Kahl, C.; et al. Impact of BRAF and RAS mutations on first-line efficacy of FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab: Analysis of the FIRE-3 (AIO KRK-0306) study. Eur. J. Cancer 2017, 79, 50–60. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Martini, G.; Troiani, T.; Cardone, C.; Vitiello, P.; Sforza, V.; Ciardiello, D.; Napolitano, S.; Della Corte, C.M.; Morgillo, F.; Raucci, A.; et al. Present and future of metastatic colorectal cancer treatment: A review of new candidate targets. World J. Gastroenterol. 2017, 23, 4675–4688. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, R.; Caporale, M.; Pietrantonio, F.; De Braud, F.; Negri, F.V.; Giuliani, F.; Pusceddu, V.; Demurtas, L.; Restivo, A.; Fontanella, C.; et al. Second-line angiogenesis inhibition in metastatic colorectal cancer patients: Straightforward or overcrowded? Crit. Rev. Oncol. Hematol. 2016, 100, 99–106. [Google Scholar] [CrossRef]
- Sandhu, J.; Lavingia, V.; Fakih, M. Systemic treatment for metastatic colorectal cancer in the era of precision medicine. J. Surg. Oncol. 2019, 119, 564–582. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Stintzing, S.; Wirapati, P.; Lenz, H.J.; Neureiter, D.; Fischer von Weikersthal, L.; Decker, T.; Kiani, A.; Kaiser, F.; Al-Batran, S.; Heintges, T.; et al. Consensus molecular subgroups (CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial. Ann. Oncol. 2019, 30, 1796–1803. [Google Scholar] [CrossRef] [Green Version]
- Lenz, H.J.; Ou, F.S.; Venook, A.P.; Hochster, H.S.; Niedzwiecki, D.; Goldberg, R.M.; Mayer, R.J.; Bertagnolli, M.M.; Blanke, C.D.; Zemla, T.; et al. Impact of Consensus Molecular Subtype on Survival in Patients with Metastatic Colorectal Cancer: Results From CALGB/SWOG 80405 (Alliance). J. Clin. Oncol. 2019, 37, 1876–1885. [Google Scholar] [CrossRef]
- Mody, K.; Baldeo, C.; Bekaii-Saab, T. Antiangiogenic Therapy in Colorectal Cancer. Cancer J. 2018, 24, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Saltz, L.B.; Clarke, S.; Díaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Lichinitser, M.; Yang, T.S.; Rivera, F.; et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: A randomized phase III study. J. Clin. Oncol. 2008, 26, 2013–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, D.; Lang, I.; Marcuello, E.; Lorusso, V.; Ocvirk, J.; Shin, D.B.; Jonker, D.; Osborne, S.; Andre, N.; Waterkamp, D.; et al. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously untreated metastatic colorectal cancer (AVEX): An open-label, randomised phase 3 trial. Lancet Oncol. 2013, 14, 1077–1085. [Google Scholar] [CrossRef]
- Loupakis, F.; Cremolini, C.; Masi, G.; Lonardi, S.; Zagonel, V.; Salvatore, L.; Cortesi, E.; Tomasello, G.; Ronzoni, M.; Spadi, R.; et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N. Engl. J. Med. 2014, 371, 1609–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giantonio, B.J.; Catalano, P.J.; Meropol, N.J.; O’Dwyer, P.J.; Mitchell, E.P.; Alberts, S.R.; Schwartz, M.A.; Benson III, A.B. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: Results from the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol. 2007, 25, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Sugrue, M.M.; Purdie, D.M.; Dong, W.; Sargent, D.; Hedrick, E.; Kozloff, M. Bevacizumab Beyond First Progression Is Associated withProlonged Overall Survival in Metastatic Colorectal Cancer: Results From a Large Observational Cohort Study (BRiTE). J. Clin. Oncol. 2008, 26, 5326–5334. [Google Scholar] [CrossRef]
