
Extracellular Inorganic Phosphate-Induced Release of Reactive Oxygen Species: Roles in Physiological Processes and Disease Development

Abstract
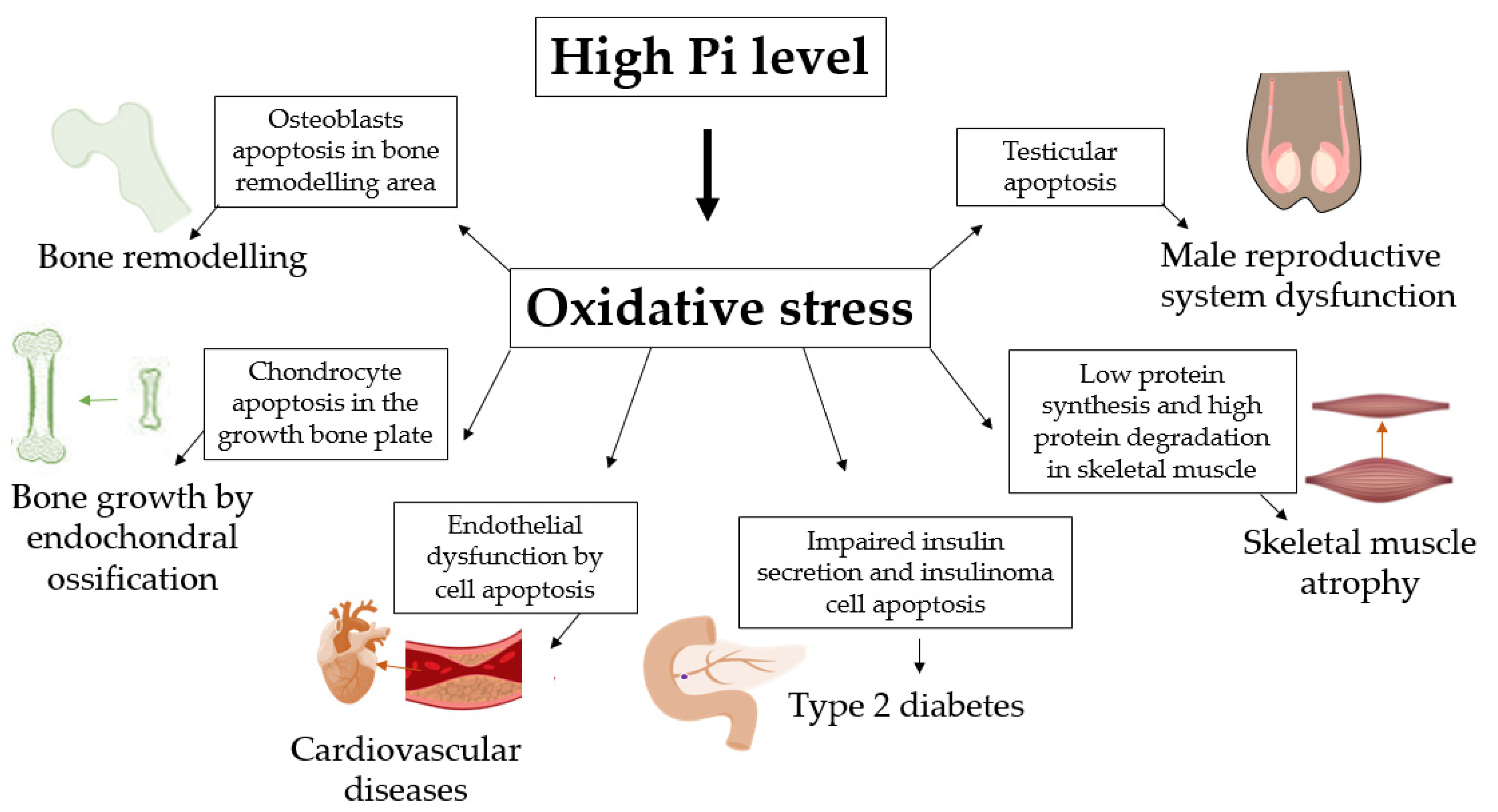
:1. Introduction
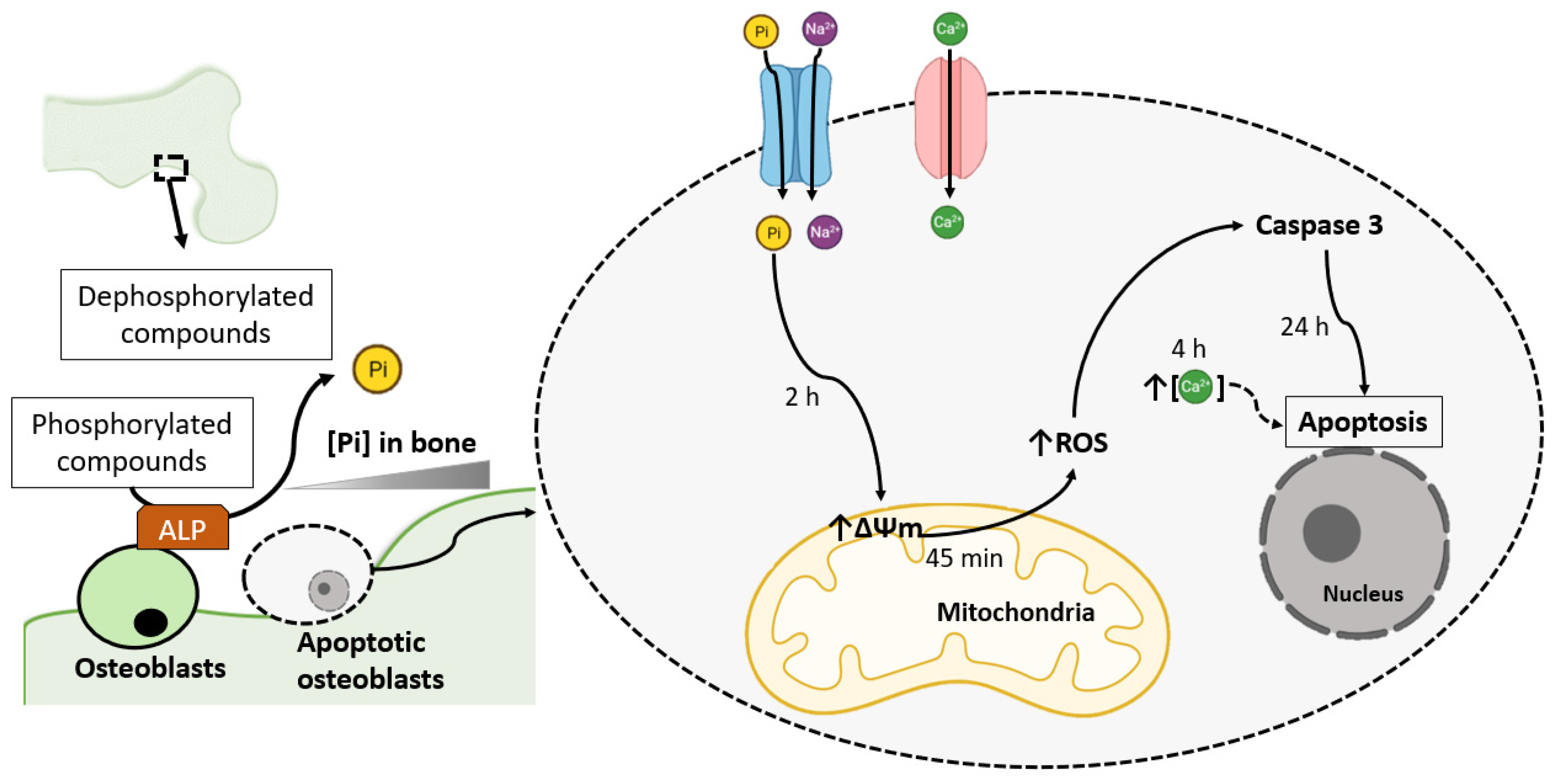
2. Pi-Induced ROS Production Promotes Osteoblast Apoptosis
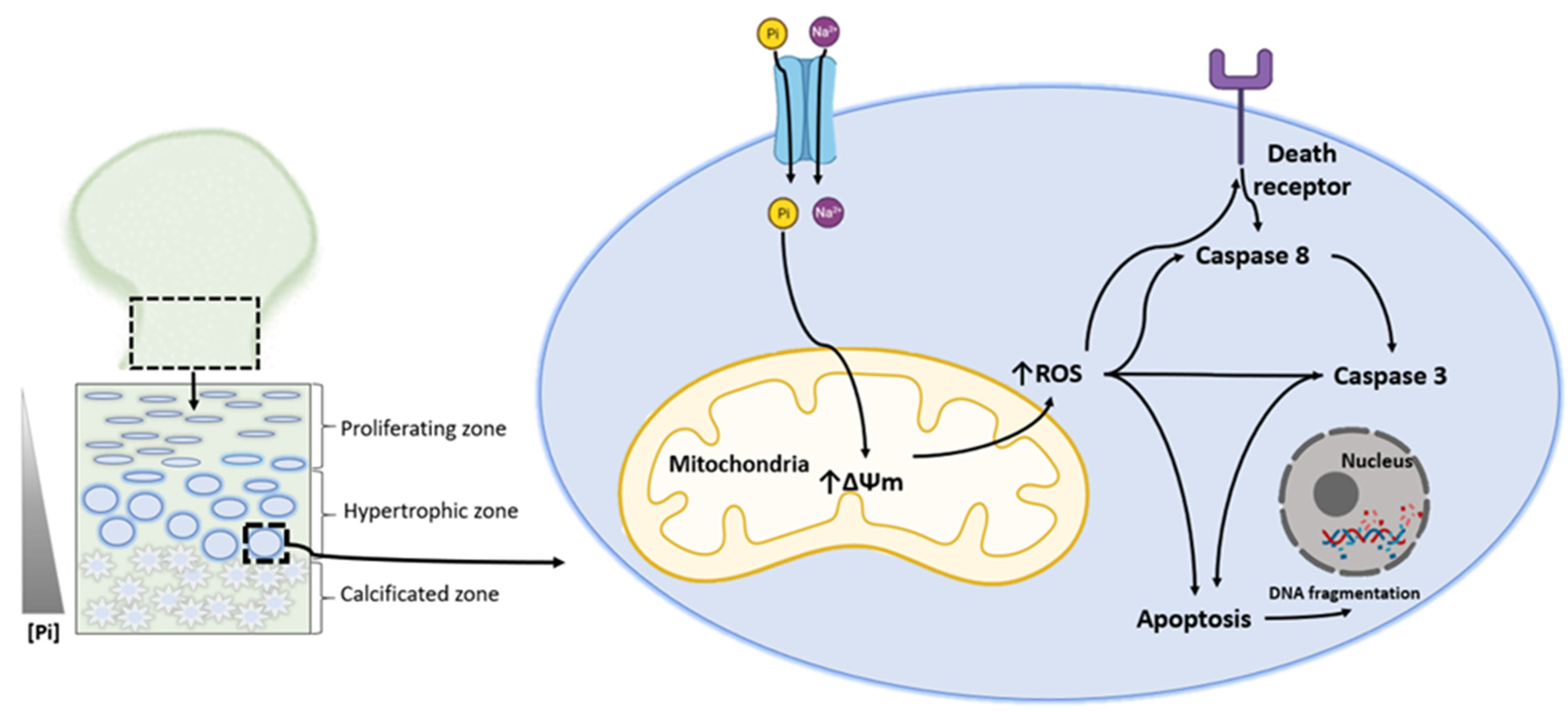
3. Phosphate-Induced Chondrocyte Apoptosis Is Mediated by Ros Production
4. Endothelial Dysfunction and Cardiovascular Disease
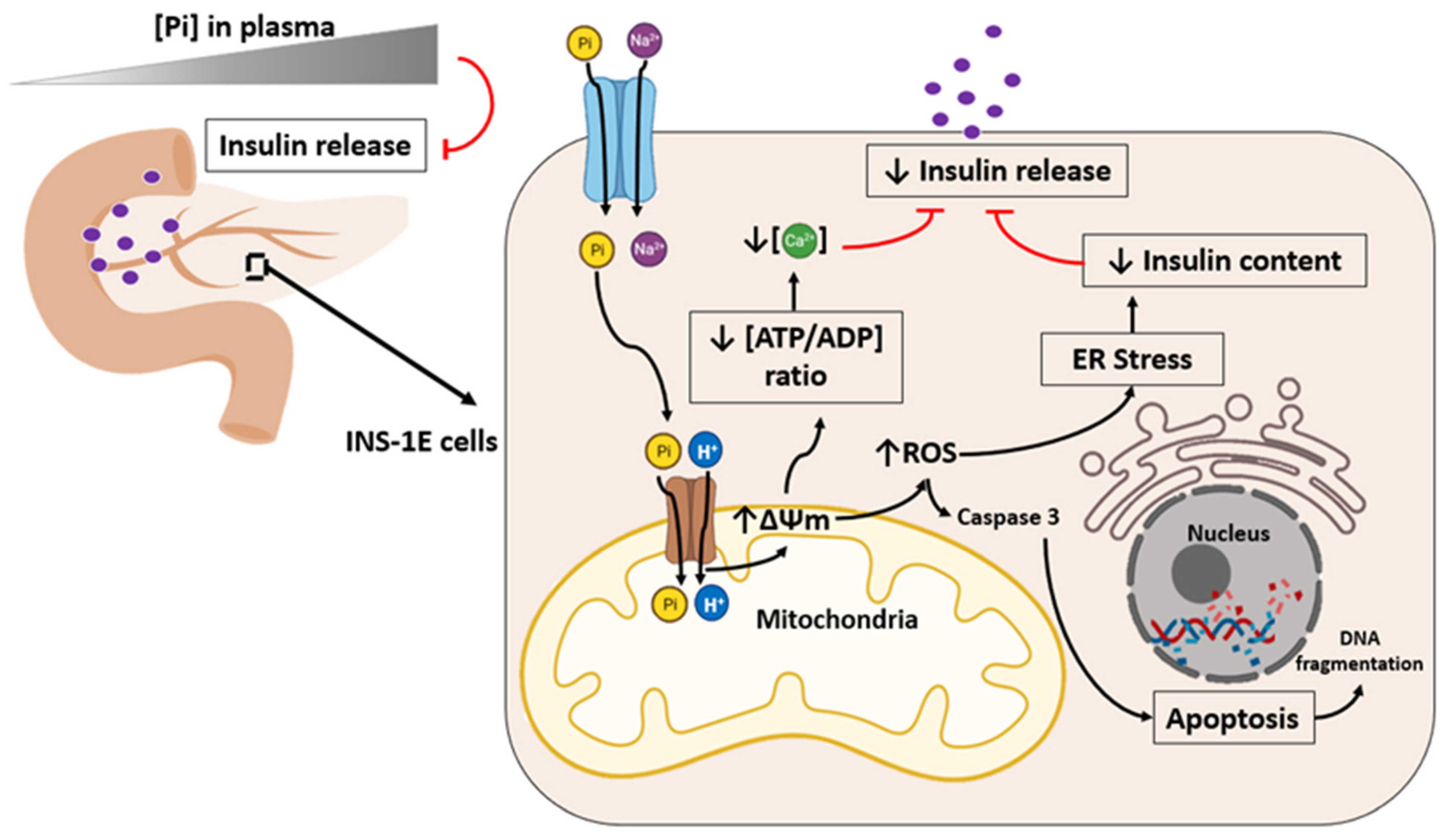
5. High Phosphate Induces Impaired Insulin Secretion
6. Skeletal Muscle Atrophy and Suppressed Myogenic Differentiation
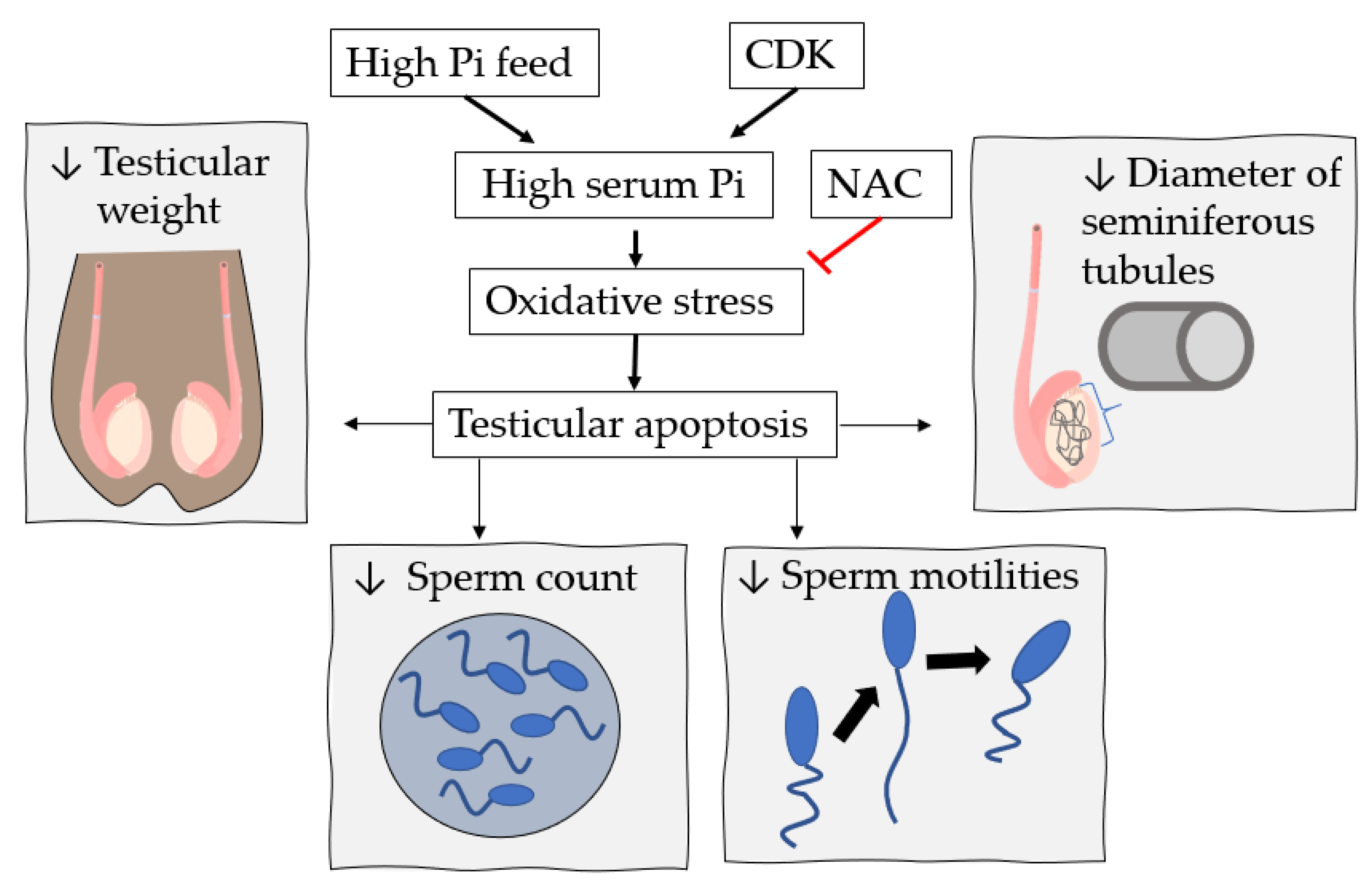
7. High Pi Diet and Male Reproductive System Dysfunction
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takeda, E.; Taketani, Y.; Morita, K.; Tatsumi, S.; Katai, K.; Nii, T.; Yamamoto, H.; Miyamoto, K.-I. Molecular mechanisms of mammalian inorganic phosphate homeostasis. Adv. Enzym. Regul. 2000, 40, 285–302. [Google Scholar] [CrossRef]
- Levi, M.; Gratton, E.; Forster, I.C.; Hernando, N.; Wagner, C.A.; Biber, J.; Sorribas, V.; Murer, H. Mechanisms of phosphate transport. Nat. Rev. Nephrol. 2019, 15, 482–500. [Google Scholar] [CrossRef]
- Lacerda-Abreu, M.A.; Russo-Abrahão, T.; Meyer-Fernandes, J.R. The roles of sodium-independent inorganic phosphate transporters in inorganic phosphate homeostasis and in cancer and other diseases. Int. J. Mol. Sci. 2020, 21, 9298. [Google Scholar] [CrossRef]
- Lacerda-Abreu, M.A.; Russo-Abrahão, T.; Cosentino-Gomes, D.; Nascimento, M.T.C.; Carvalho-Kelly, L.F.; Gomes, T.; Rodrigues, M.F.; König, S.; Rumjanek, F.D.; Monteiro, R.Q.; et al. H+-dependent inorganic phosphate transporter in breast cancer cells: Possible functions in the tumor microenvironment. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2180–2188. [Google Scholar] [CrossRef]
- McClure, S.T.; Chang, A.R.; Selvin, E.; Rebholz, C.M.; Appel, L.J. Dietary sources of phosphorus among adults in the united states: Results from NHANES 2001–2014. Nutrients 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Michigami, T.; Kawai, M.; Yamazaki, M.; Ozono, K. Phosphate as a signaling molecule and its sensing mechanism. Physiol. Rev. 2018, 98, 2317–2348. [Google Scholar] [CrossRef] [PubMed]
- Chande, S.; Bergwitz, C. Role of phosphate sensing in bone and mineral metabolism. Nat. Rev. Endocrinol. 2018, 14, 637–655. [Google Scholar] [CrossRef] [PubMed]
- Lacerda-Abreu, M.A.; Russo-Abrahão, T.; Monteiro, R.Q.; Rumjanek, F.D.; Meyer-Fernandes, J.R. Inorganic phosphate transporters in cancer: Functions, molecular mechanisms and possible clinical applications. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 291–298. [Google Scholar] [CrossRef]
- Christov, M.; Jüppner, H. Phosphate homeostasis disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 685–706. [Google Scholar] [CrossRef]
