
Targeting Inflammation after Myocardial Infarction: A Therapeutic Opportunity for Extracellular Vesicles?



Abstract
:1. Introduction
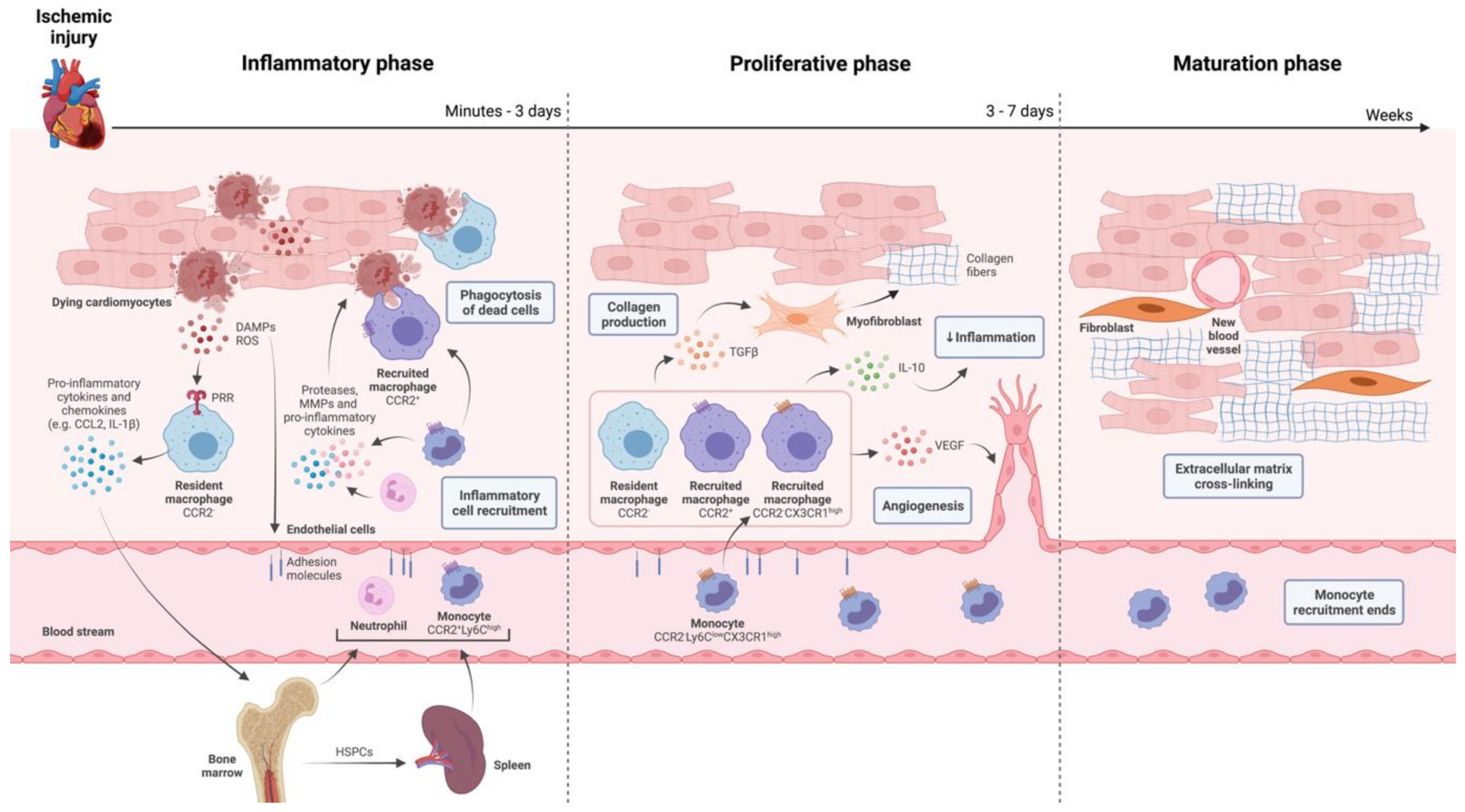
2. The Innate Immune System in Ischemic Injury and Repair of the Heart
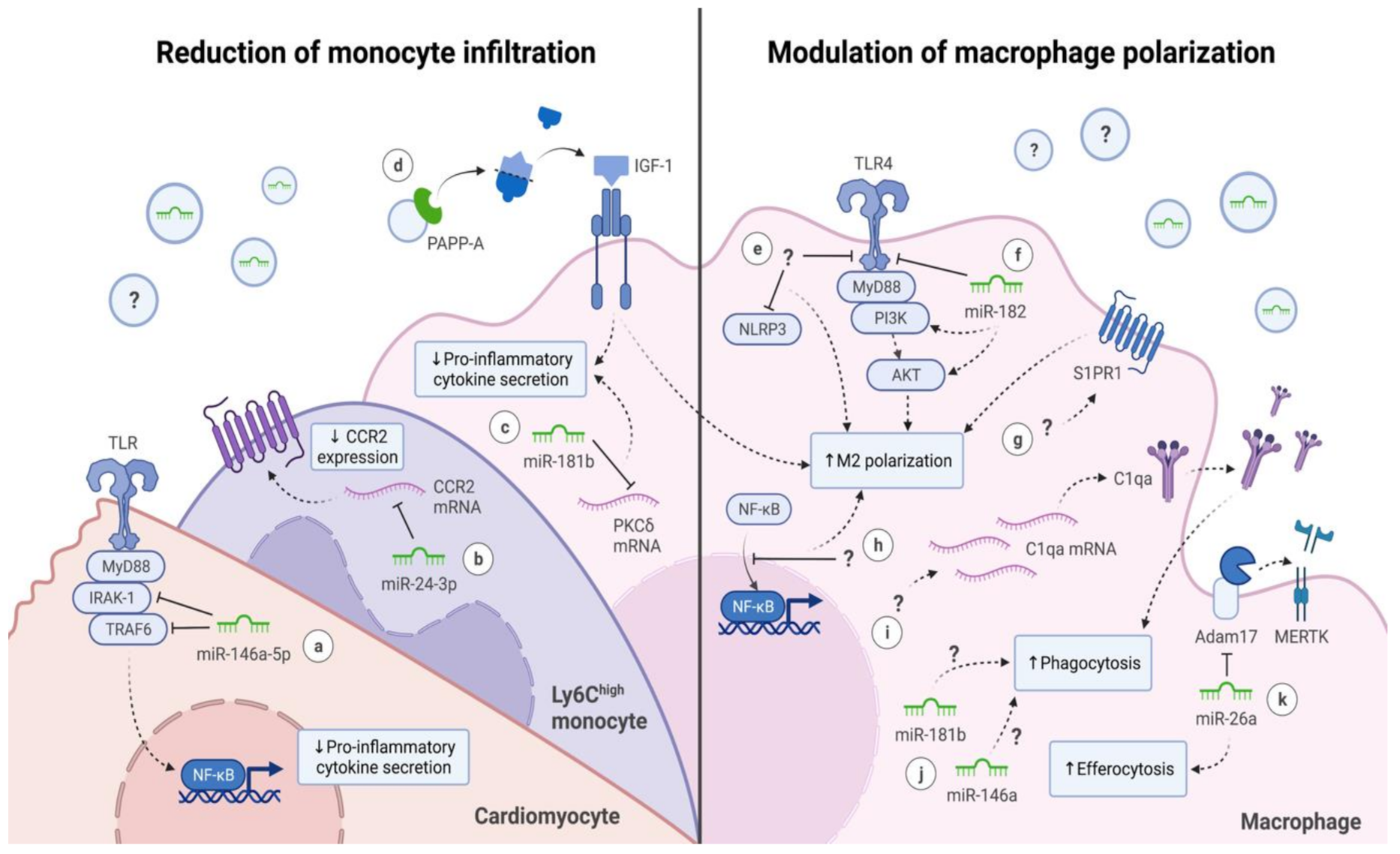
3. Extracellular Vesicles (EVs) and Their Interaction with Monocytes and Macrophages
3.1. Reduction of Monocyte Infiltration
3.2. Modulation of Macrophage Polarization
4. Improving EV-Based Therapies for Immunomodulation in MI Treatment
4.1. Modifying EV Cargo
4.2. Enhancing Cell-Specific Targeting
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update from the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Regulation of the Inflammatory Response in Cardiac Repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef]
- Ueland, T.; Gullestad, L.; Nymo, S.H.; Yndestad, A.; Aukrust, P.; Askevold, E.T. Inflammatory cytokines as biomarkers in heart failure. Clin. Chim. Acta 2015, 443, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Panahi, M.; Papanikolaou, A.; Torabi, A.; Zhang, J.-G.; Khan, H.; Vazir, A.; Hasham, M.; Cleland, J.G.F.; Rosenthal, N.; Harding, S.; et al. Immunomodulatory interventions in myocardial infarction and heart failure: A systematic review of clinical trials and meta-analysis of IL-1 inhibition. Cardiovasc. Res. 2018, 114, 1445–1461. [Google Scholar] [CrossRef]
- O’Gara, P.T.; Kushner, F.G.; Ascheim, D.D.; Casey, D.E.; Chung, M.K.; De Lemos, J.A.; Ettinger, S.M.; Fang, J.C.; Fesmire, F.M.; Franklin, B.A.; et al. 2013 ACCF/AHA Guideline for the Management of ST-Elevation Myocardial Infarction: Executive Summary. Circulation 2013, 127, 529–555. [Google Scholar] [CrossRef] [Green Version]
- Leuschner, F.; Panizzi, P.; Chico-Calero, I.; Lee, W.W.; Ueno, T.; Cortez-Retamozo, V.; Waterman, P.; Gorbatov, R.; Marinelli, B.; Iwamoto, Y.; et al. Angiotensin-Converting Enzyme Inhibition Prevents the Release of Monocytes from Their Splenic Reservoir in Mice With Myocardial Infarction. Circ. Res. 2010, 107, 1364–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grisanti, L.A.; De Lucia, C.; Thomas, T.P.; Stark, A.; Strony, J.T.; Myers, V.D.; Beretta, R.; Yu, D.; Sardu, C.; Marfella, R.; et al. Prior β-blocker treatment decreases leukocyte responsiveness to injury. JCI Insight 2019, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Cornel, J.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy with Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Achilli, F.; Pontone, G.; Bassetti, B.; Squadroni, L.; Campodonico, J.; Corrada, E.; Facchini, C.; Mircoli, L.; Esposito, G.; Scarpa, D.; et al. G-CSF for Extensive STEMI. Circ. Res. 2019, 125, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.M.; D’Amario, D.; Cannata, F.; Graziani, F.; Borovac, J.A.; Leone, G.; De Stefano, V.; Basile, E.; Siracusano, A.; Galiuto, L.; et al. The Effects of Granulocyte Colony-Stimulating Factor in Patients with a Large Anterior Wall Acute Myocardial Infarction to Prevent Left Ventricular Remodeling: A 10-Year Follow-Up of the RIGENERA Study. J. Clin. Med. 2020, 9, 1214. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; MacLellan, W.R. Stem cells and the heart—the road ahead. Science 2020, 367, 854–855. [Google Scholar] [CrossRef] [PubMed]
- Toma, C.; Pittenger, M.F.; Cahill, K.S.; Byrne, B.J.; Kessler, P.D. Human Mesenchymal Stem Cells Differentiate to a Cardiomyocyte Phenotype in the Adult Murine Heart. Circulation 2002, 105, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Messina, E.; De Angelis, L.; Frati, G.; Morrone, S.; Chimenti, S.; Fiordaliso, F.; Salio, M.; Battaglia, M.; Latronico, M.; Coletta, M.; et al. Isolation and Expansion of Adult Cardiac Stem Cells from Human and Murine Heart. Circ. Res. 2004, 95, 911–921. [Google Scholar] [CrossRef] [Green Version]
- Passier, R.; Van Laake, L.W.; Mummery, C.L. Stem-cell-based therapy and lessons from the heart. Nat. Cell Biol. 2008, 453, 322–329. [Google Scholar] [CrossRef]
- Van den Akker, F.; Feyen, D.A.; Van den Hoogen, P.; Van Laake, L.W.; Van Eeuwijk, E.C.; Hoefer, I.; Pasterkamp, G.; Chamuleau, S.A.; Grundeman, P.F.; Doevendans, P.A.; et al. Intramyocardial stem cell injection: Go(ne) with the flow. Eur. Heart J. 2016, 38, ehw056. [Google Scholar] [CrossRef] [Green Version]
