
LncRNA AERRIE Is Required for Sulfatase 1 Expression, but Not for Endothelial-to-Mesenchymal Transition

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Endothelial Cells Stimulated with TGF-β2 and IL1β Undergo EndMT
2.2. Inhibition of lncRNA AERRIE Does Not Regulate EndMT Markers
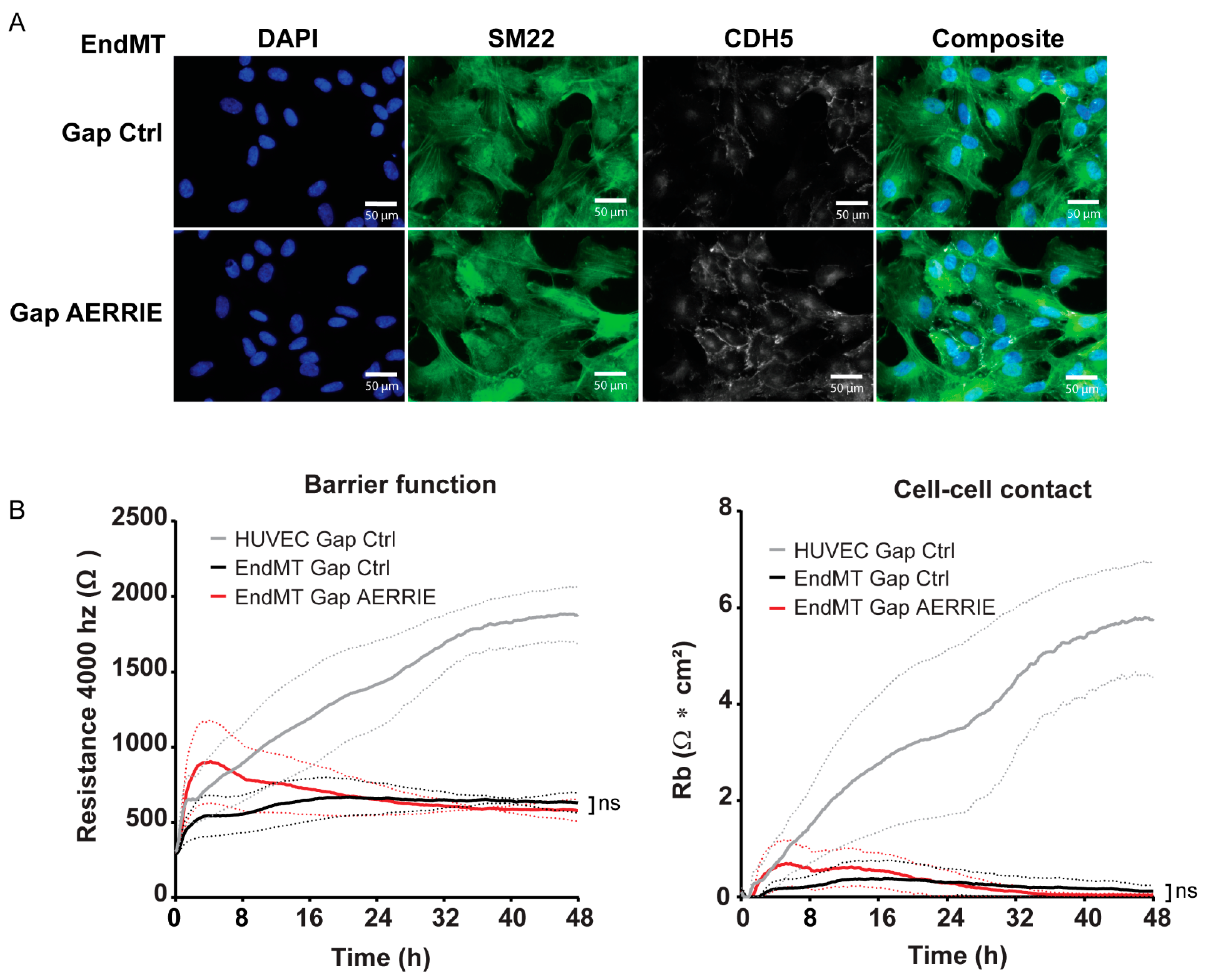
2.3. AERRIE Does Not Regulate Barrier Function in HUVECs Undergoing EndMT
2.4. Overexpression of AERRIE Does Not Affect EndMT Marker Levels or Barrier Function
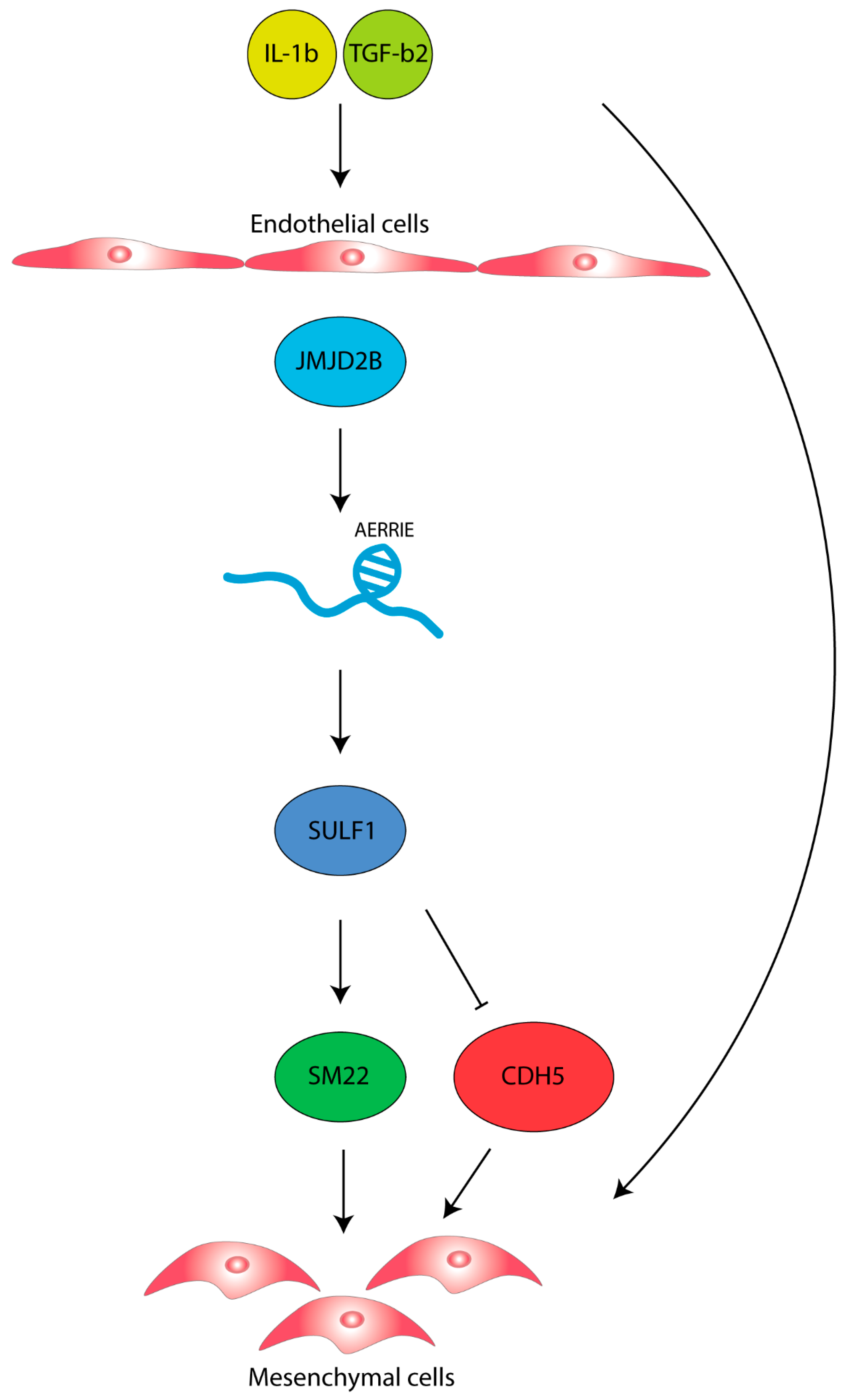
2.5. AERRIE Is Regulated by the EndMT Regulator JMJD2B and Is Required for SULF1 Expression
3. Discussion
4. Methods
4.1. Cell Culture
4.2. RT-qPCR
4.3. SiRNAs-GapmeRs
4.4. Lentiviral Constructs
4.5. Endothelial Barrier and Wound Healing
4.6. Western Blot Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dejana, E.; Hirschi, K.K.; Simons, M. The molecular basis of endothelial cell plasticity. Nat. Commun. 2017, 8, 14361. [Google Scholar] [CrossRef] [Green Version]
- Arciniegas, E.; Neves, C.Y.; Carrillo, L.M.; Zambrano, E.A.; Ramírez, R. Endothelial-Mesenchymal Transition Occurs during Embryonic Pulmonary Artery Development. Endothelium 2005, 12, 193–200. [Google Scholar] [CrossRef]
- Welch-Reardon, K.M.; Wu, N.; Hughes, C.C. A Role for Partial Endothelial–Mesenchymal Transitions in Angiogenesis? Arter. Thromb. Vasc. Biol. 2015, 35, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Wylie-Sears, J.; Levine, R.A.; Bischoff, J. Losartan inhibits endothelial-to-mesenchymal transformation in mitral valve endothelial cells by blocking transforming growth factor-β-induced phosphorylation of ERK. Biochem. Biophys. Res. Commun. 2014, 446, 870–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Friehs, I.; Hu, T.Z.; Melnychenko, I.; Tampe, B.; Alnour, F.; Iascone, M.; Kalluri, R.; Zeisberg, M.; Del Nido, P.J.; et al. Endocardial Fibroelastosis Is Caused by Aberrant Endothelial to Mesenchymal Transition. Circ. Res. 2015, 116, 857–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Péchoux, C.; Bogaard, H.J.; Dorfmüller, P.; Remy, S.; Lecerf, F.; Planté, S.; et al. Endothelial-to-Mesenchymal Transition in Pulmonary Hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Investig. 2015, 125, 4514–4528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.-Y.; Schwartz, M.A.; Simons, M. Endothelial-to-Mesenchymal Transition, Vascular Inflammation, and Atherosclerosis. Front. Cardiovasc. Med. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Mendoza, F.A.; Jimenez, S.A. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. J. Clin. Med. 2016, 5, 45. [Google Scholar] [CrossRef]