- Jacquillet, G.; Unwin, R. Physiological regulation of phosphate by vitamin D, parathyroid hormone (PTH) and phosphate (Pi). Pflügers Archiv Eur. J. Physiol. 2019, 471, 83–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, E.H.; Gonin, J.M. Disorders of phosphorus metabolism. Nephrol. Secrets 2012, 551–559. [Google Scholar] [CrossRef]
- Kestenbaum, B.; Sampson, J.N.; Rudser, K.D.; Patterson, D.J.; Seliger, S.L.; Young, B.; Sherrard, D.J.; Andress, D.L. Serum phosphate levels and mortality risk among people with chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 520–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanudel, M.R. Evaluation and treatment of disorders of phosphate balance. Curr. Treat. Options Pediatr. 2020, 6, 227–240. [Google Scholar] [CrossRef]
- Francisqueti, F.V.; Chiaverini, L.C.; Santos, K.C.; Minatel, I.O.; Ronchi, C.B.; Ferron, A.J.; Ferreira, A.L.; Corrêa, C.R. The role of oxidative stress on the pathophysiology of metabolic syndrome. Rev. Assoc. Med. Bras. 2017, 63, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Oliveira, G.A.; Kowaltowski, A.J. Phosphate increases mitochondrial reactive oxygen species release. Free Radic. Res. 2004, 38, 1113–1118. [Google Scholar] [CrossRef]
- Serna, J.; Bergwitz, C. Importance of dietary phosphorus for bone metabolism and healthy aging. Nutrients 2020, 12, 3001. [Google Scholar] [CrossRef]
- Pinedo, M.G.; Alon, U.S. Phosphate homeostasis and its role in bone health. Pediatr. Nephrol. 2012, 27, 2039–2048. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, T.; Taguchi, M.; Osaki, T.; Fukumoto, S.; Fujita, T. Phosphate enhances reactive oxygen species production and suppresses osteoblastic differentiation. J. Bone Miner. Metab. 2013, 32, 393–399. [Google Scholar] [CrossRef]
- Hori, M.; Kinoshita, Y.; Taguchi, M.; Fukumoto, S. Phosphate enhances Fgf23 expression through reactive oxygen species in UMR-106 cells. J. Bone Miner. Metab. 2016, 34, 132–139. [Google Scholar] [CrossRef]
- Meleti, Z.; Shapiro, I.M.; Adams, C.S. Inorganic phosphate induces apoptosis of osteoblast-like cells in culture. Bone 2000, 27, 359–366. [Google Scholar] [CrossRef]
- Adams, C.S.; Mansfield, K.; Perlot, R.L.; Shapiro, I.M. Matrix regulation of skeletal cell apoptosis. Role of calcium and phosphate ions. J. Biol. Chem. 2001, 276, 20316–20322. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.S.; Shapiro, I.M. Mechanisms by which extracellular matrix components induce osteoblast apoptosis. Connect Tissue Res. 2003, 44, 230–239. [Google Scholar] [CrossRef]
- Sun, M.M.; Beier, F. Chondrocyte hypertrophy in skeletal development, growth, and disease. Birth Defects Res. Part C Embryo Today Rev. 2014, 102, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Wuthier, R.E. Electrolytes of isolated epiphyseal chondrocytes, matrix vesicles, and extracellular fluid. Calcif. Tissue Res. 1977, 23, 125–133. [Google Scholar] [CrossRef]