- Grothey, A.; Flick, E.D.; Cohn, A.L.; Bekaii-Saab, T.S.; Bendell, J.C.; Kozloff, M.; Roach, N.; Mun, Y.; Fish, S.; Hurwitz, H.I. Bevacizumab exposure beyond first disease progression in patients with metastatic colorectal cancer: Analyses of the ARIES observational cohort study. Pharmacoepidemiol. Drug Saf. 2014, 23, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Bennouna, J.; Sastre, J.; Arnold, D.; Österlund, P.; Greil, R.; Van Cutsem, E.; von Moos, R.; Viéitez, J.M.; Bouché, O.; Borg, C.; et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): A randomized phase 3 trial. Lancet Oncol. 2013, 14. [Google Scholar] [CrossRef]
- Masi, G.; Salvatore, L.; Boni, L.; Loupakis, F.; Cremolini, C.; Fornaro, L.; Schirripa, M.; Cupini, S.; Barbara, C.; Safina, V.; et al. Continuation or reintroduction of bevacizumab beyond progression to first-line therapy in metastatic colorectal cancer: Final results of the randomized BEBYP trial. Ann. Oncol. 2015, 26, 724–730. [Google Scholar] [CrossRef]
- Ciombor, K.K.; Berlin, J. Aflibercept—A decoy VEGF receptor. Curr. Oncol. Rep. 2014, 16, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, G.; Febbraro, A.; Venditti, M.; Campidoglio, S.; Olivieri, N.; Raieta, K.; Parcesepe, P.; Imbriani, G.C.; Remo, A.; Pancione, M. Targeting angiogenesis and tumor microenvironment inmetastatic colorectal cancer: Role of aflibercept. Gastroenterol. Res. Pract. 2014, 2014, 526178. [Google Scholar] [CrossRef] [Green Version]
- Ruff, P.; Ferry, D.R.; Lakomỳ, R.; Prausová, J.; Van Hazel, G.A.; Hoff, P.M.; Cunningham, D.; Arnold, D.; Schmoll, H.J.; Moiseyenko, V.M.; et al. Time course of safety and efficacy of aflibercept in combination with FOLFIRI in patients with metastatic colorectal cancer who progressed on previous oxaliplatin-based therapy. Eur. J. Cancer 2015, 51, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Ruff, P.; Van Cutsem, E.; Lakomy, R.; Prausova, J.; van Hazel, G.A.; Moiseyenko, V.M.; Soussan-Lazard, K.; Dochy, E.; Magherini, E.; Macarulla, T.; et al. Observed benefit and safety of aflibercept in elderly patients with metastatic colorectal cancer: An age-based analysis from the randomized placebo-controlled phase III VELOUR trial. J. Geriatr. Oncol. 2018, 9, 32–39. [Google Scholar] [CrossRef]
- Tabernero, J.; Van Cutsem, E.; Lakomy, R.; Prausova, J.; Ruff, P.; Guy, A.; van Hazel, G.A.; Moiseyenko, V.M.; Ferry, D.R.; McKendrick, J.J.; et al. Aflibercept versus placebo in combination with fluorouracil, leucovorin and irinotecan in the treatment of previously treated metastatic colorectal cancer: Prespecified subgroup analyses from the VELOUR trial. Eur. J. Cancer. 2014, 50, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Hoff, P.M.; Van Hazel, G.; Cunningham, D. The consistency of effect of zv-aflibercept (Z) in the bevacizumab (B) pre-treated subgroup of patients (pts) in the velour trial stratified by first-line progression ≥ 9 months (mos) versus < 9 mos. J. Clin. Oncol. 2014, 32, 3639. [Google Scholar]
- Van Cutsem, E.; Joulain, F.; Hoff, P.M.; Mitchell, E.; Ruff, P.; Lakomý, R.; Prausová, J.; Moiseyenko, V.M.; van Hazel, G.; Cunningham, D.; et al. Aflibercept Plus FOLFIRI vs. Placebo Plus FOLFIRI in Second-Line Metastatic Colorectal Cancer: A Post Hoc Analysis of Survival from the Phase III VELOUR Study Subsequent to Exclusion of Patients who had Recurrence During or within 6 Months of Completing Adjuvant Oxaliplatin-Based Therapy. Target. Oncol. 2016, 11, 383–400. [Google Scholar] [CrossRef]
- Tabernero, J.; Macarulla, T.; Humblet, Y.; ten Tije, A.J.; Kroening, H.; Gravalos, C.; Le-Guennec, S.; Dochy, E.; Van Cutsem, E. Aflibercept/FOLFIRI vs. Placebo/FOLFIRI in Metastatic Colorectal Cancer: A Post-Hoc Analysis of Median Overall Survival in VELOUR From the Retrospectively Assessed Time of Starting First-Line Treatment. Ann. Oncol. 2014, 25 (Suppl. S2), ii104. [Google Scholar]
