
TGF-Beta as a Master Regulator of Diabetic Nephropathy



Abstract
:1. Introduction
2. TGF-β Signaling
3. Activation of TGF-β Signaling in DN
4. Diverse Role of TGF-β/Smad Signaling in DN
4.1. Active Versus Latent TGF-β1 in DN
4.2. TGF-β Receptors
4.3. Smad3 vs. Smad2
4.4. Smad4
4.5. Smad7
5. Role of TGF-β/Smad3-Dependent miRNAs and Long Non-Coding RNAs in DN
5.1. miRNAs
5.2. lncRNAs
6. Treatment of DN by Targeting TGF-β Signaling
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chang, A.S.; Hathaway, C.K.; Smithies, O.; Kakoki, M. Transforming growth factor-beta1 and diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2016, 310, F689–F696. [Google Scholar] [CrossRef] [Green Version]
- Bank, N. Mechanisms of diabetic hyperfiltration. Kidney Int. 1991, 40, 792–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Zuydam, N.R.; Ahlqvist, E.; Sandholm, N.; Deshmukh, H.; Rayner, N.W.; Abdalla, M.; Ladenvall, C.; Ziemek, D.; Fauman, E.; Robertson, N.R.; et al. A Genome-Wide Association Study of Diabetic Kidney Disease in Subjects with Type 2 Diabetes. Diabetes 2018, 67, 1414–1427. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Scott, W.R.; Lympany, P.A.; Rippin, J.D.; Gill, G.V.; Barnett, A.H.; Bain, S.C.; the Warren 3/UK GoKind Study Group. The TGF-beta 1 gene codon 10 polymorphism contributes to the genetic predisposition to nephropathy in Type 1 diabetes. Diabet. Med. 2005, 22, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Hansen, P.M.; Karlsen, A.E.; Langdahl, B.L.; Johannesen, J.; Nerup, J. TGF-beta1 gene mutations in insulin-dependent diabetes mellitus and diabetic nephropathy. J. Am. Soc. Nephrol. 1998, 9, 2302–2307. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y.; Poon, P.; Chow, K.M.; Szeto, C.C.; Cheung, M.K.; Li, P.K. Association of transforming growth factor-beta (TGF-beta) T869C (Leu 10Pro) gene polymorphisms with type 2 diabetic nephropathy in Chinese. Kidney Int. 2003, 63, 1831–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, M.R.; El-Gamel, A.; Hasleton, P.; Turner, D.M.; Sinnott, P.J.; Hutchinson, I.V. Genotypic variation in the transforming growth factor-beta1 gene: Association with transforming growth factor-beta1 production, fibrotic lung disease, and graft fibrosis after lung transplantation. Transplantation 1998, 66, 1014–1020. [Google Scholar] [CrossRef]
- Ng, D.P.; Warram, J.H.; Krolewski, A.S. TGF-beta 1 as a genetic susceptibility locus for advanced diabetic nephropathy in type 1 diabetes mellitus: An investigation of multiple known DNA sequence variants. Am. J. Kidney Dis. 2003, 41, 22–28. [Google Scholar] [CrossRef] [PubMed]
- McKnight, A.J.; Savage, D.A.; Patterson, C.C.; Sadlier, D.; Maxwell, A.P. Resequencing of genes for transforming growth factor beta1 (TGFB1) type 1 and 2 receptors (TGFBR1, TGFBR2), and association analysis of variants with diabetic nephropathy. BMC Med. Genet. 2007, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Jakus, V.; Sapak, M.; Kostolanska, J. Circulating TGF-beta1, glycation, and oxidation in children with diabetes mellitus type 1. Exp Diabetes Res 2012, 2012, 510902. [Google Scholar] [CrossRef] [Green Version]
- Huseynova, G.R.; Azizova, G.I.; Efendiyev, A.M. Quantitative changes in serum IL-8, TNF-α and TGF-β1 levels depending on compensation stage in type 2 diabetic patients. Int. J. Diabetes Metab. 2009, 17, 59–62. [Google Scholar] [CrossRef]
- Yadav, H.; Quijano, C.; Kamaraju, A.K.; Gavrilova, O.; Malek, R.; Chen, W.; Zerfas, P.; Zhigang, D.; Wright, E.C.; Stuelten, C.; et al. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metab. 2011, 14, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.; Rashed, L. Estimation of transforming growth factor-beta 1 as a marker of renal injury in type II diabetes mellitus. Saudi Med. J. 2007, 28, 519–523. [Google Scholar] [PubMed]
- Shaker, Y.M.; Soliman, H.A.; Ezzat, E.; Hussein, N.S.; Ashour, E.; Donia, A.; Eweida, S.M. Serum and urinary transforming growth factor beta 1 as biochemical markers in diabetic nephropathy patients. Beni-Suef Univ. J. Basic Appl. Sci. 2014, 3, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Hathaway, C.K.; Gasim, A.M.; Grant, R.; Chang, A.S.; Kim, H.S.; Madden, V.J.; Bagnell, C.R., Jr.; Jennette, J.C.; Smithies, O.; Kakoki, M. Low TGFbeta1 expression prevents and high expression exacerbates diabetic nephropathy in mice. Proc. Natl. Acad. Sci. USA 2015, 112, 5815–5820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, A.; Attisano, L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-beta/Smad signaling in renal fibrosis. Front Physiol. 2015, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef] [Green Version]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Wang, W.; Huang, X.R.; Li, A.G.; Liu, F.; Li, J.H.; Truong, L.D.; Wang, X.J.; Lan, H.Y. Signaling mechanism of TGF-beta1 in prevention of renal inflammation: Role of Smad7. J. Am. Soc. Nephrol. 2005, 16, 1371–1383. [Google Scholar] [CrossRef]