- Howell, D.S.; Pita, J.C.; Marquez, J.F.; Madruga, J.E. Partition of calcium, phosphate, and protein in the fluid phase aspirated at calcifying sites in epiphyseal cartilage. J. Clin. Investig. 1968, 47, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Rajpurohit, R.; Mansfield, K.; Ohyama, K.; Ewert, D.; Shapiro, I.M. Chondrocyte death is linked to development of a mitochondrial membrane permeability transition in the growth plate. J. Cell. Physiol. 1999, 179, 287–296. [Google Scholar] [CrossRef]
- Mansfield, K.; Rajpurohit, R.; Shapiro, I.M. Extracellular phosphate ions cause apoptosis of terminally differentiated epiphyseal chondrocytes. J. Cell. Physiol. 1999, 179, 276–286. [Google Scholar] [CrossRef]
- Mansfield, K.; Teixeira, C.C.; Adams, C.S.; Shapiro, I.M. Phosphate ions mediate chondrocyte apoptosis through a plasma membrane transporter mechanism. Bone 2001, 28, 1–8. [Google Scholar] [CrossRef]
- Mansfield, K.; Pucci, B.; Adams, C.S.; Shapiro, I.M. Induction of apoptosis in skeletal tissues: Phosphate-mediated chick chondrocyte apoptosis is calcium dependent. Calcif. Tissue Int. 2003, 73, 161–172. [Google Scholar] [CrossRef]
- Teixeira, C.C.; Mansfield, K.; Hertkorn, C.; Ischiropoulos, H.; Shapiro, I.M. Phosphate-induced chondrocyte apoptosis is linked to nitric oxide generation. Am. J. Physiol. Cell Physiol. 2001, 281, C833–C839. [Google Scholar] [CrossRef]
- Adams, C.S.; Shapiro, I.M. The fate of the terminally differentiated chondrocyte: Evidence for microenvironmental regulation of chondrocyte apoptosis. Crit. Rev. Oral Biol. Med. 2002, 13, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Ghafourifar, P.; Bringold, U.; Klein, S.D.; Richter, C. Mitochondrial nitric oxide synthase, oxidative stress and apoptosis. Neurosignals 2001, 10, 57–65. [Google Scholar] [CrossRef]
- Giachelli, C.M. Vascular calcification: In vitro evidence for the role of inorganic phosphate. J. Am. Soc. Nephrol. 2003, 14, S300–S304. [Google Scholar] [CrossRef] [Green Version]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef]
- Huang, M.; Zheng, L.; Xu, H.; Tang, D.; Lin, L.; Zhang, J.; Li, C.; Wang, W.; Yuan, Q.; Tao, L.; et al. Oxidative stress contributes to vascular calcification in patients with chronic kidney disease. J. Mol. Cell. Cardiol. 2020, 138, 256–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.T.; Shao, Y.D.; Liu, Y.Z.; Xiao, X.; Cheng, Z.B.; Qu, S.L.; Huang, L.; Zhang, C. Oxidative stress in vascular calcification. Clin. Chim. Acta 2021, 519, 101–110. [Google Scholar] [CrossRef]
- Kirkman, D.L.; Robinson, A.T.; Rossman, M.J.; Seals, D.R.; Edwards, D.G. Mitochondrial contributions to vascular endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2080–H2100. [Google Scholar] [CrossRef] [PubMed]