- Wysoczynski, M.; Khan, A.; Bolli, R. New Paradigms in Cell Therapy. Circ. Res. 2018, 123, 138–158. [Google Scholar] [CrossRef]
- Gnecchi, M.; He, H.; Liang, O.D.; Melo, L.G.; Morello, F.; Mu, H.; Noiseux, N.; Zhang, L.; Pratt, R.E.; Ingwall, J.S.; et al. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat. Med. 2005, 11, 367–368. [Google Scholar] [CrossRef]
- Gnecchi, M.; He, H.; Noiseux, N.; Liang, O.D.; Zhang, L.; Morello, F.; Mu, H.; Melo, L.G.; Pratt, R.E.; Ingwall, J.S.; et al. Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. FASEB J. 2006, 20, 661–669. [Google Scholar] [CrossRef]
- Uemura, R.; Xu, M.; Ahmad, N.; Ashraf, M. Bone Marrow Stem Cells Prevent Left Ventricular Remodeling of Ischemic Heart Through Paracrine Signaling. Circ. Res. 2006, 98, 1414–1421. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Sluijter, J.P.G.; Davidson, S.M.; Boulanger, C.M.; Buzás, E.I.; De Kleijn, D.P.V.; Engel, F.B.; Giricz, Z.; Hausenloy, D.J.; Kishore, R.; Lecour, S.; et al. Extracellular vesicles in diagnostics and therapy of the ischaemic heart: Position Paper from the Working Group on Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 19–34. [Google Scholar] [CrossRef]
- Akker, F.V.D.; De Jager, S.C.A.; Sluijter, J.P.G. Mesenchymal Stem Cell Therapy for Cardiac Inflammation: Immunomodulatory Properties and the Influence of Toll-Like Receptors. Mediat. Inflamm. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Vagnozzi, R.J.; Maillet, M.; Sargent, M.A.; Khalil, H.; Johansen, A.K.Z.; Schwanekamp, J.A.; York, A.J.; Huang, V.; Nahrendorf, M.; Sadayappan, S.; et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nat. Cell Biol. 2020, 577, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Loyer, X.; Zlatanova, I.; Devue, C.; Yin, M.; Howangyin, K.-Y.; Klaihmon, P.; Guerin, C.L.; Kheloufi, M.; Vilar, J.; Zannis, K.; et al. Intra-Cardiac Release of Extracellular Vesicles Shapes Inflammation Following Myocardial Infarction. Circ. Res. 2018, 123, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Silvis, M.J.M.; Dengler, S.E.K.G.; Odille, C.A.; Mishra, M.; Van Der Kaaij, N.P.; Doevendans, P.A.; Sluijter, J.P.G.; De Kleijn, D.P.V.; De Jager, S.C.A.; Bosch, L.; et al. Damage-Associated Molecular Patterns in Myocardial Infarction and Heart Transplantation: The Road to Translational Success. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Gwechenberger, M.; Mendoza, L.H.; Youker, K.; Frangogiannis, N.; Smith, C.W.; Michael, L.H.; Entman, M.L. Cardiac Myocytes Produce Interleukin-6 in Culture and in Viable Border Zone of Reperfused Infarctions. Circulation 1999, 99, 546–551. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G.; Dewald, O.; Xia, Y.; Ren, G.; Haudek, S.; Leucker, T.; Kraemer, D.; Taffet, G.; Rollins, B.J.; Entman, M.L. Critical Role of Monocyte Chemoattractant Protein-1/CC Chemokine Ligand 2 in the Pathogenesis of Ischemic Cardiomyopathy. Circulation 2007, 115, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Cook-Mills, J.M.; Marchese, M.E.; Valencia, H.A. Vascular Cell Adhesion Molecule-1 Expression and Signaling During Disease: Regulation by Reactive Oxygen Species and Antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.-L.; Kohler, R.; Chudnovskiy, A.; Waterman, P.; et al. Identification of Splenic Reservoir Monocytes and Their Deployment to Inflammatory Sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuschner, F.; Rauch, P.; Ueno, T.; Gorbatov, R.; Marinelli, B.; Lee, W.W.; Dutta, P.; Wei, Y.; Robbins, C.; Iwamoto, Y.; et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 2012, 209, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Wan, E.; Yeap, X.Y.; Dehn, S.; Terry, R.L.; Novak, M.L.; Zhang, S.; Iwata, S.; Han, X.; Homma, S.; Drosatos, K.; et al. Enhanced Efferocytosis of Apoptotic Cardiomyocytes Through Myeloid-Epithelial-Reproductive Tyrosine Kinase Links Acute Inflammation Resolution to Cardiac Repair After Infarction. Circ. Res. 2013, 113, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- King, K.R.; Aguirre, A.D.; Ye, Y.-X.; Sun, Y.; Roh, J.D.; Ng, R.; Kohler, R.H.; Arlauckas, S.P.; Iwamoto, Y.; Savol, A.; et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017, 23, 1481–1487. [Google Scholar] [CrossRef]