- Chau, I.; Joulain, F.; Iqbal, S.U.; Bridgewater, J. A VELOUR post hoc subset analysis: Prognostic groups and treatment outcomes in patients with metastatic colorectal cancer treated with aflibercept and FOLFIRI. BMC Cancer 2014, 14, 605. [Google Scholar] [CrossRef] [Green Version]
- Wirapati, P.; Pomella, V.; Vandenbosch, B.; Kerr, P.; Maiello, E.; Grahame, M.J.; Curca, R.D.; Karthasus, M.; Bidgewater, J.A.; Mihailov, A.C.; et al. VELOUR Trial biomarkers update: Impact of RAS, BRAF and sideness on aflibercept activity. J. Clin. Oncol. 2017, 28, iii151–iii152. [Google Scholar]
- Riechelmann, R.P.; Srimuninnimit, V.; Bordonaro, R.; Kavan, P.; Di Bartolomeo, M.; Maiello, E.; Cicin, I.; García-Alfonso, P.; Chau, I.; Fedyanin, M.Y.; et al. Aflibercept Plus FOLFIRI for Second-line Treatment of Metastatic Colorectal Cancer: Observations from the Global Aflibercept Safety and Health-Related Quality-of-Life Program (ASQoP). Clin. Colorectal. Cancer 2019, 18, 183–191.e3. [Google Scholar] [CrossRef] [Green Version]
- Debeuckelaere, C.; Murgioni, S.; Lonardi, S.; Girardi, N.; Alberti, G.; Fano, C.; Gallimberti, S.; Magro, C.; Ahcene-Djaballah, S.; Daniel, F.; et al. Ramucirumab: The long and winding road toward being an option for mCRC treatment. Expert Opin. Biol. Ther. 2019, 19, 399–409. [Google Scholar] [CrossRef]
- Verdaguer, H.; Tabernero, J.; Macarulla, T. Ramucirumab in metastatic colorectal cancer: Evidence to date and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Obermannová, R.; Van Cutsem, E.; Yoshino, T.; Bodoky, G.; Prausová, J.; Garcia-Carbonero, R.; Ciuleanu, T.; Garcia Alfonso, P.; Portnoy, D.; Cohn, A.; et al. Subgroup analysis in RAISE: A randomized, double-blind phase 3 study of irinotecan, folinic acid, and 5-fluorouracil (FOLFIRI) plus ramucirumab or placebo in patients with metastatic colorectal carcinoma progression. Ann. Oncol. 2016, 27, 2082–2090. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Portnoy, D.C.; Obermannová, R.; Bodoky, G.; Prausová, J.; Garcia-Carbonero, R.; Ciuleanu, T.; García-Alfonso, P.; Cohn, A.L.; Van Cutsem, E.; et al. Biomarker analysis beyond angiogenesis: RAS/RAF mutation status, tumour sidedness, and second-line ramucirumab efficacy in patients with metastatic colorectal carcinoma from RAISE-a global phase III study. Ann. Oncol. 2019, 30, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grothey, A.; Yoshino, T.; Bodoky, G.; Ciuleanu, T.; Garcia-Carbonero, R.; García-Alfonso, P.; Van Cutsem, E.; Muro, K.; Mytelka, D.S.; Li, L.; et al. Association of baseline absolute neutrophil counts and survival in patients with metastatic colorectal cancer treated with second-line antiangiogenic therapies: Exploratory analyses of the RAISE trial and validation in an electronic medical record data set. ESMO Open 2018, 3, e000347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, T.; Obermannová, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.; Portnoy, D.C.; Kim, T.W.; Hsu, Y.; Ferry, D.; Nasroulah, F.; et al. Baseline carcinoembryonic antigen as a predictive factor of ramucirumab efficacy in RAISE, a second-line metastatic colorectal carcinoma phase III trial. Eur. J. Cancer 2017, 78, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornasier, G.; Francescon, S.; Baldo, P. An Update of Efficacy and Safety of Cetuximab in Metastatic Colorectal Cancer: A Narrative Review. Adv. Ther. 2018, 35, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Lerchenmüller, C.; Kahl, C.; Seipelt, G.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab Monotherapy and Cetuximab plus rinotecan in Irinotecan-Refractory Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Sobrero, A.F.; Maurel, J.; Fehrenbacher, L.; Scheithauer, W.; Abubakr, Y.A.; Lutz, M.P.; Vega-Villegas, M.E.; Eng, C.; Steinhauer, E.U.; Prausova, J.; et al. EPIC: Phase III Trial of Cetuximab Plus Irinotecan after Fluoropyrimidine and Oxaliplatin Failure in Patients with Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 26, 2311–2319. [Google Scholar] [CrossRef] [Green Version]