- Lopez-Casillas, F.; Wrana, J.L.; Massague, J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell 1993, 73, 1435–1444. [Google Scholar] [CrossRef]
- Bernabeu, C.; Lopez-Novoa, J.M.; Quintanilla, M. The emerging role of TGF-beta superfamily coreceptors in cancer. Biochim. Biophys. Acta 2009, 1792, 954–973. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y.; Chung, A.C.K. Transforming growth factor-beta and Smads. Contrib. Nephrol. 2011, 170, 75–82. [Google Scholar] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Choi, M.E. TGF-beta-activated kinase-1: New insights into the mechanism of TGF-beta signaling and kidney disease. Kidney Res. Clin. Pract. 2012, 31, 94–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.R.; Zhang, Y.; Wrana, J.L. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y. Transforming growth factor-beta/Smad signalling in diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 2012, 39, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Pankewycz, O.G.; Guan, J.X.; Bolton, W.K.; Gomez, A.; Benedict, J.F. Renal TGF-beta regulation in spontaneously diabetic NOD mice with correlations in mesangial cells. Kidney Int. 1994, 46, 748–758. [Google Scholar] [CrossRef] [Green Version]
- Isono, M.; Mogyorosi, A.; Han, D.C.; Hoffman, B.B.; Ziyadeh, F.N. Stimulation of TGF-beta type II receptor by high glucose in mouse mesangial cells and in diabetic kidney. Am. J. Physiol. Renal. Physiol. 2000, 278, F830–F838. [Google Scholar] [CrossRef]
- Hong, S.W.; Isono, M.; Chen, S.; Iglesias-De La Cruz, M.C.; Han, D.C.; Ziyadeh, F.N. Increased glomerular and tubular expression of transforming growth factor-beta1, its type II receptor, and activation of the Smad signaling pathway in the db/db mouse. Am. J. Pathol. 2001, 158, 1653–1663. [Google Scholar] [CrossRef]
- Isono, M.; Chen, S.; Hong, S.W.; Iglesias-de la Cruz, M.C.; Ziyadeh, F.N. Smad pathway is activated in the diabetic mouse kidney and Smad3 mediates TGF-beta-induced fibronectin in mesangial cells. Biochem. Biophys. Res. Commun. 2002, 296, 1356–1365. [Google Scholar] [CrossRef]
- Wolf, G.; Sharma, K.; Chen, Y.; Ericksen, M.; Ziyadeh, F.N. High glucose-induced proliferation in mesangial cells is reversed by autocrine TGF-beta. Kidney Int. 1992, 42, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, B.B.; Sharma, K.; Zhu, Y.; Ziyadeh, F.N. Transcriptional activation of transforming growth factor-beta1 in mesangial cell culture by high glucose concentration. Kidney Int. 1998, 54, 1107–1116. [Google Scholar] [CrossRef] [Green Version]
- Han, D.C.; Isono, M.; Hoffman, B.B.; Ziyadeh, F.N. High glucose stimulates proliferation and collagen type I synthesis in renal cortical fibroblasts: Mediation by autocrine activation of TGF-beta. J. Am. Soc. Nephrol. 1999, 10, 1891–1899. [Google Scholar] [CrossRef]
- Rocco, M.V.; Chen, Y.; Goldfarb, S.; Ziyadeh, F.N. Elevated glucose stimulates TGF-beta gene expression and bioactivity in proximal tubule. Kidney Int. 1992, 41, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Skorczewski, J.; Feng, X.; Mei, L.; Murphy-Ullrich, J.E. Glucose up-regulates thrombospondin 1 gene transcription and transforming growth factor-beta activity through antagonism of cGMP-dependent protein kinase repression via upstream stimulatory factor 2. J. Biol. Chem. 2004, 279, 34311–34322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy-Ullrich, J.E.; Suto, M.J. Thrombospondin-1 regulation of latent TGF-beta activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018, 68–69, 28–43. [Google Scholar] [CrossRef]
- Tan, A.L.; Forbes, J.M.; Cooper, M.E. AGE, RAGE, and ROS in diabetic nephropathy. Semin. Nephrol. 2007, 27, 130–143. [Google Scholar] [CrossRef]
- Li, J.H.; Huang, X.R.; Zhu, H.J.; Oldfield, M.; Cooper, M.; Truong, L.D.; Johnson, R.J.; Lan, H.Y. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: Implications for diabetic renal and vascular disease. FASEB J. 2004, 18, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Soulis, T.; Thallas, V.; Panagiotopoulos, S.; Long, D.M.; Vasan, S.; Wagle, D.; Jerums, G.; Cooper, M.E. Renoprotective effects of a novel inhibitor of advanced glycation. Diabetologia 2001, 44, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Chung, A.C.; Zhang, H.; Kong, Y.Z.; Tan, J.J.; Huang, X.R.; Kopp, J.B.; Lan, H.Y. Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J. Am. Soc. Nephrol. 2010, 21, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Vita, J.; Sanchez-Lopez, E.; Esteban, V.; Ruperez, M.; Egido, J.; Ruiz-Ortega, M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 2005, 111, 2509–2517. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Huang, X.R.; Canlas, E.; Oka, K.; Truong, L.D.; Deng, C.; Bhowmick, N.A.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 2006, 98, 1032–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Chung, A.C.; Huang, X.R.; Lan, H.Y. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: The role of Smad3. Hypertension 2009, 54, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigert, C.; Brodbeck, K.; Klopfer, K.; Haring, H.U.; Schleicher, E.D. Angiotensin II induces human TGF-beta 1 promoter activation: Similarity to hyperglycaemia. Diabetologia 2002, 45, 890–898. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, G.H.; Pratt, R.E.; Dzau, V.J. Vascular smooth muscle cell hypertrophy vs. hyperplasia. Autocrine transforming growth factor-beta 1 expression determines growth response to angiotensin II. J. Clin. Investig. 1992, 90, 456–461. [Google Scholar] [CrossRef]
- Rosario, R.F.; Prabhakar, S. Lipids and diabetic nephropathy. Curr. Diab. Rep. 2006, 6, 455–462. [Google Scholar] [CrossRef]
- Su, Y.; Chen, Q.; Ju, Y.; Li, W.; Li, W. Palmitate induces human glomerular mesangial cells fibrosis through CD36-mediated transient receptor potential canonical channel 6/nuclear factor of activated T cell 2 activation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158793. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.B.; Yu, M.R.; Yang, Y.; Jiang, Z.; Ha, H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14, S241–S245. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Ramos, M.; Mora, I.; de Frutos, S.; Garesse, R.; Rodriguez-Puyol, M.; Olmos, G.; Rodriguez-Puyol, D. Intracellular redox equilibrium is essential for the constitutive expression of AP-1 dependent genes in resting cells: Studies on TGF-beta1 regulation. Int. J. Biochem. Cell Biol. 2012, 44, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Kopp, J.B.; Factor, V.M.; Mozes, M.; Nagy, P.; Sanderson, N.; Bottinger, E.P.; Klotman, P.E.; Thorgeirsson, S.S. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab. Investig. 1996, 74, 991–1003. [Google Scholar] [PubMed]
- Krag, S.; Osterby, R.; Chai, Q.; Nielsen, C.B.; Hermans, C.; Wogensen, L. TGF-beta1-induced glomerular disorder is associated with impaired concentrating ability mimicking primary glomerular disease with renal failure in man. Lab. Investig. 2000, 80, 1855–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wogensen, L.; Nielsen, C.B.; Hjorth, P.; Rasmussen, L.M.; Nielsen, A.H.; Gross, K.; Sarvetnick, N.; Ledet, T. Under control of the Ren-1c promoter, locally produced transforming growth factor-beta1 induces accumulation of glomerular extracellular matrix in transgenic mice. Diabetes 1999, 48, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Kakoki, M.; Pochynyuk, O.M.; Hathaway, C.M.; Tomita, H.; Hagaman, J.R.; Kim, H.S.; Zaika, O.L.; Mamenko, M.; Kayashima, Y.; Matsuki, K.; et al. Primary aldosteronism and impaired natriuresis in mice underexpressing TGFbeta1. Proc. Natl. Acad. Sci. USA 2013, 110, 5600–5605. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Han, H.J. Troglitazone ameliorates high glucose-induced EMT and dysfunction of SGLTs through PI3K/Akt, GSK-3beta, Snail1, and beta-catenin in renal proximal tubule cells. Am. J. Physiol. Renal. Physiol. 2010, 298, F1263–F1275. [Google Scholar] [CrossRef] [Green Version]
- Russo, L.M.; del Re, E.; Brown, D.; Lin, H.Y. Evidence for a role of transforming growth factor (TGF)-beta1 in the induction of postglomerular albuminuria in diabetic nephropathy: Amelioration by soluble TGF-beta type II receptor. Diabetes 2007, 56, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.R.; Chung, A.C.; Wang, X.J.; Lai, K.N.; Lan, H.Y. Mice overexpressing latent TGF-beta1 are protected against renal fibrosis in obstructive kidney disease. Am. J. Physiol. Renal. Physiol. 2008, 295, F118–F127. [Google Scholar] [CrossRef]
- Yang, X.; Letterio, J.J.; Lechleider, R.J.; Chen, L.; Hayman, R.; Gu, H.; Roberts, A.B.; Deng, C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999, 18, 1280–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.G.; Wang, D.; Feng, X.H.; Wang, X.J. Latent TGFbeta1 overexpression in keratinocytes results in a severe psoriasis-like skin disorder. EMBO J. 2004, 23, 1770–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Fan, C.; Cai, D.; Zhang, Y.; Zuo, R.; Zhu, L.; Cao, Y.; Zhang, J.; Liu, C.; Chen, Y.; et al. Contribution of TGF-Beta-Mediated NLRP3-HMGB1 Activation to Tubulointerstitial Fibrosis in Rat with Angiotensin II-Induced Chronic Kidney Disease. Front Cell Dev. Biol. 2020, 8, 1. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Chun, J.; Vilaysane, A.; Clark, S.; French, G.; Bracey, N.A.; Trpkov, K.; Bonni, S.; Duff, H.J.; et al. Inflammasome-independent NLRP3 augments TGF-beta signaling in kidney epithelium. J. Immunol. 2013, 190, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Han, W.; Song, S.; Du, Y.; Liu, C.; Chen, N.; Wu, H.; Shi, Y.; Duan, H. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol. Cell Endocrinol. 2018, 478, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Huang, X.R.; Xiao, J.; Chen, H.Y.; Zhong, X.; Chung, A.C.; Lan, H.Y. Diverse roles of TGF-beta receptor II in renal fibrosis and inflammation in vivo and in vitro. J. Pathol. 2012, 227, 175–188. [Google Scholar] [CrossRef]