- Takeda, E.; Taketani, Y.; Nashiki, K.; Nomoto, M.; Shuto, E.; Sawada, N.; Yamamoto, H.; Isshiki, M. A novel function of phosphate-mediated intracellular signal transduction pathways. Adv. Enzym. Regul. 2006, 46, 154–161. [Google Scholar] [CrossRef]
- Shuto, E.; Taketani, Y.; Tanaka, R.; Harada, N.; Isshiki, M.; Sato, M.; Nashiki, K.; Amo, K.; Yamamoto, H.; Higashi, Y.; et al. Dietary phosphorus acutely impairs endothelial function. J. Am. Soc. Nephrol. 2009, 20, 1504–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.M.; Xu, M.J.; Cai, Y.; Zhao, G.; Guan, Y.; Kong, W.; Tang, C.; Wang, X. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011, 79, 1071–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Kim, H.J.; Lee, K.; Kim, J.M.; Kim, H.S.; Kim, J.R.; Ha, C.M.; Choi, Y.K.; Lee, S.J.; Kim, J.Y.; et al. α-Lipoic acid attenuates vascular calcification via reversal of mitochondrial function and restoration of Gas6/Axl/Akt survival pathway. J. Cell. Mol. Med. 2012, 16, 273–286. [Google Scholar] [CrossRef]
- Di Marco, G.S.; Hausberg, M.; Hillebrand, U.; Rustemeyer, P.; Wittkowski, W.; Lang, D.; Pavenstädt, H. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am. J. Physiol. Renal Physiol. 2008, 294, F1381–F1387. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Nguyen, T.T.; Ly, D.D.; Xia, J.B.; Qi, X.F.; Lee, I.K.; Cha, S.K.; Park, K.S. Oxidative stress by Ca2+ overload is critical for phosphate-induced vascular calcification. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H1302–H1312. [Google Scholar] [CrossRef]
- Hu, W.; Jiang, S.; Liao, Y.; Li, J.; Dong, F.; Guo, J.; Wang, X.; Fei, L.; Cui, Y.; Ren, X.; et al. High phosphate impairs arterial endothelial function through AMPK-related pathways in mouse resistance arteries. Acta Physiol. 2021, 231, e13595. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef]
- Wollheim, C.B. Beta-cell mitochondria in the regulation of insulin secretion: A new culprit in type II diabetes. Diabetologia 2000, 43, 265–277. [Google Scholar] [CrossRef]
- Wiederkehr, A.; Wollheim, C.B. Minireview: Implication of mitochondria in insulin secretion and action. Endocrinology 2006, 147, 2643–2649. [Google Scholar] [CrossRef] [Green Version]
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 2008, 1778, 1978–2021. [Google Scholar] [CrossRef] [Green Version]
- Wohlrab, H. Molecular aspects of inorganic phosphate transport in mitochondria. Biochim. Biophys. Acta 1986, 853, 115–134. [Google Scholar] [CrossRef]
- Quan, X.; Das, R.; Xu, S.; Cline, G.W.; Wiederkehr, A.; Wollheim, C.B.; Park, K.S. Mitochondrial phosphate transport during nutrient stimulation of INS-1E insulinoma cells. Mol. Cell. Endocrinol. 2013, 381, 198–209. [Google Scholar] [CrossRef]