- Ciardiello, F.; Normanno, N.; Martinelli, E.; Troiani, T.; Pisconti, S.; Cardone, C.; Nappi, A.; Bordonaro, A.R.; Rachiglio, M.; Lambiase, M.; et al. Cetuximab continuation after first progression in metastatic colorectal cancer (CAPRI-GOIM): A randomized phase II trial of FOLFOX plus cetuximab versus FOLFOX. Ann. Oncol. 2016, 27, 1055–1061. [Google Scholar] [CrossRef] [Green Version]
- Battaglin, F.; Dadduzio, V.; Bergamo, F.; Manai, C.; Schirripa, M.; Lonardi, S.; Zagonel, V.; Loupakis, F. Anti-EGFR monoclonal antibody panitumumab for the treatment of patients with metastatic colorectal cancer: An overview of current practice and future perspectives. Expert Opin. Biol. Ther. 2017, 17, 1297–1308. [Google Scholar] [CrossRef]
- Peeters, M.; Price, T.J.; Cervantes, A.; Sobrero, A.F.; Ducreux, M.; Hotko, Y.; André, T.; Chan, E.; Lordick, F.; Punt, C.J.; et al. Randomized Phase III Study of Panitumumab with Fluorouracil, Leucovorin, and Irinotecan (FOLFIRI) Compared with FOLFIRI Alone As Second-Line Treatment in Patients with Metastatic Colorectal Cancer. J. Clin. Oncol. 2010, 28, 4706–4713. [Google Scholar] [CrossRef]
- Seymour, M.T.; Brown, S.R.; Middleton, G.; Maughan, T.; Richman, S.; Gwyther, S.; Lowe, C.; Seligmann, J.F.; Wadsley, J.; Maisey, N.; et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): A prospectively stratified randomized trial. Lancet Oncol. 2013, 14, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Barras, D.; Missiaglia, E.; Wirapati, P.; Sieber, O.M.; Jorissen, R.N.; Love, C.; Molloy, P.L.; Jones, I.T.; McLaughlin, S.; Gibbs, P.; et al. BRAF V600E Mutant Colorectal Cancer Subtypes Based on Gene Expression. Clin Cancer Res. 2017, 23, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, I.; Hirota, T.; Shinozaki, E. BRAF Mutation in Colorectal Cancers: From Prognostic Marker to Targetable Mutation. Cancers 2020, 12, 3236. [Google Scholar] [CrossRef]
- Sanz-Garcia, E.; Argiles, G.; Elez, E.; Tabernero, J. BRAF mutant colorectal cancer: Prognosis, treatment, and new perspectives. Ann. Oncol. 2017, 28, 2648–2657. [Google Scholar] [CrossRef]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. Non-V600BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients with Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736, Erratum in 2018, 379, 1585. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, M.; Tian, F.; Mariadason, J.M.; Tsao, C.C.; Lemos, R., Jr.; Dayyani, F.; Gopal, Y.N.; Jiang, Z.Q.; Wistuba, I.I.; Tang, X.M.; et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin. Cancer Res. 2013, 19, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejci, P.; Aklian, A.; Kaucka, M.; Sevcikova, E.; Prochazkova, J.; Masek, J.K.; Mikolka, P.; Pospisilova, T.; Spoustova, T.; Weis, M.; et al. Receptor tyrosine kinases activate canonical WNT/β-catenin signaling via MAP kinase/LRP6 pathway and direct β-catenin phosphorylation. PLoS ONE 2012, 7, e35826. [Google Scholar] [CrossRef]
- Lake, D.; Corrêa, S.A.; Müller, J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef] [Green Version]
- Hazar-Rethinam, M.; Kleyman, M.; Han, G.C.; Liu, D.; Ahronian, L.G.; Shahzade, H.A.; Chen, L.; Parikh, A.R.; Allen, J.N.; Clark, J.W.; et al. Convergent Therapeutic Strategies to Overcome the Heterogeneity of Acquired Resistance in BRAFV600E Colorectal Cancer. Cancer Discov. 2018, 8, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Morris, V.K.; El Osta, B.; Sorokin, A.V.; Janku, F.; Fu, S.; Overman, M.J.; Piha-Paul, S.; Subbiah, V.; Kee, B.; et al. Phase IB Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAFV600E Mutation. Cancer Discov. 2016, 6, 1352–1365. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, R.; Cercek, A.; O’Reilly, E.M.; Reidy, D.L.; Kemeny, N.; Wolinsky, T.; Capanu, M.; Gollub, M.J.; Rosen, N.; Berger, M.F.; et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin. Cancer Res. 2015, 21, 1313–1320. [Google Scholar] [CrossRef] [Green Version]