- Kim, H.W.; Kim, B.C.; Song, C.Y.; Kim, J.H.; Hong, H.K.; Lee, H.S. Heterozygous mice for TGF-betaIIR gene are resistant to the progression of streptozotocin-induced diabetic nephropathy. Kidney Int. 2004, 66, 1859–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Ren, G.L.; Wei, B.; Jin, J.; Huang, X.R.; Shao, W.; Li, J.; Meng, X.M.; Lan, H.Y. Conditional knockout of TGF-betaRII /Smad2 signals protects against acute renal injury by alleviating cell necroptosis, apoptosis and inflammation. Theranostics 2019, 9, 8277–8293. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.A.; Kim, H.T.; Cho, I.S.; Sheen, Y.Y.; Kim, D.K. IN-1130, a novel transforming growth factor-beta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 2006, 70, 1234–1243. [Google Scholar] [CrossRef] [Green Version]
- Grygielko, E.T.; Martin, W.M.; Tweed, C.; Thornton, P.; Harling, J.; Brooks, D.P.; Laping, N.J. Inhibition of gene markers of fibrosis with a novel inhibitor of transforming growth factor-beta type I receptor kinase in puromycin-induced nephritis. J. Pharmacol. Exp. Ther. 2005, 313, 943–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juarez, P.; Vilchis-Landeros, M.M.; Ponce-Coria, J.; Mendoza, V.; Hernandez-Pando, R.; Bobadilla, N.A.; Lopez-Casillas, F. Soluble betaglycan reduces renal damage progression in db/db mice. Am. J. Physiol. Renal. Physiol. 2007, 292, F321–F329. [Google Scholar] [CrossRef] [Green Version]
- Gerrits, T.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Baelde, H.J.; Scharpfenecker, M. Endoglin Promotes Myofibroblast Differentiation and Extracellular Matrix Production in Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, 7713. [Google Scholar] [CrossRef]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Huang, X.R.; Chung, A.C.; Qin, W.; Shao, X.; Igarashi, P.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1477–1487. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.A.; Croatt, A.J.; Warner, G.M.; Grande, J.P. Genetic deficiency of Smad3 protects against murine ischemic acute kidney injury. Am. J. Physiol. Renal. Physiol. 2011, 301, F436–F442. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, M.; Maezawa, Y.; Yokote, K.; Joh, K.; Kobayashi, K.; Kawamura, H.; Nishimura, M.; Roberts, A.B.; Saito, Y.; Mori, S. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem. Biophys. Res. Commun. 2003, 305, 1002–1007. [Google Scholar] [CrossRef]
- Wang, A.; Ziyadeh, F.N.; Lee, E.Y.; Pyagay, P.E.; Sung, S.H.; Sheardown, S.A.; Laping, N.J.; Chen, S. Interference with TGF-beta signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am. J. Physiol. Renal. Physiol. 2007, 293, F1657–F1665. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Sheng, J.; You, Y.K.; Huang, X.R.; Ma, R.C.W.; Wang, Q.; Lan, H.Y. Deletion of Smad3 prevents renal fibrosis and inflammation in type 2 diabetic nephropathy. Metabolism 2020, 103, 154013. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Wang, L.; Tang, P.M.; Wang, H.L.; Li, J.C.; Xu, B.H.; Xue, V.W.; Tan, R.Z.; Jin, N.; Chan, T.F.; et al. Smad3 deficiency promotes beta cell proliferation and function in db/db mice via restoring Pax6 expression. Theranostics 2021, 11, 2845–2859. [Google Scholar] [CrossRef]
- Loeffler, I.; Liebisch, M.; Allert, S.; Kunisch, E.; Kinne, R.W.; Wolf, G. FSP1-specific SMAD2 knockout in renal tubular, endothelial, and interstitial cells reduces fibrosis and epithelial-to-mesenchymal transition in murine STZ-induced diabetic nephropathy. Cell Tissue Res. 2018, 372, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Saxena, A.; Su, Y.; Frangogiannis, N.G. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1363–H1372. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Huang, X.R.; Xiao, J.; Chung, A.C.; Qin, W.; Chen, H.Y.; Lan, H.Y. Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 2012, 81, 266–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Sun, Y.B.Y.; Chen, W.; Fan, J.; Li, S.; Qu, X.; Chen, Q.; Chen, R.; Zhu, D.; Zhang, J.; et al. Smad4 promotes diabetic nephropathy by modulating glycolysis and OXPHOS. EMBO Rep. 2020, 21, e48781. [Google Scholar] [CrossRef]
- Hou, C.C.; Wang, W.; Huang, X.R.; Fu, P.; Chen, T.H.; Sheikh-Hamad, D.; Lan, H.Y. Ultrasound-microbubble-mediated gene transfer of inducible Smad7 blocks transforming growth factor-beta signaling and fibrosis in rat remnant kidney. Am. J. Pathol. 2005, 166, 761–771. [Google Scholar] [CrossRef]