- Yoshimatsu, Y.; Kimuro, S.; Pauty, J.; Takagaki, K.; Nomiyama, S.; Inagawa, A.; Maeda, K.; Podyma-Inoue, K.A.; Kajiya, K.; Matsunaga, Y.T.; et al. TGF-beta and TNF-alpha cooperatively induce mesenchymal transition of lymphatic endothelial cells via activation of Activin signals. PLoS ONE 2020, 15, e0232356. [Google Scholar] [CrossRef]
- Mahler, G.J.; Farrar, E.J.; Butcher, J.T. Inflammatory Cytokines Promote Mesenchymal Transformation in Embryonic and Adult Valve Endothelial Cells. Arter. Thromb. Vasc. Biol. 2013, 33, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; Hilaire, C.S.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D.; et al. TGF- Signaling Mediates Endothelial-to-Mesenchymal Transition (EndMT) During Vein Graft Remodeling. Sci. Transl. Med. 2014, 6, 227ra34. [Google Scholar] [CrossRef] [Green Version]
- Evrard, S.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.-R.; D’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 2016, 7, 11853. [Google Scholar] [CrossRef]
- Medici, D.; Potenta, S.; Kalluri, R. Transforming growth factor-β2 promotes Snail-mediated endothelial–mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem. J. 2011, 437, 515–520. [Google Scholar] [CrossRef]
- Hiepen, C.; Jatzlau, J.; Hildebrandt, S.; Kampfrath, B.; Goktas, M.; Murgai, A.; Camacho, J.L.C.; Haag, R.; Ruppert, C.; Sengle, G.; et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol. 2019, 17, e3000557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, A.; Attisano, L. The TGFbeta Superfamily Signaling Pathway. Wiley Interdiscip. Rev. Dev. Biol. 2012, 2, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Kokudo, T.; Suzuki, Y.; Yoshimatsu, Y.; Yamazaki, T.; Watabe, T.; Miyazono, K. Snail is required for TGFβ-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 2008, 121, 3317–3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.J.; Park, J.H.; Shin, J.U.; Noh, H.; Lew, D.H.; Yang, W.I.; Yun, C.O.; Lee, K.H.; Lee, J.H. Endothelial-to-mesenchymal transition induced by Wnt 3a in keloid pathogenesis. Wound Repair Regen. 2015, 23, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Zawieja, D.C.; Davis, M.J.; Muthuchamy, M. MicroRNA signature of inflamed lymphatic endothelium and role of miR-9 in lymphangiogenesis and inflammation. Am. J. Physiol. Physiol. 2015, 309, C680–C692. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Zhang, Y.; Tang, Y.; Li, Q. MALAT1 Modulates TGF-β1-Induced Endothelial-to-Mesenchymal Transition through Downregulation of miR-145. Cell. Physiol. Biochem. 2017, 42, 357–372. [Google Scholar] [CrossRef]