- Kopetz, S.; Guthrie, K.A.; Morris, V.K.; Lenz, H.J.; Magliocco, A.M.; Maru, D.; Yan, Y.; Lanman, R.; Manyam, G.; Hong, D.S.; et al. Randomized Trial of Irinotecan and Cetuximab with or without Vemurafenib in BRAF-Mutant Metastatic Colorectal Cancer (SWOG S1406). J. Clin. Oncol. 2021, 39, 285–294. [Google Scholar] [CrossRef]
- Corcoran, R.B.; André, T.; Atreya, C.E.; Schellens, J.H.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF(V600E)-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef] [Green Version]
- Drug combo beneficial in colorectal cancer. Cancer Discov. 2015, 5, 102. [CrossRef] [Green Version]
- van Geel, R.M.J.M.; Tabernero, J.; Elez, E.; Bendell, J.C.; Spreafico, A.; Schuler, M.; Yoshino, T.; Delord, J.P.; Yamada, Y.; Lolkema, M.P.; et al. A Phase Ib Dose-Escalation Study of Encorafenib and Cetuximab with or without Alpelisib in Metastatic BRAF-Mutant Colorectal Cancer. Cancer Discov. 2017, 7, 610–619. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Geel, R.V.; Guren, T.K.; Yaeger, R.D.; Spreafico, A.; Faris, J.E.; Yoshino, T.; Yamada, Y.; Kim, T.W.; Bendell, J.C.; et al. Phase 2 results: Encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in pa- tients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). J. Clin. Oncol. 2016, 34, 3544. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Huijberts, S.; Grothey, A.; Yaeger, R.; Cuyle, P.J.; Elez, E.; Fakih, M.; Montagut, C.; Peeters, M.; Yoshino, T.; et al. Binimetinib, Encorafenib, and Cetuximab Triplet Therapy for Patients with BRAF V600E-Mutant Metastatic Colorectal Cancer: Safety Lead-In Results From the Phase III BEACON Colorectal Cancer Study. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.S.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib plus cetuximab with or without binimetinib for BRAF V600E-mutant metastatic colorectal cancer: Quality-of-life results from a randomized, three-arm, phase III study versus the choice of either irinotecan or FOLFIRI plus cetuximab (BEACON CRC). J. Clin. Oncol. 2020, 38. [Google Scholar] [CrossRef]
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J. Clin. Oncol. 2021, 39, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Battaglin, F.; Naseem, M.; Lenz, H.J.; Salem, M.E. Microsatellite instability in colorectal cancer: Overview of its clinical significance and novel perspectives. Clin. Adv. Hematol. Oncol. 2018, 16, 735–745. [Google Scholar]
- Harada, S.; Morlote, D. Molecular Pathology of Colorectal Cancer. Adv. Anat. Pathol. 2020, 27, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Picard, E.; Verschoor, C.P.; Ma, G.W.; Pawelec, G. Relationships Between Immune Landscapes, Genetic Subtypes and Responses to Immunotherapy in Colorectal Cancer. Front. Immunol. 2020, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, D.; Vitiello, P.P.; Cardone, C.; Martini, G.; Troiani, T.; Martinelli, E.; Ciardiello, F. Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy. Cancer Treat. Rev. 2019, 76, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Sinicrope, F.A.; Shi, Q.; Allegra, C.J.; Smyrk, T.C.; Thibodeau, S.N.; Goldberg, R.M.; Meyers, J.P.; Pogue-Geile, K.L.; Yothers, G.; Sargent, D.J.; et al. Association of DNA Mismatch Repair and Mutations in BRAF and KRAS with Survival after Recurrence in Stage III Colon Cancers: A Secondary Analysis of 2 Randomized Clinical Trials. JAMA Oncol. 