- Lorenzo, C.; Hanley, A.J.; Rewers, M.J.; Haffner, S.M. Calcium and phosphate concentrations and future development of type 2 diabetes: The insulin resistance atherosclerosis study. Diabetologia 2014, 57, 1366–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadda, G.Z.; Hajjar, S.M.; Perna, A.F.; Zhou, X.J.; Lipson, L.G.; Massry, S.G. On the mechanism of impaired insulin secretion in chronic renal failure. J. Clin. Investig. 1991, 87, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Utsugi, T.; Ohno, T.; Ohyama, Y.; Uchiyama, T.; Saito, Y.; Matsumura, Y.; Aizawa, H.; Itoh, H.; Kurabayashi, M.; Kawazu, S.; et al. Decreased insulin production and increased insulin sensitivity in the klotho mutant mouse, a novel animal model for human aging. Metabolism 2000, 49, 1118–1123. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Quan, X.; Hwang, K.H.; Xu, S.; Das, R.; Choi, S.K.; Wiederkehr, A.; Wollheim, C.B.; Cha, S.K.; Park, K.S. Mitochondrial oxidative stress mediates high-phosphate-induced secretory defects and apoptosis in insulin-secreting cells. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E933–E941. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.; Quan, X.; Xu, S.; Das, R.; Cha, S.K.; Kong, I.D.; Shong, M.; Wollheim, C.B.; Park, K.S. Intracellular alkalinization by phosphate uptake via type III sodium-phosphate co-transporter participates in high-phosphate-induced mitochondrial oxidative stress and defective insulin secretion. FASEB J. 2016, 30, 3979–3988. [Google Scholar] [CrossRef] [Green Version]
- Furrer, R.; Handschin, C. Muscle wasting diseases: Novel targets and treatments. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 315–339. [Google Scholar] [CrossRef]
- Tsao, C.W.; Hsu, Y.J.; Chang, T.C.; Wu, S.T.; Cha, T.L.; Liu, C.Y. A high phosphorus diet impairs testicular function and spermatogenesis in male mice with chronic kidney disease. Nutrients 2020, 12, 2624. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ichihara, G.; Sahabudeen, S.M.; Kato, A.; Yamaguchi, T.; Imanaka-Yoshida, K.; Yoshida, T.; Yamada, Y.; Ichihara, S. Rats with metabolic syndrome resist the protective effects of N-acetyl l-cystein against impaired spermatogenesis induced by high-phosphorus/zinc-free diet. Exp. Toxicol. Pathol. 2013, 65, 1173–1182. [Google Scholar] [CrossRef]
- Chung, L.H.; Liu, S.T.; Huang, S.M.; Salter, D.M.; Lee, H.S.; Hsu, Y.J. High phosphate induces skeletal muscle atrophy and suppresses myogenic differentiation by increasing oxidative stress and activating Nrf2 signaling. Aging 2020, 12, 21446–21468. [Google Scholar] [CrossRef]
- Back, S.H.; Kang, S.W.; Han, J.; Chung, H.T. Endoplasmic reticulum stress in the β-cell pathogenesis of type 2 diabetes. Exp. Diabetes Res. 2011, 2012, 618396. [Google Scholar] [CrossRef] [Green Version]