- Saxena, A.; Carninci, P. Long non-coding RNA modifies chromatin: Epigenetic silencing by long non-coding RNAs. BioEssays 2011, 33, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Kugel, J.F.; Goodrich, J.A. Non-coding RNAs: Key regulators of mammalian transcription. Trends Biochem. Sci. 2012, 37, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A Long Noncoding RNA Controls Muscle Differentiation by Functioning as a Competing Endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Sun, T.; Hacisuleyman, E.; Fei, T.; Wang, X.; Brown, M.; Rinn, J.L.; Lee, M.G.S.; Chen, Y.; Kantoff, P.W.; et al. Integrative analyses reveal a long noncoding RNA-mediated sponge regulatory network in prostate cancer. Nat. Commun. 2016, 7, 10982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nat. Cell Biol. 2012, 491, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Vidal, N.; I Bink, D.; A Boon, R. Long noncoding RNA in cardiac aging and disease. J. Mol. Cell Biol. 2019, 11, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zou, Y.G.; Xue, Y.Z.; Wang, X.H.; Gao, H.; Dong, H.W.; Zhang, Q. Long non-coding RNA H19 protects acute myocardial infarction through activating autophagy in mice. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5647–5651. [Google Scholar] [CrossRef]
- Liu, L.; An, X.; Li, Z.; Song, Y.; Li, L.; Zuo, S.; Liu, N.; Yang, G.; Wang, H.; Cheng, X.; et al. The H19 long noncoding RNA is a novel negative regulator of cardiomyocyte hypertrophy. Cardiovasc. Res. 2016, 111, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, P.; Sommer, J.; Theodorou, K.; Kirchhof, L.; Fischer, A.; Li, Y.; Perisic, L.; Hedin, U.; Maegdefessel, L.; Dimmeler, S.; et al. Long non-coding RNA H19 regulates endothelial cell aging via inhibition of STAT3 signalling. Cardiovasc. Res. 2018, 115, 230–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Zhao, Z.-A.; Liu, J.; Hao, K.; Yu, Y.; Han, X.; Li, J.; Wang, Y.; Lei, W.; Dong, N.; et al. Long noncoding RNA Meg3 regulates cardiomyocyte apoptosis in myocardial infarction. Gene Ther. 2018, 25, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, M.-T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the Cardiac Fibroblast–Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Zha, F.; Qu, X.; Tang, B.; Li, J.; Wang, Y.; Zheng, P.; Ji, T.; Zhu, C.; Bai, S. Long non-coding RNA MEG3 promotes fibrosis and inflammatory response in diabetic nephropathy via miR-181a/Egr-1/TLR4 axis. Aging 2019, 11, 3716–3730. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.; Hofmann, P.; Michalik, K.M.; Lozano-Vidal, N.; Berghäuser, D.; Fischer, A.; Knau, A.; Jaé, N.; Schürmann, C.; Dimmeler, S. Long Noncoding RNA Meg3 Controls Endothelial Cell Aging and Function: Implications for Regenerative Angiogenesis. J. Am. Coll. Cardiol. 2016, 68, 2589–2591. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, W.; Lin, C.-H.; Yang, J.; Shang, C.; Nuernberg, S.T.; Jin, K.K.; Xu, W.; Lin, C.-Y.; Lin, C.-J.; et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nat. Cell Biol. 2014, 514, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Liu, F.; Zhou, L.