2017, 3, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condelli, V.; Calice, G.; Cassano, A.; Basso, M.; Rodriquenz, M.G.; Zupa, A.; Maddalena, F.; Crispo, F.; Pietrafesa, M.; Aieta, M.; et al. Novel Epigenetic Eight-Gene Signature Predictive of Poor Prognosis and MSI-Like Phenotype in Human Metastatic Colorectal Carcinomas. Cancers 2021, 13, 158. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Sharfman, W.H.; Drake, C.G.; Wollner, I.; Taube, J.M.; Anders, R.A.; Xu, H.; Yao, S.; Pons, A.; Chen, L.; et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin. Cancer Res. 2013, 19, 462–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.; Kemberling, H.; Eyring, A.; Azad, N.S.; Laheru, D.; Donehower, R.C.; Crocenzi, T.S.; et al. Programmed death-1 blockade in mismatch repair deficient colorectal cancer. J. Clin. Oncol. 2016, 34, 103. [Google Scholar] [CrossRef]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatelite instability–high/mismatch repair–deficient metastatic colorectal cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Andre, T.; Lonardi, S.; Wong, M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.; Van Cutsem, E.; McDermott, R.S.; Hill, A.G.; et al. Nivolumab+ipilimumab combination in patients with DNA mismatch repair-deficient/microsatellite instability-high (dMMR/MSI-H) metastatic colorectal cancer (mCRC): First report of the full cohort from CheckMate-142. J. Clin. Oncol. 2018, 36, 553. [Google Scholar] [CrossRef]
- Morse, M.A.; Overman, M.J.; Hartman, L.; Khoukaz, T.; Brutcher, E.; Lenz, H.J.; Atasoy, A.; Shangguan, T.; Zhao, H.; El-Rayes, B. Safety of Nivolumab plus Low-Dose Ipilimumab in Previously Treated Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer. Oncologist 2019, 24, 1453–1461. [Google Scholar] [CrossRef] [Green Version]
- Lenz, H.J.; Lonardi, S.; Zagonel, V.; Van Cutsem, E.; Limon, M.L.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; Garcia-Alfonso, P.; Neyns, B.; et al. Nivolumab plus low-dose ipilimumab as first-line therapy in microsatellite insta- bility-high/DNA mismatch repair deficient metastatic colorectal cancer: Clinical up-date. J. Clin. Oncol. 2020, 38, 11. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Modest, D.P.; Pant, S.; Sartore-Bianchi, A. Treatment sequencing in metastatic colorectal cancer. Eur. J. Cancer 2019, 109, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Tumors of the Colon. 2020. Available online: https://www.aiom.it/linee-guida-aiom-2020-tumori-del-colon/ (accessed on 16 April 2021).
- Chebib, R.; Verlingue, L.; Cozic, N.; Faron, M.; Burtin, P.; Boige, V.; Hollebecque, A.; Malka, D. Angiogenesis inhibition in the second-line treatment of metastatic colorectal cancer: A systematic review and pooled analysis. Semin. Oncol. 2017, 44, 114–128. [Google Scholar] [CrossRef]
- Arjaans, M.; Schröder, C.P.; Oosting, S.F.; Dafni, U.; Kleibeuker, J.E.; de Vries, E.G. VEGF pathway targeting agents, vessel normalization and tumor drug uptake: From bench to bedside. Oncotarget 2016, 7, 21247–21258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.P.; Sotomayor, P.; Carrasco-Avino, G.; Corvalan, A.H.; Owen, G.I. Escaping Antiangiogenic Therapy: Strategies Employed by Cancer Cells. Int. J. Mol. Sci. 2016, 17, 1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.; Thijssen, V.L.; Griffioen, A.W. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macarulla, T.; Montagut, C.; Sánchez-Martin, F.J.; Granja, M.; Verdaguer, H.; Sastre, J.; Tabernero, J. The role of PIGF blockade in the treatment of colorectal cancer: Overcoming the pitfalls. Expert Opin. Biol. Ther. 2020, 20, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Paccard, C.; Chiron, M.; Dochy, E.; Van Cutsem, E. Placental Growth Factor and the angiogenic environment based on analysis of baseline plasma biomarkers from the VELOUR trial. J. Clin. Oncol. 