- Burgos-Morón, E.; Abad-Jiménez, Z.; Marañón, A.M.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: The battle continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, S.K.; Choi, K.; Kim, S.K.; Lee, G.I.; Cho, I.C. Phosphorus as predictive factor for erectile dysfunction in middle aged men: A cross sectional study in Korea. Investig. Clin. Urol. 2016, 57, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Banjoko, S.O.; Adeseolu, F.O. Seminal plasma pH, inorganic phosphate, total and ionized calcium concentrations in the assessment of human spermatozoa function. J. Clin. Diagn. Res. 2013, 7, 2483–2486. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| High Phosphorus 1 | Low Phosphorus 2 |
|---|---|
| Grain Products. | Sugar, Sweeteners & Beverages. |
| Meat, Poultry, Fish & Mixtures. | Vegetables and fruits. |
| Milk & Milk Products. | Legumes, Nuts, Seeds and eggs. |
| Fats, Oils & Salad Dressings. |
| Cell Type or Tissue Sample | Results Observed | Pi Treatment | Pharmacological Effect |
|---|---|---|---|
| Osteoblastic murine MC3T3-E1 cells | Increased ROS production by NOX 1 and 4 [20]. | 5 mM Pi for 20–42 h [20]. | Inhibition by NOX inhibitors (0.5 mM apocynin and 10 μM DPI) and PFA (0.5–1 mM) [20]. |
| Low osteoblastic markers expression (ALP, osteocalcin, and runt-related transcription factor 2) [20]. | 5 mM Pi for 48 h [20]. | - | |
| Rat osteoblastic cell line (UMR-106). | Increased ROS production [21]. | 5 mM Pi for 48 h [21]. | Inhibition by 0.5 mM apocynin [21]. |
| Increased FGF-23 release [21]. | 5 mM Pi for 48 h [21]. | Inhibition by 0.5 mM apocynin [21]. | |
| Osteoblast-like cells (human bone) | Increased mitochondrial membrane potential [22,23]. | 7 mM Pi for 24 h [22]. 5 mM Pi and 2.9 mM Ca2+ for 2 h [23]. | - |
| Increased ROS production [22]. | 8 mM Pi and 2.9 mM Ca2+ for 45 min [24]. | - | |
| Increased intracellular Ca2+ [23]. | 5 mM Pi and 2.9 mM Ca2+ for 4 h [23]. | - | |
| Increased cell death [22,23]. | 7 mM Pi for 24, 48 and 96 h [22]. 5 mM Pi and 2.9 mM Ca2+ for 24 h [23]. | Inhibition by Pi transporter inhibitor (PFA, 5 mM) [22,23] and apoptosis inhibitors (aurintricarboxylic acid, ATA or DEVD-CHO) [21]. | |
| Increased cell apoptosis [20,21]. | 7 mM Pi for 48 h [20]. 3 mM Pi and 2.9 mM Ca2+ for 24 h [21]. | - |
| Results Observed | Pi Treatment | Pharmacologic Effect |
|---|---|---|
| A marked early increase in mitochondrial membrane potential until 135 min and a small decrease after 150 min [31]. | 3 mM Pi and 2.8 mM Ca2+ [31]. | - |
| Decreased mitochondrial membrane potential [29,32]. | 7 mM Pi overnight [28]. | - |
| 5 mM Pi for 24 h [32]. | Inhibition by Pi transporter inhibitor (5 mM PFA) and 5 mM NOS inhibitors (L-NAME and L-NMMA) [32]. | |
| Increased ROS production after 75 min [31]. | 3 mM Pi and 2.8 mM Ca2+ [31]. | - |