-Y.; Long, B.; Yuan, S.-M.; Wang, Y.; Liu, C.-Y.; Sun, T.; Zhang, X.-J.; Li, P.-F. The Long Noncoding RNA CHRF Regulates Cardiac Hypertrophy by Targeting miR-489. Circ. Res. 2014, 114, 1377–1388. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.P.; Bink, D.I.; Stanicek, L.; Van Bergen, A.; Van Leeuwen, E.; Tran, Y.; Matic, L.; Hedin, U.; Wittig, I.; Dimmeler, S.; et al. Long Non-coding RNA Aerrie Controls DNA Damage Repair via YBX1 to Maintain Endothelial Cell Function. Front. Cell Dev. Biol. 2021, 8. [Google Scholar] [CrossRef]
- Wang, W.; Xu, S.; Di, Y.; Zhang, Z.; Li, Q.; Guo, K.; Lv, Y.; Wang, B. Novel role of LINC01013/miR-6795-5p/FMNL3 axis in the regulation of hepatocellular carcinoma stem cell features. Acta Biochim. Biophys. Sin. 2021. [Google Scholar] [CrossRef]
- Yang, H.; Cao, Y.; Zhang, J.; Liang, Y.; Su, X.; Zhang, C.; Liu, H.; Han, X.; Ge, L.; Fan, Z. DLX5 and HOXC8 enhance the chondrogenic differentiation potential of stem cells from apical papilla via LINC01013. Stem Cell Res. Ther. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.-H.; Lu, P.-H.; Lin, Y.-H.; Tsai, M.-M.; Lin, Y.-W.; Yeh, C.-T.; Lin, K.-H. The long non-coding RNA LINC01013 enhances invasion of human anaplastic large-cell lymphoma. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Glaser, S.F.; Heumüller, A.W.; Tombor, L.S.; Hofmann, P.; Muhly-Reinholz, M.; Fischer, A.; Günther, S.; Kokot, K.E.; Okada, H.; Hassel, D.; et al. The histone demethylase JMJD2B regulates endothelial-to-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2020, 117, 4180–4187. [Google Scholar] [CrossRef] [Green Version]
- Kole, R.; Krainer, A.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fodor, B.D.; Kubicek, S.; Yonezawa, M.; O’Sullivan, R.J.; Sengupta, R.; Perez-Burgos, L.; Opravil, S.; Mechtler, K.; Schotta, G.; Jenuwein, T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006, 20, 1557–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Sun, L.; Li, Q.; Liang, J.; Yu, W.; Yi, X.; Yang, X.; Li, Y.; Han, X.; Zhang, Y.; et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 7541–7546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto-Tomita, M.; Uchimura, K.; Werb, Z.; Hemmerich, S.; Rosen, S.D. Cloning and Characterization of Two Extracellular Heparin-degrading Endosulfatases in Mice and Humans. J. Biol. Chem. 2002, 277, 49175–49185. [Google Scholar] [CrossRef] [Green Version]
- Esko, J.D.; Lindahl, U. Molecular diversity of heparan sulfate. J. Clin. Investig. 2001, 108, 169–173. [Google Scholar] [CrossRef]
- Nakato, H.; Kimata, K. Heparan sulfate fine structure and specificity of proteoglycan functions. Biochim. Biophys. Acta (BBA) Gen. Subj. 2002, 1573, 312–318. [Google Scholar] [CrossRef]