2017, 35 (Suppl. 4S), 592. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Paccard, C.; Chiron, M.; Tabernero, J. Impact of Prior Bevacizumab Treatment on VEGF-A and PlGF Levels and Outcome Following Second-Line Aflibercept Treatment: Biomarker Post Hoc Analysis of the VELOUR Trial. Clin. Cancer Res. 2020, 26, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Hozak, R.R.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.E.; Portnoy, D.C.; Prausová, J.; et al. Analysis of angiogenesis biomarkers for ramucirumab efficacy in patients with metastatic colorectal cancer from RAISE, a global, randomized, double-blind, phase III study. Ann. Oncol. 2018, 29, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Zeichner, S.B.; Kohn, C.G.; Goldstein, D.A. Economics of ramucirumab for metastatic colorectal cancer. Expert Rev. Pharm. Outcomes Res. 2016, 16, 733–745. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Cremolini, C.; Marmorino, F.; Loupakis, F.; Masi, G.; Antoniotti, C.; Salvatore, L.; Schirripa, M.; Boni, L.; Zagonel, V.; Lonardi, S.; et al. TRIBE-2: A phase III, randomized, open-label, strategy trial in unresectable metastatic colorectal cancer patients by the GONO group. BMC Cancer 2017, 17, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremolini, C.; Antoniotti, C.; Rossini, D.; Lonardi, S.; Loupakis, F.; Pietrantonio, F.; Bordonaro, R.; Latiano, T.P.; Tamburini, E.; Santini, D.; et al. Upfront FOLFOXIRI plus bevacizumab and reintroduction after progression versus mFOLFOX6 plus bevacizumab followed by FOLFIRI plus bevacizumab in the treatment of patients with metastatic colorectal cancer (TRIBE2): A multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2020, 21, 497–507. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.B.; Lee, J.; Tejpar, S.; Sartore-Bianchi, A.; Hechtman, J.F.; Christiansen, J.; Novara, L.; Tebbutt, N.; et al. ALK, ROS1, and NTRK Rearrangements in Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, H.; Shiomi, A.; Kusuhara, M.; Kagawa, H.; Yamakawa, Y.; Hatakeyama, K.; Kawabata, T.; Oishi, T.; Urakami, K.; Nagashima, T.; et al. Clinicopathological and mutational analyses of colorectal cancer with mutations in the POLE gene. Cancer Med. 2019, 8, 4587–4597. [Google Scholar] [CrossRef] [Green Version]
- Guler, I.; Askan, G.; Klostergaard, J.; Sahin, I.H. Precision medicine for metastatic colorectal cancer: An evolving era. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 919–931. [Google Scholar] [CrossRef]
- Borrero-Palacios, A.; Cebrián, A.; Gómez Del Pulgar, M.T.; García-Carbonero, R.; Garcia-Alfonso, P.; Aranda, E.; Elez, E.; López-López, R.; Cervantes, A.; Valladares, M.; et al. Combination of KIR2DS4 and FcγRIIa polymorphisms predicts the response to cetuximab in KRAS mutant metastatic colorectal cancer. Sci. Rep. 2019, 9, 2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.H.; Kuo, C.C.; Hsiao, C.W.; Chen, C.Y.; Hu, J.M.; Hsu, C.H.; Chou, Y.C.; Lin, Y.W.; Shih, Y.L. A Novel Prognostic DNA Methylation Panel for Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 4672. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Drug | Type of Molecule | Mechanism of Action | 2nd Line Labelling |
|---|---|---|---|
| Bevacizumab | Humanized MoAb | Binding VEGF-A | Plus standard CHT |
| Aflibercept | Recombinant Fusion Protein | VEGFR decoy binding VEGF-A/B; PlGF | Plus FOLFIRI in OXA pretreated |
| Ramucirumab | Fully Human MoAb | Binding VEGFR2 | Plus FOLFIRI in OXA + FP + BEV pretreated |
| Cetuximab | Human-Mouse Chimeric MoAb | Binding EGFR | Plus IRI in RAS wt |
| Panitumumab | Fully Human MoAb | Binding EGFR | Plus FOLFIRI in RAS wt |
| Encorafenib | Threonine and Serine kinase Inhibitor | BRAF gene Inhibition | Plus CET in BRAFV600E mt |