- Ka, S.M.; Huang, X.R.; Lan, H.Y.; Tsai, P.Y.; Yang, S.M.; Shui, H.A.; Chen, A. Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J. Am. Soc. Nephrol. 2007, 18, 1777–1788. [Google Scholar] [CrossRef]
- Lan, H.Y.; Mu, W.; Tomita, N.; Huang, X.R.; Li, J.H.; Zhu, H.J.; Morishita, R.; Johnson, R.J. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J. Am. Soc. Nephrol. 2003, 14, 1535–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.Y.; Zhou, L.; Huang, X.R.; Fu, P.; Lan, H.Y. Smad7 protects against chronic aristolochic acid nephropathy in mice. Oncotarget 2015, 6, 11930–11944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.X.; Li, Y.Q.; Huang, X.R.; Wei, L.H.; Zhang, Y.; Feng, M.; Meng, X.M.; Chen, H.Y.; Shi, Y.J.; Lan, H.Y. Smad7 inhibits AngII-mediated hypertensive nephropathy in a mouse model of hypertension. Clin. Sci. (Lond.) 2014, 127, 195–208. [Google Scholar] [CrossRef]
- Chung, A.C.; Huang, X.R.; Zhou, L.; Heuchel, R.; Lai, K.N.; Lan, H.Y. Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol. Dial. Transplant. 2009, 24, 1443–1454. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.X.; Li, Y.Q.; Huang, X.R.; Wei, L.; Chen, H.Y.; Shi, Y.J.; Heuchel, R.L.; Lan, H.Y. Disruption of Smad7 promotes ANG II-mediated renal inflammation and fibrosis via Sp1-TGF-beta/Smad3-NF.kappaB-dependent mechanisms in mice. PLoS ONE 2013, 8, e53573. [Google Scholar]
- Gu, Y.Y.; Liu, X.S.; Huang, X.R.; Yu, X.Q.; Lan, H.Y. Diverse Role of TGF-beta in Kidney Disease. Front Cell Dev. Biol. 2020, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.Y.; Hou, C.C.; Wang, W.; Huang, X.R.; Lan, H.Y. Blockade of NFkappaB activation and renal inflammation by ultrasound-mediated gene transfer of Smad7 in rat remnant kidney. Kidney Int. Suppl. 2005, 67, S83–S91. [Google Scholar] [CrossRef] [Green Version]
- Ka, S.M.; Yeh, Y.C.; Huang, X.R.; Chao, T.K.; Hung, Y.J.; Yu, C.P.; Lin, T.J.; Wu, C.C.; Lan, H.Y.; Chen, A. Kidney-targeting Smad7 gene transfer inhibits renal TGF-beta/MAD homologue (SMAD) and nuclear factor kappaB (NF-kappaB) signalling pathways, and improves diabetic nephropathy in mice. Diabetologia 2012, 55, 509–519. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Huang, X.R.; Wang, W.; Li, J.H.; Heuchel, R.L.; Chung, A.C.; Lan, H.Y. The protective role of Smad7 in diabetic kidney disease: Mechanism and therapeutic potential. Diabetes 2011, 60, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Filipowicz, W. RNAi: The nuts and bolts of the RISC machine. Cell 2005, 122, 17–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, P.M.; Zhang, Y.Y.; Mak, T.S.; Tang, P.C.; Huang, X.R.; Lan, H.Y. Transforming growth factor-beta signalling in renal fibrosis: From Smads to non-coding RNAs. J. Physiol. 2018, 596, 3493–3503. [Google Scholar] [CrossRef]
- Zhou, Q.; Guo, H.; Yu, C.; Huang, X.R.; Liang, L.; Zhang, P.; Yu, J.; Zhang, J.; Chan, T.F.; Ma, R.C.W.; et al. Identification of Smad3-related transcriptomes in type-2 diabetic nephropathy by whole transcriptome RNA sequencing. J. Cell Mol. Med. 2021, 25, 2052–2068. [Google Scholar] [CrossRef] [PubMed]
- Zarjou, A.; Yang, S.; Abraham, E.; Agarwal, A.; Liu, G. Identification of a microRNA signature in renal fibrosis: Role of miR-21. Am. J. Physiol. Renal. Physiol. 2011, 301, F793–F801. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Chung, A.C.; Chen, H.Y.; Meng, X.M.; Lan, H.Y. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1668–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, W.; Chung, A.C.; Huang, X.R.; Meng, X.M.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 2011, 22, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chung, A.C.; Huang, X.R.; Dong, Y.; Yu, X.; Lan, H.Y. Identification of novel long noncoding RNAs associated with TGF-beta/Smad3-mediated renal inflammation and fibrosis by RNA sequencing. Am. J. Pathol. 2014, 184, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Chung, A.C.; Chen, H.Y.; Dong, Y.; Meng, X.M.; Li, R.; Yang, W.; Hou, F.F.; Lan, H.Y. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia 2013, 56, 663–674. [Google Scholar] [CrossRef]
- Kato, M.; Zhang, J.; Wang, M.; Lanting, L.; Yuan, H.; Rossi, J.J.; Natarajan, R. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc. Natl. Acad. Sci. USA 2007, 104, 3432–3437. [Google Scholar] [CrossRef] [Green Version]
- Chung, A.C.; Huang, X.R.; Meng, X.; Lan, H.Y. miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1317–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wang, Y.; Minto, A.W.; Wang, J.; Shi, Q.; Li, X.; Quigg, R.J. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J 2008, 22, 4126–4135. [Google Scholar] [CrossRef] [PubMed]