- Yue, X.; Li, X.; Nguyen, H.T.; Chin, D.R.; Sullivan, D.E.; Lasky, J.A. Transforming Growth Factor-β1 Induces Heparan Sulfate 6-O-Endosulfatase 1 Expression In Vitro and In Vivo. J. Biol. Chem. 2008, 283, 20397–20407. [Google Scholar] [CrossRef] [Green Version]
- Uchimura, K.; Morimoto-Tomita, M.; Bistrup, A.; Li, J.; Lyon, M.; Gallagher, J.; Werb, Z.; Rosen, S.D. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: Effects on VEGF, FGF-1, and SDF-1. BMC Biochem. 2006, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Fleenor, B.S.; Marshall, K.D.; Rippe, C.; Seals, U.R. Replicative Aging Induces Endothelial to Mesenchymal Transition in Human Aortic Endothelial Cells: Potential Role of Inflammation. J. Vasc. Res. 2012, 49, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: From cardiovascular development to disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef] [Green Version]
- Medici, D.; Olsen, B.R. The role of endothelial-mesenchymal transition in heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Duffhues, G.; Williams, E.; Benderitter, P.; Orlova, V.; Van Wijhe, M.; De Vinuesa, A.G.; Kerr, G.; Caradec, J.; Lodder, K.; De Boer, H.C.; et al. Development of Macrocycle Kinase Inhibitors for ALK2 Using Fibrodysplasia Ossificans Progressiva-Derived Endothelial Cells. JBMR Plus 2019, 3, e10230. [Google Scholar] [CrossRef]
- Neumann, P.; Jaé, N.; Knau, A.; Glaser, S.F.; Fouani, Y.; Rossbach, O.; Krüger, M.; John, D.; Bindereif, A.; Grote, P.; et al. The lncRNA GATA6-AS epigenetically regulates endothelial gene expression via interaction with LOXL2. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Graham, K.; Murphy, J.I.; Dhoot, G.K. SULF1/SULF2 reactivation during liver damage and tumour growth. Histochem. Cell Biol. 2016, 146, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korf-Klingebiel, M.; Reboll, M.R.; Grote, K.; Schleiner, H.; Wang, Y.; Wu, X.; Klede, S.; Mikhed, Y.; Nobre, A.; Bauersachs, J.; et al. Heparan Sulfate–Editing Extracellular Sulfatases Enhance VEGF Bioavailability for Ischemic Heart Repair. Circ. Res. 2019, 125, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Frese, M.-A.; Milz, F.; Dick, M.; Lamanna, W.C.; Dierks, T. Characterization of the Human Sulfatase Sulf1 and Its High Affinity Heparin/Heparan Sulfate Interaction Domain. J. Biol. Chem. 2009, 284, 28033–28044. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Fu, L.; Kong, X.; Xu, J.; Wang, Z.; Ma, X.; Akiyama, Y.; Chen, Y.; Fang, J. Jumonji domain-containing protein 2B silencing induces DNA damage response via STAT3 pathway in colorectal cancer. Br. J. Cancer 2014, 110, 1014–1026. [Google Scholar] [CrossRef] [Green Version]