| Pembrolizumab | Humanized MoAb | Binding PD-1 | MSI-H or dMMR FP, OXA and IRI pretreated |
| Nivolumab * | Fully Human MoAb | Binding PD-1 | MSI-H or dMMR FP, OXA and IRI pretreated |
| Study [Ref.] | Pts | Study Arms | RR (%) | PFS (Months) | HR (95% CI) | OS (Months) | HR (95% CI) |
|---|---|---|---|---|---|---|---|
| E3200 [41] | 829 | FOLFOX4 + BEV vs. FOLFOX4 vs. BEV | 22.7 vs. 8.6 vs. 3.3 p < 0.0001 | 7.3 vs. 4.7 vs. 2.7 p < 0.0001 | 0.61 (NA) | 12.9 vs. 10.8 vs. 10.2 p < 0.0011 | 0.75 (NA) |
| ML18147 [44] | 820 | BEV + CHT vs. CHT alone | 6 vs. 4 | 5.7 vs. 4.1 p < 0.0001 | 0.68 (0.59–0.78) | 11.2 vs. 9.8 p < 0.0062 | 0.81 (0·69–0·94) |
| BEBYP [45] | 185 | CHT + BEV vs. CHT | 21 vs. 17 p = 0.124 | 6.8 vs. 5.0 p = 0.010 | 0.70 (0.52–0.95) | 15.5 vs. 14.1 p = 0.043 | 0.77 (0.56–1.06) |
| VELOUR [12] | 1226 | AFL + FOLFIRI vs. PBO + FOLFIRI | 19.8 vs. 11.1 p < 0.0001 | 6.90 vs. 4.67 p < 0.0001 | 0.758 (0.661–0.869) | 13.50 vs. 12.06 p < 0.0032 | 0.817 (0.713–0.937) |
| RAISE [13] | 1072 | RAM + FOLFIRI vs. PBO + FOLFIRI | 13.4 vs. 12.5 p = 0.63 | 5.7 vs. 4.5 p = 0.0005 | 0.793 (0.697–0.903) | 11.7 vs. 13.3 p < 0.0219 | 0.844 (0.730–0.976) |
| EPIC [66] | 1298 | IRI + CET vs. IRI | 16.4 vs. 4.2 p = 0.0001 | 4 vs. 2.6 p = 0.0001 | 0.692 (0.617–0.776) | 10.7 vs. 10 p = 0.71 | 0.975 (0.854–1.114) |
| BOND [65] ° | 329 | IRI + CET vs. CET | 22.9 vs. 10.8 p = 0.007 | 4.1 vs. 1.5 p = 0.001 | 0.54 (0.42–0.71) | 8.6 vs. 6.9 p = 0.48 | 0.91 (0.68–1.21) |
| STUDY 181 [69] | 1186 | FOLFIRI + PAN vs. FOLFIRI | 35 vs. 10 § p < 0.0001 | 5.9 vs. 3.9 § p = 0.004 | 0.73 (0.59–0.90) | 14.5 vs. 12.5 § p = 0.12 | 0.85 (0.70–1.04) |
| PICCOLO [70] | 460ç | IRI + PAN vs. IRI | 34 vs. 12 p < 0.0001 | not available ^ p = 0.015 | 0.78 (0.65–0.95) | 10.9 vs. 10.4 p = 0.91 | 1.01 (0.83–1.23) |
| BEACON [25] | 665 | ENC + BIN + CET vs. ENC + CET vs. FOLFIRI + CET | 26 vs. 20 vs. 2 p < 0.001 | 4.3 vs. 4.2 vs. 1.5 p < 0.001 | 0.40 * (0.31–0.52) | 9.0 vs. 8.4 vs. 5.4 p < 0.001 | 0.60 * (0.45–0.79) |
| KEYNOTE ** 164 [102,103] | 124 | PEMBRO | 33 | 4.1 | NA | 31.4 | NA |
| CHECKMATE 142 ** [106] | 119 | NIVO + IPI | 55 | NR | NA | NR | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giordano, G.; Parcesepe, P.; Bruno, G.; Piscazzi, A.; Lizzi, V.; Remo, A.; Pancione, M.; D’Andrea, M.R.; De Santis, E.; Coppola, L.; et al. Evidence-Based Second-Line Treatment in RAS Wild-Type/Mutated Metastatic Colorectal Cancer in the Precision Medicine Era. Int. J. Mol. Sci. 2021, 22, 7717. https://doi.org/10.3390/ijms22147717
Giordano G, Parcesepe P, Bruno G, Piscazzi A, Lizzi V, Remo A, Pancione M, D’Andrea MR, De Santis E, Coppola L, et al. Evidence-Based Second-Line Treatment in RAS Wild-Type/Mutated Metastatic Colorectal Cancer in the Precision Medicine Era. International Journal of Molecular Sciences. 2021; 22(14):7717. https://doi.org/10.3390/ijms22147717
Chicago/Turabian StyleGiordano, Guido, Pietro Parcesepe, Giuseppina Bruno, Annamaria Piscazzi, Vincenzo Lizzi, Andrea Remo, Massimo Pancione, Mario Rosario D’Andrea, Elena De Santis, Luigi Coppola, and et al. 2021. "Evidence-Based Second-Line Treatment in RAS Wild-Type/Mutated Metastatic Colorectal Cancer in the Precision Medicine Era" International Journal of Molecular Sciences 22, no. 14: 7717. https://doi.org/10.3390/ijms22147717
APA StyleGiordano, G., Parcesepe, P., Bruno, G., Piscazzi, A., Lizzi, V., Remo, A., Pancione, M., D’Andrea, M. R., De Santis, E., Coppola, L., Pietrafesa, M., Fersini, A., Ambrosi, A., & Landriscina, M. (2021). Evidence-Based Second-Line Treatment in RAS Wild-Type/Mutated Metastatic Colorectal Cancer in the Precision Medicine Era. International Journal of Molecular Sciences, 22(14), 7717. https://doi.org/10.3390/ijms22147717