- Forte, E.; Furtado, M.B.; Rosenthal, N. The interstitium in cardiac repair: Role of the immune–stromal cell interplay. Nat. Rev. Cardiol. 2018, 15, 601–616. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential Contribution of Monocytes to Heart Macrophages in Steady-State and After Myocardial Infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Nahrendorf, M.; Pittet, M.J.; Swirski, F. Monocytes: Protagonists of Infarct Inflammation and Repair after Myocardial Infarction. Circulation 2010, 121, 2437–2445. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.-L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [Green Version]
- Nahrendorf, M. Myeloid cell contributions to cardiovascular health and disease. Nat. Med. 2018, 24, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Tsujioka, H.; Imanishi, T.; Ikejima, H.; Kuroi, A.; Takarada, S.; Tanimoto, T.; Kitabata, H.; Okochi, K.; Arita, Y.; Ishibashi, K.; et al. Impact of Heterogeneity of Human Peripheral Blood Monocyte Subsets on Myocardial Salvage in Patients with Primary Acute Myocardial Infarction. J. Am. Coll. Cardiol. 2009, 54, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Van der Laan, A.M.; Hirsch, A.; Robbers, L.F.; Nijveldt, R.; Lommerse, I.; Delewi, R.; van der Vleuten, P.A.; Biemond, B.J.; Zwaginga, J.J.; van der Giessen, W.J.; et al. A proinflammatory monocyte response is associated with myocardial injury and impaired functional outcome in patients with ST-segment elevation myocardial infarction: Monocytes and myocardial infarction. Am. Heart J. 2012, 163, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Mounier, R.; Théret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Göransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. AMPKα1 Regulates Macrophage Skewing at the Time of Resolution of Inflammation during Skeletal Muscle Regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2016, 38, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurora, A.B.; Porrello, E.; Tan, W.; Mahmoud, A.I.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; Olson, E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 2014, 124, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Ben-Mordechai, T.; Holbova, R.; Landa-Rouben, N.; Harel-Adar, T.; Feinberg, M.S.; Abd-Elrahman, I.; Blum, G.; Epstein, F.H.; Silman, Z.; Cohen, S.; et al. Macrophage Subpopulations Are Essential for Infarct Repair with and Without Stem Cell Therapy. J. Am. Coll. Cardiol. 2013, 62, 1890–1901. [Google Scholar] [CrossRef] [Green Version]
- De Couto, G.; Liu, W.; Tseliou, E.; Sun, B.; Makkar, N.; Kanazawa, H.; Arditi, M.; Marbán, E. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J. Clin. Investig. 2015, 125, 3147–3162. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Zhu, Y.; Meng, X.; Zhang, C.; Yang, Y.; Bei, Y. Immunomodulation by Exosomes in Myocardial Infarction. J. Cardiovasc. Transl. Res. 2018, 12, 28–36. [Google Scholar] [CrossRef]
- Bebelman, M.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef]
- Witwer, K.W.; Théry, C. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J. Extracell. Vesicles 2019, 8, 1648167. [Google Scholar] [CrossRef]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.K.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.E.; Timmers, L.; Van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc. Res. 2014, 103, 530–541. [Google Scholar] [CrossRef]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.S.; Benedict, C.; et al. Embryonic Stem Cell-Derived Exosomes Promote Endogenous Repair Mechanisms and Enhance Cardiac Function Following Myocardial Infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doğan, A. Embryonic Stem Cells in Development and Regenerative Medicine; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 1–15. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Martin, B.J. Mesenchymal Stem Cells and Their Potential as Cardiac Therapeutics. Circ. Res. 2004, 95, 9–20. [Google Scholar] [CrossRef]