- Al-Kafaji, G.; Al-Muhtaresh, H.A. Expression of microRNA377 and microRNA192 and their potential as bloodbased biomarkers for early detection of type 2 diabetic nephropathy. Mol. Med. Rep. 2018, 18, 1171–1180. [Google Scholar] [PubMed] [Green Version]
- Du, B.; Ma, L.M.; Huang, M.B.; Zhou, H.; Huang, H.L.; Shao, P.; Chen, Y.Q.; Qu, L.H. High glucose down-regulates miR-29a to increase collagen IV production in HK-2 cells. FEBS Lett. 2010, 584, 811–816. [Google Scholar] [CrossRef] [Green Version]
- Tung, C.W.; Ho, C.; Hsu, Y.C.; Huang, S.C.; Shih, Y.H.; Lin, C.L. MicroRNA-29a Attenuates Diabetic Glomerular Injury through Modulating Cannabinoid Receptor 1 Signaling. Molecules 2019, 24, 264. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Zhong, X.; Huang, X.R.; Meng, X.M.; You, Y.; Chung, A.C.; Lan, H.Y. MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol. Ther. 2014, 22, 842–853. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huang, X.R.; Wei, L.H.; Chung, A.C.; Yu, C.M.; Lan, H.Y. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol. Ther. 2014, 22, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, M.; Tang, P.M.; Huang, X.R.; Sun, S.F.; You, Y.K.; Xiao, J.; Lv, L.L.; Xu, A.P.; Lan, H.Y. TGF-beta Mediates Renal Fibrosis via the Smad3-Erbb4-IR Long Noncoding RNA Axis. Mol. Ther. 2018, 26, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.F.; Tang, P.M.K.; Feng, M.; Xiao, J.; Huang, X.R.; Li, P.; Ma, R.C.W.; Lan, H.Y. Novel lncRNA Erbb4-IR Promotes Diabetic Kidney Injury in db/db Mice by Targeting miR-29b. Diabetes 2018, 67, 731–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Y.; Tang, P.M.; Tang, P.C.; Xiao, J.; Huang, X.R.; Yu, C.; Ma, R.C.W.; Lan, H.Y. LRNA9884, a Novel Smad3-Dependent Long Noncoding RNA, Promotes Diabetic Kidney Injury in db/db Mice via Enhancing MCP-1-Dependent Renal Inflammation. Diabetes 2019, 68, 1485–1498. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Koh, P.; Winbanks, C.; Coughlan, M.T.; McClelland, A.; Watson, A.; Jandeleit-Dahm, K.; Burns, W.C.; Thomas, M.C.; Cooper, M.E.; et al. miR-200a Prevents renal fibrogenesis through repression of TGF-beta2 expression. Diabetes 2011, 60, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Park, J.T.; Kato, M.; Lanting, L.; Castro, N.; Nam, B.Y.; Wang, M.; Kang, S.W.; Natarajan, R. Repression of let-7 by transforming growth factor-beta1-induced Lin28 upregulates collagen expression in glomerular mesangial cells under diabetic conditions. Am. J. Physiol. Renal. Physiol. 2014, 307, F1390–F1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Jha, J.C.; Hagiwara, S.; McClelland, A.D.; Jandeleit-Dahm, K.; Thomas, M.C.; Cooper, M.E.; Kantharidis, P. Transforming growth factor-beta1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 2014, 85, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Chau, B.N.; Xin, C.; Hartner, J.; Ren, S.; Castano, A.P.; Linn, G.; Li, J.; Tran, P.T.; Kaimal, V.; Huang, X.; et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 2012, 4, 121ra18. [Google Scholar] [CrossRef] [Green Version]
- Putta, S.; Lanting, L.; Sun, G.; Lawson, G.; Kato, M.; Natarajan, R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 458–469. [Google Scholar] [CrossRef] [Green Version]
- Saadi, G.; Meligi, A.E.; El-Ansary, M.; Alkemary, A.; Ahmed, G. Evaluation of microRNA-192 in patients with diabetic nephropathy. Egypt. J. Int. Med. 2019, 31, 122–128. [Google Scholar] [CrossRef]
- Krupa, A.; Jenkins, R.; Luo, D.D.; Lewis, A.; Phillips, A.; Fraser, D. Loss of MicroRNA-192 promotes fibrogenesis in diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 438–447. [Google Scholar] [CrossRef]
- Wang, J.; Chu, E.S.; Chen, H.Y.; Man, K.; Go, M.Y.; Huang, X.R.; Lan, H.Y.; Sung, J.J.; Yu, J. microRNA-29b prevents liver fibrosis by attenuating hepatic stellate cell activation and inducing apoptosis through targeting PI3K/AKT pathway. Oncotarget 2015, 6, 7325–7338. [Google Scholar] [CrossRef]
- Xiao, J.; Meng, X.M.; Huang, X.R.; Chung, A.C.; Feng, Y.L.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. miR-29 inhibits bleomycin-induced pulmonary fibrosis in mice. Mol. Ther. 2012, 20, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanasaki, K. The role of renal dipeptidyl peptidase-4 in kidney disease: Renal effects of dipeptidyl peptidase-4 inhibitors with a focus on linagliptin. Clin. Sci. 2018, 132, 489–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.P.; Shi, S.; Kanasaki, M.; Nagai, T.; Kitada, M.; He, J.; Nakamura, Y.; Ishigaki, Y.; Kanasaki, K.; Koya, D. Effect of Antifibrotic MicroRNAs Crosstalk on the Action of N-acetyl-seryl-aspartyl-lysyl-proline in Diabetes-related Kidney Fibrosis. Sci. Rep. 2016, 6, 29884. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Luo, M.L.; Song, E.; Zhou, Z.; Ma, T.; Wang, J.; Jia, N.; Wang, G.; Nie, S.; Liu, Y.; et al. Long noncoding RNA lnc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-beta/Smad3 pathway. Sci. Transl. Med. 2018, 10, e2039. [Google Scholar] [CrossRef] [Green Version]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Panchapakesan, U.; Gross, S.; Komala, M.G.; Pegg, K.; Pollock, C.A. DPP4 inhibition in human kidney proximal tubular cells—renoprotection in diabetic nephropathy? Diabetes 2013, 63, 1829–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravid, M.; Lang, R.; Rachmani, R.; Lishner, M. Long-term renoprotective effect of angiotensin-converting enzyme inhibition in non-insulin-dependent diabetes mellitus. A 7-year follow-up study. Arch. Intern. Med. 1996, 156, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Parving, H.H.; Andersen, S.; Jacobsen, P.; Christensen, P.K.; Rossing, K.; Hovind, P.; Rossing, P.; Tarnow, L. Angiotensin receptor blockers in diabetic nephropathy: Renal and cardiovascular end points. Semin. Nephrol. 2004, 24, 147–157. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, Z.; Zhang, X.; Zhao, J.; Zhou, X.; Xu, Q.; Shang, H.; Dong, J.; Liao, L. Efficacy of statins in patients with diabetic nephropathy: A meta-analysis of randomized controlled trials. Lipids Health Dis. 2016, 15, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytvyn, Y.; Godoy, L.C.; Scholtes, R.A.; van Raalte, D.H.; Cherney, D.Z. Mineralocorticoid Antagonism and Diabetic Kidney Disease. Curr. Diab. Rep. 2019, 19, 4. [Google Scholar] [CrossRef]
- Georgianos, P.I.; Agarwal, R. Endothelin A receptor antagonists in diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 338–344. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Brosius, F.C., 3rd; Adler, S.G.; Kretzler, M.; Mehta, R.L.; Tumlin, J.A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M.E.; et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: Results from a Phase 2 randomized controlled clinical trial. Nephrol. Dial. Transplant. 2018, 33, 1950–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.P.; Li, J.; Kitada, M.; Fujita, H.; Yamada, Y.; Goodwin, J.E.; Kanasaki, K.; Koya, D. SIRT3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death Dis 2018, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Jin, Y.; Guo, J.; Ziyadeh, F.N. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes 1996, 45, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Ziyadeh, F.N.; Hoffman, B.B.; Han, D.C.; Iglesias-De La Cruz, M.C.; Hong, S.W.; Isono, M.; Chen, S.; McGowan, T.A.; Sharma, K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 8015–8020. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Iglesias-de la Cruz, M.C.; Jim, B.; Hong, S.W.; Isono, M.; Ziyadeh, F.N. Reversibility of established diabetic glomerulopathy by anti-TGF-beta antibodies in db/db mice. Biochem. Biophys. Res. Commun. 2003, 300, 16–22. [Google Scholar] [CrossRef]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti-TGF-beta1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Li, M.O.; Wan, Y.Y.; Flavell, R.A. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 2007, 26, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Petersen, M.; Thorikay, M.; Deckers, M.; van Dinther, M.; Grygielko, E.T.; Gellibert, F.; de Gouville, A.C.; Huet, S.; ten Dijke, P.; Laping, N.J. Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008, 73, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Kanasaki, K.; Shi, S.; Kanasaki, M.; He, J.; Nagai, T.; Nakamura, Y.; Ishigaki, Y.; Kitada, M.; Srivastava, S.P.; Koya, D. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes 2014, 63, 2120–2131. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Wang, H.; Wu, Z.; Zhong, X.; Zhu, M.; Zhang, Y.; Tan, R.; Liu, Y.; Li, J.; Wang, L. Specific Inhibitor of Smad3 (SIS3) Attenuates Fibrosis, Apoptosis, and Inflammation in Unilateral Ureteral Obstruction Kidneys by Inhibition of Transforming Growth Factor beta (TGF-beta)/Smad3 Signaling. Med. Sci. Monit. 2018, 24, 1633–1641. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, X.M.; Huang, X.R.; Lan, H.Y. The preventive and therapeutic implication for renal fibrosis by targetting TGF-beta/Smad3 signaling. Clin. Sci. 2018, 132, 1403–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Qu, X.; Yao, J.; Caruana, G.; Ricardo, S.D.; Yamamoto, Y.; Yamamoto, H.; Bertram, J.F. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 2612–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Chen, X.C.; Li, Z.H.; Wu, H.L.; Jing, K.P.; Huang, X.R.; Ye, L.; Wei, B.; Lan, H.Y.; Liu, H.F. SMAD3 promotes autophagy dysregulation by triggering lysosome depletion in tubular epithelial cells in diabetic nephropathy. Autophagy 2020, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Zhou, H.; Setia, O.; Liu, B.; Kanasaki, K.; Koya, D.; Dardik, A.; Fernandez-Hernando, C.; Goodwin, J. Loss of endothelial glucocorticoid receptor accelerates diabetic nephropathy. Nat. Commun. 2021, 12, 2368. [Google Scholar] [CrossRef]