- Castellini, L.; Moon, E.J.; Razorenova, O.V.; Krieg, A.J.; Von Eyben, R.; Giaccia, A.J. KDM4B/JMJD2B is a p53 target gene that modulates the amplitude of p53 response after DNA damage. Nucleic Acids Res. 2017, 45, 3674–3692. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.-P.; Sandhu, D.S.; Moser, C.D.; Cazanave, S.C.; Oseini, A.M.; Shire, A.M.; Shridhar, V.; Sanderson, S.O.; Roberts, L.R. Additive effect of apicidin and doxorubicin in sulfatase 1 expressing hepatocellular carcinoma in vitro and in vivo. J. Hepatol. 2009, 50, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-Y.; Yeh, B.-W.; Chan, T.-C.; Yang, K.-F.; Li, W.-M.; Huang, C.-N.; Ke, H.-L.; Li, C.-C.; Yeh, H.-C.; Liang, P.-I.; et al. Sulfatase-1 overexpression indicates poor prognosis in urothelial carcinoma of the urinary bladder and upper tract. Oncotarget 2017, 8, 47216–47229. [Google Scholar] [CrossRef] [PubMed]
- Platel, V.; Faure, S.; Corre, I.; Clere, N. Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J. Oncol. 2019, 2019, 1–13. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Harvey, L.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.-J.; Perez, V.A.D.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, S.; Guo, H.; Cheng, Y.; Zhou, Z.; Zhang, W.; Han, B.; Luo, W.; Wang, J.; Xie, W.; Chao, J. circHECTD1 promotes the silica-induced pulmonary endothelial-mesenchymal transition via HECTD1. Cell Death Dis. 2018, 9, 396. [Google Scholar] [CrossRef] [Green Version]
- Moonen, J.R.A.; Lee, E.S.; Schmidt, M.; Maleszewska, M.; Koerts, J.A.; Brouwer, L.A.; Van Kooten, T.G.; Van Luyn, M.J.; Zeebregts, C.J.; Krenning, G.; et al. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc. Res. 2015, 108, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, T.P.; van Bergen, A.S.; Kremer, V.; Glaser, S.F.; Dimmeler, S.; Boon, R.A. LncRNA AERRIE Is Required for Sulfatase 1 Expression, but Not for Endothelial-to-Mesenchymal Transition. Int. J. Mol. Sci. 2021, 22, 8088. https://doi.org/10.3390/ijms22158088
Pham TP, van Bergen AS, Kremer V, Glaser SF, Dimmeler S, Boon RA. LncRNA AERRIE Is Required for Sulfatase 1 Expression, but Not for Endothelial-to-Mesenchymal Transition. International Journal of Molecular Sciences. 2021; 22(15):8088. https://doi.org/10.3390/ijms22158088
Chicago/Turabian StylePham, Tan Phát, Anke S. van Bergen, Veerle Kremer, Simone F. Glaser, Stefanie Dimmeler, and Reinier A. Boon. 2021. "LncRNA AERRIE Is Required for Sulfatase 1 Expression, but Not for Endothelial-to-Mesenchymal Transition" International Journal of Molecular Sciences 22, no. 15: 8088. https://doi.org/10.3390/ijms22158088
APA StylePham, T. P., van Bergen, A. S., Kremer, V., Glaser, S. F., Dimmeler, S., & Boon, R. A. (2021). LncRNA AERRIE Is Required for Sulfatase 1 Expression, but Not for Endothelial-to-Mesenchymal Transition. International Journal of Molecular Sciences, 22(15), 8088. https://doi.org/10.3390/ijms22158088