- Smits, A.M.; van Vliet, P.; Metz, C.H.; Korfage, T.; Sluijter, J.P.; Doevendans, P.A.; Goumans, M.-J. Human cardiomyocyte progenitor cells differentiate into functional mature cardiomyocytes: An in vitro model for studying human cardiac physiology and pathophysiology. Nat. Protoc. 2009, 4, 232–243. [Google Scholar] [CrossRef]
- Le, T.Y.L.; Chong, J. Cardiac progenitor cells for heart repair. Cell Death Discov. 2016, 2, 16052. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhu, J.; Zhang, C.; Wang, J.; Yue, F.; Jia, X.; Liu, H. Stem cell-derived extracellular vesicles for myocardial infarction: A meta-analysis of controlled animal studies. Aging 2019, 11, 1129–1150. [Google Scholar] [CrossRef]
- Vrijsen, K.R.; Maring, J.A.; Chamuleau, S.A.J.; Verhage, V.; Mol, E.A.; Deddens, J.C.; Metz, C.H.G.; Lodder, K.; Van Eeuwijk, E.C.M.; Van Dommelen, S.M.; et al. Exosomes from Cardiomyocyte Progenitor Cells and Mesenchymal Stem Cells Stimulate Angiogenesis Via EMMPRIN. Adv. Healthc. Mater. 2016, 5, 2555–2565. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.D.; French, K.; Ghosh-Choudhary, S.; Maxwell, J.T.; Brown, M.E.; Platt, M.O.; Searles, C.D.; Davis, M.E. Identification of Therapeutic Covariant MicroRNA Clusters in Hypoxia-Treated Cardiac Progenitor Cell Exosomes Using Systems Biology. Circ. Res. 2015, 116, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, B.; Bansal, S.S.; Ismahil, M.A.; Hamid, T.; Rokosh, G.; Mack, M.; Prabhu, S.D. CCR2+ Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl. Sci. 2018, 3, 230–244. [Google Scholar] [CrossRef]
- Lima Correa, B.; El Harane, N.; Gomez, I.; Rachid Hocine, H.; Vilar, J.; Desgres, M.; Bellamy, V.; Keirththana, K.; Guillas, C.; Perotto, M.; et al. Extracellular vesicles from human cardiovascular progenitors trigger a reparative immune response in infarcted hearts. Cardiovasc. Res. 2020, 117, 292–307. [Google Scholar] [CrossRef]
- De Abreu, R.C.; Fernandes, H.; Martins, P.A.D.C.; Sahoo, S.; Emanueli, C.; Ferreira, L. Native and bioengineered extracellular vesicles for cardiovascular therapeutics. Nat. Rev. Cardiol. 2020, 17, 685–697. [Google Scholar] [CrossRef]
- Di Gregoli, K.; Jenkins, N.; Salter, R.; White, S.; Newby, A.C.; Johnson, J.L. MicroRNA-24 Regulates Macrophage Behavior and Retards Atherosclerosis. Arter. Thromb. Vasc. Biol. 2014, 34, 1990–2000. [Google Scholar] [CrossRef] [Green Version]
- Maegdefessel, L.; Spin, J.M.; Raaz, U.; Eken, S.; Toh, R.; Azuma, J.; Adam, M.; Nagakami, F.; Heymann, H.M.; Chernogubova, E.; et al. miR-24 limits aortic vascular inflammation and murine abdominal aneurysm development. Nat. Commun. 2014, 5, 5214. [Google Scholar] [CrossRef] [Green Version]
- Qiao, S.; Zhang, W.; Yin, Y.; Wei, Z.; Chen, F.; Zhao, J.; Sun, X.; Mu, D.; Xie, J.; Xu, B. Extracellular vesicles derived from Krüppel-Like Factor 2-overexpressing endothelial cells attenuate myocardial ischemia-reperfusion injury by preventing Ly6Chigh monocyte recruitment. Theranostics 2020, 10, 11562–11579. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Zhang, Y.; Lan, B.; Wang, J.; Zhang, Z.; Zhang, L.; Xiao, P.; Meng, Q.; Geng, Y.-J.; Yu, X.-Y.; et al. MiRNA-Sequence Indicates That Mesenchymal Stem Cells and Exosomes Have Similar Mechanism to Enhance Cardiac Repair. BioMed Res. Int. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Milano, G.; Biemmi, V.; Lazzarini, E.; Balbi, C.; Ciullo, A.; Bolis, S.; Ameri, P.; Di Silvestre, D.; Mauri, P.; Barile, L.; et al. Intravenous administration of cardiac progenitor cell-derived exosomes protects against doxorubicin/trastuzumab-induced cardiac toxicity. Cardiovasc. Res. 2020, 116, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-F.; Shu, R.; Jiang, S.-Y.; Liu, D.-L.; Ni, J.; Zhang, X.-L. MicroRNA-146 inhibits pro-inflammatory cytokine secretion through IL-1 receptor-associated kinase 1 in human gingival fibroblasts. J. Inflamm. 2013, 10, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Couto, G.; Gallet, R.; Cambier, L.; Jaghatspanyan, E.; Makkar, N.; Dawkins, J.F.; Berman, B.P.; Marbán, E. Exosomal MicroRNA Transfer into Macrophages Mediates Cellular Postconditioning. Circulation 2017, 136, 200–214. [Google Scholar] [CrossRef]