| Increased intracellular Ca2+ after 45 min [31]. | 3 mM Pi and 2.8 mM Ca2+ [31]. | - |
| Increased nitric oxide generation [32]. | 5 mM Pi for 24 h [32]. | Inhibition by 5 mM PFA and L-NAME and L-NMMA [32]. |
| Decreased glutathione levels [32]. | 5 mM Pi for 24 h [32]. | Inhibition by 5 mM PFA and 5 mM L-NAME [32]. |
| Increased cell apoptosis [28,29,32]. | 7 mM Pi overnight [28,29]. | Inhibition by 1 mM PFA and alendronate [28,29]. |
| 5 mM Pi for 24 h [32]. | Inhibition by 5 mM PFA and 5 mM L-NAME and L-NMMA [32]. |
| Cell Type or Tissue Sample | Results Observed | Pi Treatment | Pharmacologic Effect |
|---|---|---|---|
| Bovine aortic endothelial cells [40,41]. | Increased ROS production [40]. | 2.8 mM Pi for 80 min. | Inhibition by NOX inhibitor (10 μM DPI). |
| Reduced NO production [40]. | 2.8 mM Pi for 800 s. | - | |
| Increased PKC activity [41]. | 3 mM Pi for 15 min. | Inhibition by 200 μM PFA. | |
| Bovine aortic smooth muscle cells [42]. | Increased mitochondrial membrane potential. | 10 mM β-glycerophosphate (BGP) for 2 days. | - |
| Increased ROS production | 10 mM BGP for 2 days. | Inhibition by a respiratory chain inhibitor (rotenone, 10 μM) or carbonyl cyanide m-chlorophenyl hydrazone (CCCP; 10 μM). | |
| Increased smooth muscle cells calcification and bone-related markers. | 10 mM BGP for 2 days. | Inhibition by 10 μM rotenone and 10 μM CCCP. | |
| Bovine aortic smooth muscle cells from male C57BL6N mice [43]. | Decreased mitochondrial membrane potential. | 3.6 mM Pi for 4 days. | - |
| Decreased ATP production. | 3.6 mM Pi for 4 days. | Inhibition by 300 μM α-lipoic acid. | |
| Increased ROS production. | 3.6 mM Pi for 4 days. | Inhibition by 300 μM α-lipoic acid. | |
| Increased cells apoptosis. | 3.6 mM Pi for 4 days. | Inhibition by 300 μM α-lipoic acid. | |
| Increased smooth muscle calcification. | 3.6 mM Pi for 4 days. | Inhibition by 300 μM α-lipoic acid. | |
| Human endothelial cells (EAhy926 cells and GM-7373 cells) [44]. | Decreased mitochondrial membrane potential. | 2.5–5 mM Pi and 2.8 mM Ca2+ for 2 h. | Inhibition by a Pi transporter inhibitor (1 mM PFA). |
| Increased ROS production. | 2.5 mM Pi and 2.8 mM Ca2+ for 75 min. | Inhibition by a superoxide scavenger (3-dimethyl-2-thiourea—DMTU, 10 mM). | |
| Increased cell apoptosis. | 5 mM Pi and 2.8 mM Ca2+ for 24 h. | Inhibition by a general caspase inhibitor (Z-VAD-FMK, 100 μM). | |
| Human umbilical vein endothelial cells (HUVECs) and endothelial cells of C57Bl/6 mice [46]. | Impairment of the PPARα/LKB1/AMPK/NOS pathway. | 3 mM Pi for 48 h. | Inhibition by a AMPK agonist (AIACR, 100 μM), PPARα agonist (100 μM WY-14643), PGC-1α inhibitor (SR-18292, 50 µM) and PFA (0.5 mM). |
| Decreased the mitochondrial membrane potential. | 3 mM Pi for 48 h. | Inhibition by AIACR (100 μM), SR-18292 (50 µM). | |