- Li, G.; Wang, S.; Gelehrter, T.D. Identification of glucocorticoid receptor domains involved in transrepression of transforming growth factor-beta action. J. Biol. Chem. 2003, 278, 41779–41788. [Google Scholar] [CrossRef] [Green Version]
- Nitta, K.; Shi, S.; Nagai, T.; Kanasaki, M.; Kitada, M.; Srivastava, S.P.; Haneda, M.; Kanasaki, K.; Koya, D. Oral Administration of N-Acetyl-seryl-aspartyl-lysyl-proline Ameliorates Kidney Disease in Both Type 1 and Type 2 Diabetic Mice via a Therapeutic Regimen. Biomed. Res. Int. 2016, 2016, 9172157. [Google Scholar] [CrossRef]
- Kanasaki, K.; Nagai, T.; Nitta, K.; Kitada, M.; Koya, D. N-acetyl-seryl-aspartyl-lysyl-proline: A valuable endogenous anti-fibrotic peptide for combating kidney fibrosis in diabetes. Front. Pharmacol. 2014, 5, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanasaki, K.; Koya, D.; Sugimoto, T.; Isono, M.; Kashiwagi, A.; Haneda, M. N-Acetyl-seryl-aspartyl-lysyl-proline inhibits TGF-beta-mediated plasminogen activator inhibitor-1 expression via inhibition of Smad pathway in human mesangial cells. J. Am. Soc. Nephrol. 2003, 14, 863–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.D.; Li, C.Y.; Cui, W.P.; Guo, Q.Y.; Dong, C.Q.; Zou, H.B.; Liu, S.J.; Dong, W.P.; Miao, L.N. Review of Herbal Traditional Chinese Medicine for the Treatment of Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 5749857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.C.; Su, Y.C.; Sun, M.F.; Huang, S.T. Chinese Herbal Medicine Improves the Long-Term Survival Rate of Patients with Chronic Kidney Disease in Taiwan: A Nationwide Retrospective Population-Based Cohort Study. Front. Pharmacol. 2018, 9, 1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Sun, S.; Zhang, H.; Huang, X.; Yan, M.; Dong, X.; Wen, Y.; Wang, H.; Lan, H.Y.; Li, P. Therapeutic Effects of Tangshen Formula on Diabetic Nephropathy in Rats. PLoS ONE 2016, 11, e0147693. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chen, Y.; Liu, J.; Hong, J.; Deng, Y.; Yang, F.; Jin, X.; Gao, J.; Li, J.; Fang, H.; et al. Efficacy and safety of tangshen formula on patients with type 2 diabetic kidney disease: A multicenter double-blinded randomized placebo-controlled trial. PLoS ONE 2015, 10, e0126027. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.T.; Zhang, H.J.; Lu, X.G.; Huang, X.R.; Zhang, W.K.; Wang, H.; Lan, H.Y.; Li, P. Chaihuang-Yishen granule inhibits diabetic kidney disease in rats through blocking TGF-beta/Smad3 signaling. PLoS ONE 2014, 9, e90807. [Google Scholar]
- Sun, S.F.; Zhao, T.T.; Zhang, H.J.; Huang, X.R.; Zhang, W.K.; Zhang, L.; Yan, M.H.; Dong, X.; Wang, H.; Wen, Y.M.; et al. Renoprotective effect of berberine on type 2 diabetic nephropathy in rats. Clin. Exp. Pharmacol. Physiol. 2015, 42, 662–670. [Google Scholar] [CrossRef]
- Meng, X.M.; Zhang, Y.; Huang, X.R.; Ren, G.L.; Li, J.; Lan, H.Y. Treatment of renal fibrosis by rebalancing TGF-beta/Smad signaling with the combination of asiatic acid and naringenin. Oncotarget 2015, 6, 36984–36997. [Google Scholar] [CrossRef] [Green Version]


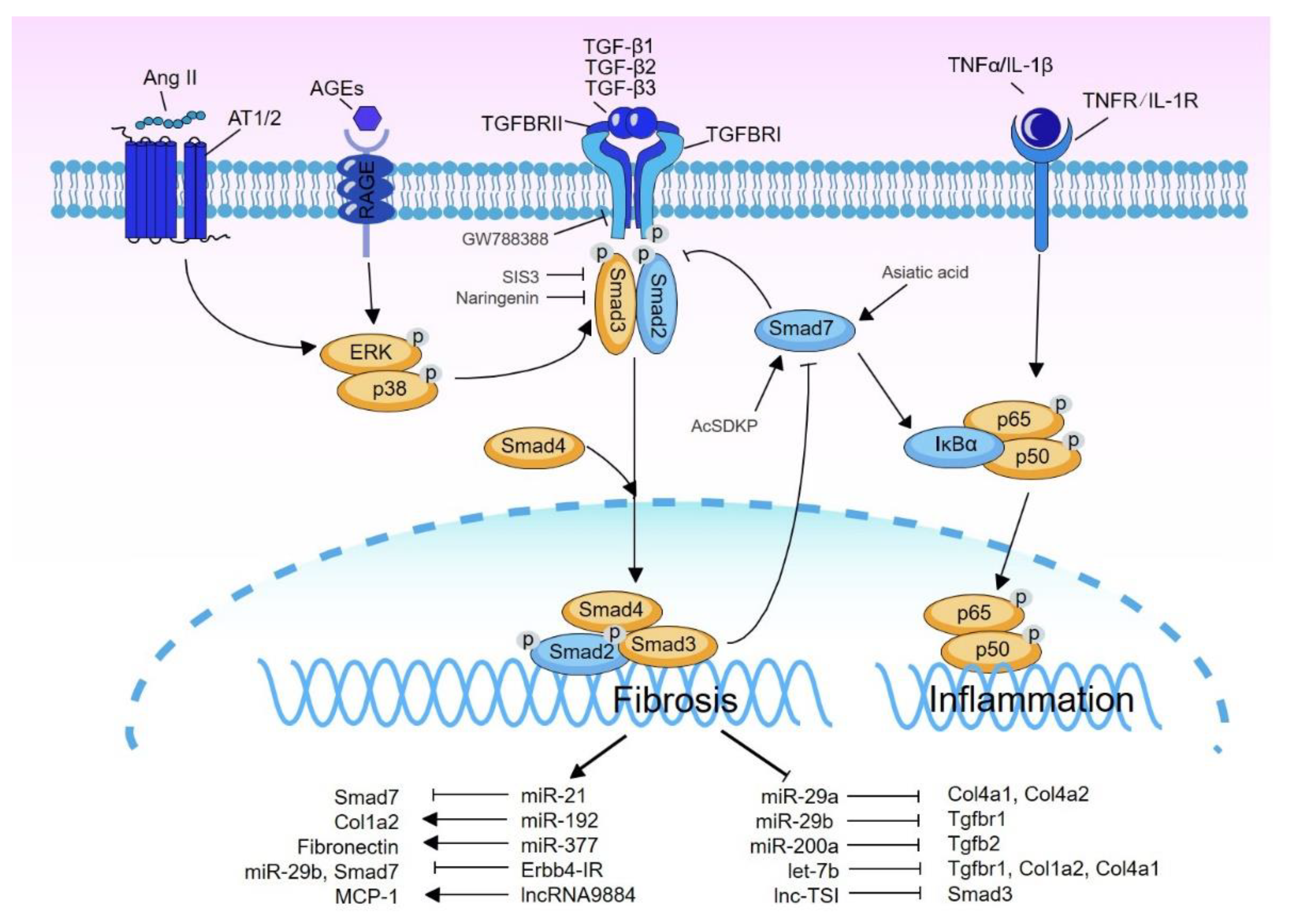{kind=link}
| miRNA/lncRNA | ND Model | Target Gene and Reference |
|---|---|---|
| Pathogenic and upregulated | ||
| miR-21 | db/db mice | Smad7 [101] |
| miR-192 | STZ-induced DN db/db mice | Col1a2 via directly targeting SIP1 [102,103] |
| miR-377 | STZ-induced DN diabetic NOD mice | unclear [104,105] |
| miR-29a | STZ-induced DN | Col4a1 and Col4a2 [106,107] |
| miR-29b | db/db mice | Tgfbr1 [108,109] |
| Erbb4-IR | db/db mice | miR-9b, Smad7 [110,111] |
| lncRNA9884 | db/db mice | MCP-1 [112] |
| Renoprotective and downregulated | ||
| miR-200a | STZ-induced DN | Tgfb2 [113] |
| let-7b | STZ-induced DN | Tgfbr1, Col1a2, Col4a1 [114,115] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Wang, H.-L.; Liu, T.-T.; Lan, H.-Y. TGF-Beta as a Master Regulator of Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 7881. https://doi.org/10.3390/ijms22157881
Wang L, Wang H-L, Liu T-T, Lan H-Y. TGF-Beta as a Master Regulator of Diabetic Nephropathy. International Journal of Molecular Sciences. 2021; 22(15):7881. https://doi.org/10.3390/ijms22157881
Chicago/Turabian StyleWang, Li, Hong-Lian Wang, Tong-Tong Liu, and Hui-Yao Lan. 2021. "TGF-Beta as a Master Regulator of Diabetic Nephropathy" International Journal of Molecular Sciences 22, no. 15: 7881. https://doi.org/10.3390/ijms22157881
APA StyleWang, L., Wang, H. -L., Liu, T. -T., & Lan, H. -Y. (2021). TGF-Beta as a Master Regulator of Diabetic Nephropathy. International Journal of Molecular Sciences, 22(15), 7881. https://doi.org/10.3390/ijms22157881