- Sun, X.; Icli, B.; Wara, A.K.; Belkin, N.; He, S.; Kobzik, L.; Hunninghake, G.M.; Vera, M.P.; Blackwell, T.S.; Baron, R.M.; et al. MicroRNA-181b regulates NF-κB–mediated vascular inflammation. J. Clin. Investig. 2012, 122, 1973–1990. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Cervio, E.; Lionetti, V.; Milano, G.; Ciullo, A.; Biemmi, V.; Bolis, S.; Altomare, C.; Matteucci, M.; Di Silvestre, D.; et al. Cardioprotection by cardiac progenitor cell-secreted exosomes: Role of pregnancy-associated plasma protein-A. Cardiovasc. Res. 2018, 114, 992–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, M.P.; Lexow, J.; Borsellino, G.; Slonimski, E.; Zarrinpashneh, E.; Poggioli, T.; Rosenthal, N. IGF-1Ea induces vessel formation after injury and mediates bone marrow and heart cross-talk through the expression of specific cytokines. Biochem. Biophys. Res. Commun. 2011, 410, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Tonkin, J.; Temmerman, L.; Sampson, R.D.; Gallego-Colon, E.; Barberi, L.; Bilbao, D.; Schneider, M.D.; Musarò, A.; Rosenthal, N. Monocyte/Macrophage-derived IGF-1 Orchestrates Murine Skeletal Muscle Regeneration and Modulates Autocrine Polarization. Mol. Ther. 2015, 23, 1189–1200. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Colon, E.; Sampson, R.; Sattler, S.; Schneider, M.D.; Rosenthal, N.; Tonkin, J. Cardiac-Restricted IGF-1Ea Overexpression Reduces the Early Accumulation of Inflammatory Myeloid Cells and Mediates Expression of Extracellular Matrix Remodelling Genes after Myocardial Infarction. Mediat. Inflamm. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Santini, M.P.; Tsao, L.; Monassier, L.; Theodoropoulos, C.; Carter, J.; Lara-Pezzi, E.; Slonimsky, E.; Salimova, E.; Delafontaine, P.; Song, Y.-H.; et al. Enhancing Repair of the Mammalian Heart. Circ. Res. 2007, 100, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Sager, H.B.; Hulsmans, M.; Lavine, K.J.; Moreira, M.B.; Heidt, T.; Courties, G.; Sun, Y.; Iwamoto, Y.; Tricot, B.; Khan, O.; et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ. Res. 2016, 119, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla, D.; Johnson, T.; Dargani, Z.T. Exosome Treatment Enhances Anti-Inflammatory M2 Macrophages and Reduces Inflammation-Induced Pyroptosis in Doxorubicin-Induced Cardiomyopathy. Cells 2019, 8, 1224. [Google Scholar] [CrossRef] [Green Version]
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef]
- Kuriakose, S.; Onyilagha, C.; Singh, R.; Olayinka-Adefemi, F.; Jia, P.; Uzonna, J.E. TLR-2 and MyD88-Dependent Activation of MAPK and STAT Proteins Regulates Proinflammatory Cytokine Response and Immunity to Experimental Trypanosoma congolense Infection. Front. Immunol. 2019, 10, 2673. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Shan, A.; Wei, Z.; Xu, B. Intravenous mesenchymal stem cell-derived exosomes ameliorate myocardial inflammation in the dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2018, 503, 2611–2618. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhang, F.; Chai, R.; Zhou, W.; Hu, M.; Liu, B.; Chen, X.; Liu, M.; Xu, Q.; Liu, N.; et al. Exosomes derived from pro-inflammatory bone marrow-derived mesenchymal stem cells reduce inflammation and myocardial injury via mediating macrophage polarization. J. Cell. Mol. Med. 2019, 23, 7617–7631. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Zhou, X.; Ge, Z.; Song, Y.; Wang, H.; Liu, X.; Zhang, D. Exosomes from adipose-derived mesenchymal stem cells ameliorate cardiac damage after myocardial infarction by activating S1P/SK1/S1PR1 signaling and promoting macrophage M2 polarization. Int. J. Biochem. Cell Biol. 2019, 114, 105564. [Google Scholar] [CrossRef]
- Zhao, J.; Li, X.; Hu, J.; Chen, F.; Qiao, S.; Sun, X.; Gao, L.; Xie, J.; Xu, B. Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovasc. Res. 2019, 115, 1205–1216. [Google Scholar] [CrossRef] [Green Version]
- Mentkowski, K.; Mursleen, A.; Snitzer, J.D.; Euscher, L.M.; Lang, J.K. CDC-derived extracellular vesicles reprogram inflammatory macrophages to an arginase 1-dependent proangiogenic phenotype. Am. J. Physiol. Circ. Physiol. 2020, 318, H1447–H1460. [Google Scholar] [CrossRef] [PubMed]