| Increased mitochondrial ROS production. | 3 mM Pi for 48 h. | Inhibition by AIACR (100 μM), SR-18292 (50 µM), cytoplasmic ROS scavenger (tempol, 100 µM) and mitochondrial ROS scavenger (mito-tempo, 100 µM). | |
| Reduced NO production. | 3 mM Pi for 48 h. | Inhibition by AIACR (100 μM), WY-14643 (100 μM). | |
| Increased relaxation in mesenteric arteries. | Mice fed high Pi diet (1.3% phosphate). | Inhibition by AIACR (100 μmol/L), compound C (1 μmol/L, AMPK inhibitor) and WY-14643 (100 μmol/L in drinking water, 2 weeks). |
| Cell Type or Tissue Sample | Results Observed | Pi Treatment | Pharmacologic Effect |
|---|---|---|---|
| Rat insulinoma cells [56]. | Increased the mitochondrial membrane potential. | 3–5 mM Pi for 30 min. | Inhibition by FCCP ionophore, butylmalonate (BMA). |
| Increased superoxide generation. | 3–5 mM Pi for 30 min. | Inhibition by BMA and mitochondrial antioxidants: mitoTEMPO (100 nM) or Mn-TBAP (0.5 mM). | |
| C2C12 skeletal muscle cells [58]. | Decreased the mitochondrial membrane potential. | 4 mM Pi for 24 h. | - |
| Increased O2 consumption. | 4 mM Pi for 24 h. | - | |
| Increased ROS generation. | 4 mM Pi for 24 h. | Strongly inhibited by cytosolic ROS scavenger (10 mM NAC). | |
| High MDA production [59,60]. | Normal diet (0.3% Pi) compared to high Pi diet (1.2%) [60]. Normal diet (0.6% Pi) compared to high Pi diet (2.0%) [59]. | Inhibition by NAC (1.5 mg/weight/day) [60]. | |
| Testicular tissue of WKY rats [60] or C57BL/6 mice [59]. | Decreased glutathione levels [61]. | High Pi diet (1.2%) [60]. | - |
| Low level of antioxidant activities (SOD, CAT and GPx) [60]. | High Pi diet (2.0%) [59]. | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lacerda-Abreu, M.A.; Meyer-Fernandes, J.R. Extracellular Inorganic Phosphate-Induced Release of Reactive Oxygen Species: Roles in Physiological Processes and Disease Development. Int. J. Mol. Sci. 2021, 22, 7768. https://doi.org/10.3390/ijms22157768
Lacerda-Abreu MA, Meyer-Fernandes JR. Extracellular Inorganic Phosphate-Induced Release of Reactive Oxygen Species: Roles in Physiological Processes and Disease Development. International Journal of Molecular Sciences. 2021; 22(15):7768. https://doi.org/10.3390/ijms22157768
Chicago/Turabian StyleLacerda-Abreu, Marco Antonio, and José Roberto Meyer-Fernandes. 2021. "Extracellular Inorganic Phosphate-Induced Release of Reactive Oxygen Species: Roles in Physiological Processes and Disease Development" International Journal of Molecular Sciences 22, no. 15: 7768. https://doi.org/10.3390/ijms22157768
APA StyleLacerda-Abreu, M. A., & Meyer-Fernandes, J. R. (2021). Extracellular Inorganic Phosphate-Induced Release of Reactive Oxygen Species: Roles in Physiological Processes and Disease Development. International Journal of Molecular Sciences, 22(15), 7768. https://doi.org/10.3390/ijms22157768