- De Couto, G.; Jaghatspanyan, E.; Deberge, M.; Liu, W.; Luther, K.; Wang, Y.; Tang, J.; Thorp, E.B.; Marbán, E. Mechanism of Enhanced MerTK-Dependent Macrophage Efferocytosis by Extracellular Vesicles. Arter. Thromb. Vasc. Biol. 2019, 39, 2082–2096. [Google Scholar] [CrossRef] [PubMed]
- Cambier, L.; De Couto, G.; Ibrahim, A.; Echavez, A.K.; Valle, J.; Liu, W.; Kreke, M.; Smith, R.R.; Marbán, L.; Marbán, E. Y RNA fragment in extracellular vesicles confers cardioprotection via modulation of IL-10 expression and secretion. EMBO Mol. Med. 2017, 9, 337–352. [Google Scholar] [CrossRef]
- López, E.; Blázquez, R.; Marinaro, F.; Álvarez, V.; Blanco, V.; Báez, C.; González, I.; Abad, A.; Moreno, B.; Sánchez-Margallo, F.M.; et al. The Intrapericardial Delivery of Extracellular Vesicles from Cardiosphere-Derived Cells Stimulates M2 Polarization during the Acute Phase of Porcine Myocardial Infarction. Stem Cell Rev. Rep. 2019, 16, 612–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Sicco, C.; Reverberi, D.; Balbi, C.; Ulivi, V.; Principi, E.; Pascucci, L.; Becherini, P.; Bosco, M.C.; Varesio, L.; Franzin, C.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Mediators of Anti-Inflammatory Effects: Endorsement of Macrophage Polarization. Stem Cells Transl. Med. 2017, 6, 1018–1028. [Google Scholar] [CrossRef]
- Qin, S.; Peng, D.; Lu, J.; Ke, Z. MiR-182-5p inhibited oxidative stress and apoptosis triggered by oxidized low-density lipoprotein via targeting toll-like receptor 4. J. Cell. Physiol. 2018, 233, 6630–6637. [Google Scholar] [CrossRef]
- Sivaraman, V.; Hausenloy, D.J.; Kolvekar, S.; Hayward, M.; Yap, J.; Lawrence, D.; Di Salvo, C.; Yellon, D.M. The divergent roles of protein kinase C epsilon and delta in simulated ischaemia–reperfusion injury in human myocardium. J. Mol. Cell. Cardiol. 2009, 46, 758–764. [Google Scholar] [CrossRef]
- Murphy, D.E.; De Jong, O.G.; Brouwer, M.; Wood, M.J.; Lavieu, G.; Schiffelers, R.M.; Vader, P. Extracellular vesicle-based therapeutics: Natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Nazari-Shafti, T.Z.; Stamm, C.; Falk, V.; Emmert, M.Y. Exosomes for Cardioprotection: Are We Ready for Clinical Translation? Eur. Heart J. 2019, 40, 953–956. [Google Scholar] [CrossRef]
- Kennedy, T.L.; Russell, A.J.; Riley, P. Experimental limitations of extracellular vesicle-based therapies for the treatment of myocardial infarction. Trends Cardiovasc. Med. 2020. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Kowal, J.; Zucchetti, A.E.; Enserink, L.; Jouve, M.; Lankar, D.; Saitakis, M.; Martin-Jaular, L.; Théry, C. Qualitative differences in T-cell activation by dendritic cell-derived extracellular vesicle subtypes. EMBO J. 2017, 36, 3012–3028. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willms, E.; Johansson, H.J.; Mäger, I.; Lee, Y.; Blomberg, K.E.M.; Sadik, M.; Alaarg, A.; Smith, C.I.E.; Lehtiö, J.; El Andaloussi, S.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef] [PubMed]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.; Hughes, S.; Cheng, H.H.; Arroyo, J.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef] [Green Version]
- Roefs, M.T.; Sluijter, J.P.; Vader, P. Extracellular Vesicle-Associated Proteins in Tissue Repair. Trends Cell Biol. 2020, 30, 990–1013. [Google Scholar] [CrossRef]
- Hayashidani, S.; Tsutsui, H.; Shiomi, T.; Ikeuchi, M.; Matsusaka, H.; Suematsu, N.; Wen, J.; Egashira, K.; Takeshita, A.; Jensen, L.O.; et al. Anti-Monocyte Chemoattractant Protein-1 Gene Therapy Attenuates Left Ventricular Remodeling and Failure After Experimental Myocardial Infarction. Circulation 2003, 108, 2134–2140. [Google Scholar] [CrossRef] [Green Version]
- Majmudar, M.D.; Keliher, E.J.; Heidt, T.; Leuschner, F.; Truelove, J.; Sena, B.F.; Gorbatov, R.; Iwamoto, Y.; Dutta, P.; Wojtkiewicz, G.; et al. Monocyte-Directed RNAi Targeting CCR2 Improves Infarct Healing in Atherosclerosis-Prone Mice. Circulation 2013, 127, 2038–2046. [Google Scholar] [CrossRef] [Green Version]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The Macrophage in Cardiac Homeostasis and Disease. J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef]
- Shiraishi, M.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 2016, 126, 2151–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilgendorf, I.; Gerhardt, L.M.S.; Tan, T.C.; Winter, C.; Holderried, T.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; et al. Ly-6C high Monocytes Depend on Nr4a1 to Balance Both Inflammatory and Reparative Phases in the Infarcted Myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courties, G.; Heidt, T.; Sebas, M.; Iwamoto, Y.; Jeon, D.; Truelove, J.; Tricot, B.; Wojtkiewicz, G.; Dutta, P.; Sager, H.B.; et al. In Vivo Silencing of the Transcription Factor IRF5 Reprograms the Macrophage Phenotype and Improves Infarct Healing. J. Am. Coll. Cardiol. 2014, 63, 1556–1566. [Google Scholar] [CrossRef]
- Rhee, A.J.; LaVine, K.J. New Approaches to Target Inflammation in Heart Failure: Harnessing Insights from Studies of Immune Cell Diversity. Annu. Rev. Physiol. 2020, 82, 1–20. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.; Melo, S.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nat. Cell Biol. 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Takahashi, Y.; Nishikawa, M.; Sano, K.; Morishita, M.; Charoenviriyakul, C.; Saji, H.; Takakura, Y. Role of Phosphatidylserine-Derived Negative Surface Charges in the Recognition and Uptake of Intravenously Injected B16BL6-Derived Exosomes by Macrophages. J. Pharm. Sci. 2017, 106, 168–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; García, E.D.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109. [Google Scholar] [CrossRef]
- Richter, M.; Vader, P.; Fuhrmann, G. Approaches to surface engineering of extracellular vesicles. Adv. Drug Deliv. Rev. 2021, 173, 416–426. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Gitz-Francois, J.J.J.M.; Schiffelers, R.M.; Vader, P. Recombinant phosphatidylserine-binding nanobodies for targeting of extracellular vesicles to tumor cells: A plug-and-play approach. Nanoscale 2018, 10, 2413–2426. [Google Scholar] [CrossRef] [Green Version]
- Dooley, K.; McConnell, R.E.; Xu, K.; Lewis, N.D.; Haupt, S.; Youniss, M.R.; Martin, S.; Sia, C.L.; McCoy, C.; Moniz, R.J.; et al. A versatile platform for generating engineered extracellular vesicles with defined therapeutic properties. Mol. Ther. 2021, 29, 1729–1743. [Google Scholar] [CrossRef]
- Piffoux, M.; Silva, A.K.A.; Wilhelm, C.; Gazeau, F.; Tareste, D. Modification of Extracellular Vesicles by Fusion with Liposomes for the Design of Personalized Biogenic Drug Delivery Systems. ACS Nano 2018, 12, 6830–6842. [Google Scholar] [CrossRef]
- Zhang, N.; Song, Y.; Huang, Z.; Chen, J.; Tan, H.; Yang, H.; Fan, M.; Li, Q.; Wang, Q.; Gao, J.; et al. Monocyte mimics improve mesenchymal stem cell-derived extracellular vesicle homing in a mouse MI/RI model. Biomaterials 2020, 255, 120168. [Google Scholar] [CrossRef]
- Nakase, I.; Futaki, S. Combined treatment with a pH-sensitive fusogenic peptide and cationic lipids achieves enhanced cytosolic delivery of exosomes. Sci. Rep. 2015, 5, 10112. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| EV Source | Experimental Model | EV Administration | EV Isolation Method | Functional EV Content | Molecular Mechanism | Biological Effect | REF |
|---|---|---|---|---|---|---|---|
| Reduction of monocyte infiltration | |||||||
| Human ESC-derived MSCs | MI mouse model (I/R) | Intravenous; 5 min before reperfusion | SEC | Unknown | Unknown | Reduced neutrophil and macrophage infiltration in the hearts and WBC count. | [51] |
| Rat bone marrow MSCs | MI rat model (PL) | Intramyocardial (2 different sites); immediately after ligation | Precipitation | miR-24-3p | Unknown | Decreased of CD68+ macrophages in the peri-infarct zone. | [67] |
| Human CDCs | MI rat and pig model (I/R) | Intramyocardial (10 sequential points in pig); 20 min after reperfusion in rat/30 min after reperfusion in pig | Ultrafiltration and PEG precipitation | miR-181b | Downregulation of protein kinase C δ | Reduced of CD68+ macrophages within infarcted tissue and increased phagocytosis capacity of macrophages. | [70] |
| Human CPCs | MI rat model (PL) | Intramyocardial (3 different sites); 60 min after ligation | UC | PAPP-A | Unknown | Decreased CD68+ macrophages within infarcted tissue. | [72] |
| Human iPSC-derived CPCs | MI mouse model (PL) | Transcutaneous (three peri-infarcted areas); 2 days (acute) or 3 weeks (chronic) after PL | UC | Unknown | Unknown | Decreased Ly6Chigh monocytes in the heart and levels of pro-inflammatory cytokines. | [62] |
| Human CPCs | Dox/Trz-induced cardiotoxicity rat model | Intravenous; Days 5, 11, and 19 | UC | miR-146a-5p | Inhibition of Traf6 and Irak1 | Reduced CD68+ macrophages infiltrates in the heart. | [68] |
| Human and mouse KLF2-overexpressing endothelial cells | MI mouse model (I/R) | Intravenous; immediately after reperfusion | UC | miR-24-3p | CCL2/CCR2 axis | Inhibited Ly6Chigh monocytes recruitment from bone marrow by inhibiting CCR2 expression. | [66] |
| Modulation of macrophage polarization | |||||||
| Mouse ESCs | Dox-induced cardiotoxicity mouse model | Intraperitoneal (3 injections in 3 different days between Dox treatment) | Precipitation (Exoquick TC) | Unknown | Inhibition of MyD88 /P38/JNK and NLRP3 pathway | Increased M2 macrophages and anti-inflammatory cytokine IL-10. | [81] |
| Mouse bone marrow MSCs | MI mouse model (I/R) | Intramyocardial (3 different sites); immediately after reperfusion | UC | miR-182 | Inhibition of TLR4/NF-κB pathway and activation of PI3K/AKT pathway | Promoted M2 polarization in macrophages. | [87] |
| Rat adipose tissue MSCs | MI rat model (PL) | Intravenous; 60 min after ligation | UC | Unknown | Activation of S1P/SK1/S1PR1 signaling | Promoted M2 polarization in macrophages. | [86] |
| Rat bone marrow MSCs | MI mouse model (PL) | Intramyocardial (4 different sites); immediately after ligation | Density-gradient UC | Unknown | Inhibition of nuclear translocation of NF-κB p65 and activation of phosphorylation of AKT1 and AKT2 | Decreased the production of pro-inflammatory cytokines and increased M2 polarization in macrophages. | [85] |
| Mouse bone marrow MSCs | Dox-induced dilated cardiomyopathy mouse model | Intravenous; 7 days after Dox treatment | UC | Unknown | Activation JAK2-STAT6 pathway | Decreased circulating pro-inflammatory cytokines and M1 macrophages in the heart, while increasing M2 macrophages. | [84] |
| Human CDCs | MI rat model (I/R) | Intramyocardial; 10 min after reperfusion | Ultrafiltration | Y RNA fragment | Unknown | Increased IL-10 secretion in macrophages. | [90] |
| Human CDCs | MI rat and mouse model (I/R) | Intramyocardial (3 different sites); 20 min after reperfusion | Ultrafiltration | miR-26a | Suppression of Adam17 and upregulation of C1qa | Inducted of C1qa and MerTK expression in macrophages, which enhances phagocytosis and efferocytosis. | [89] |
| Pig CDCs | MI pig model (I/R) | Intrapericardially; 3 days after MI | Ultrafiltration | Unknown | Unknown | Increased circulation of M2 monocytes. | [91] |
| Human CDCs | In vitro | NA | Precipitation (ExoQuick-TC) or ultrafiltration | miR-146a | Unknown | Increased phagocytosis in macrophages. | [88] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viola, M.; de Jager, S.C.A.; Sluijter, J.P.G. Targeting Inflammation after Myocardial Infarction: A Therapeutic Opportunity for Extracellular Vesicles? Int. J. Mol. Sci. 2021, 22, 7831. https://doi.org/10.3390/ijms22157831
Viola M, de Jager SCA, Sluijter JPG. Targeting Inflammation after Myocardial Infarction: A Therapeutic Opportunity for Extracellular Vesicles? International Journal of Molecular Sciences. 2021; 22(15):7831. https://doi.org/10.3390/ijms22157831
Chicago/Turabian StyleViola, Margarida, Saskia C. A. de Jager, and Joost P. G. Sluijter. 2021. "Targeting Inflammation after Myocardial Infarction: A Therapeutic Opportunity for Extracellular Vesicles?" International Journal of Molecular Sciences 22, no. 15: 7831. https://doi.org/10.3390/ijms22157831
APA StyleViola, M., de Jager, S. C. A., & Sluijter, J. P. G. (2021). Targeting Inflammation after Myocardial Infarction: A Therapeutic Opportunity for Extracellular Vesicles? International Journal of Molecular Sciences, 22(15), 7831. https://doi.org/10.3390/ijms22157831