
Pyrimethamine Modulates Interplay between Apoptosis and Autophagy in Chronic Myelogenous Leukemia Cells


 ,
,  ,
,  and
and 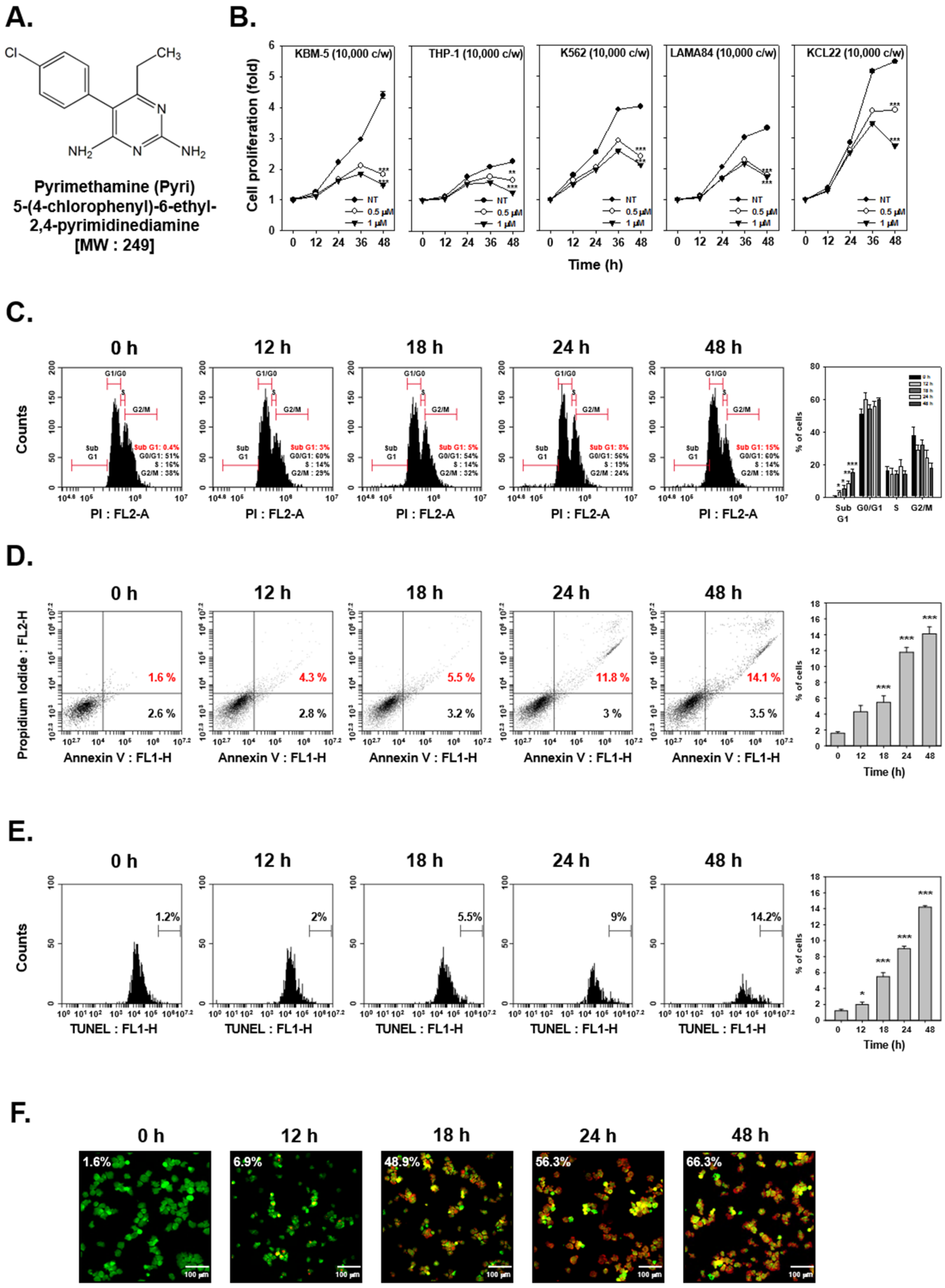{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Pyri Reduced the Viability of Human Myelogenous Leukemia Cell Lines
2.2. Pyri Stimulated the Apoptosis-Induced Cell Death
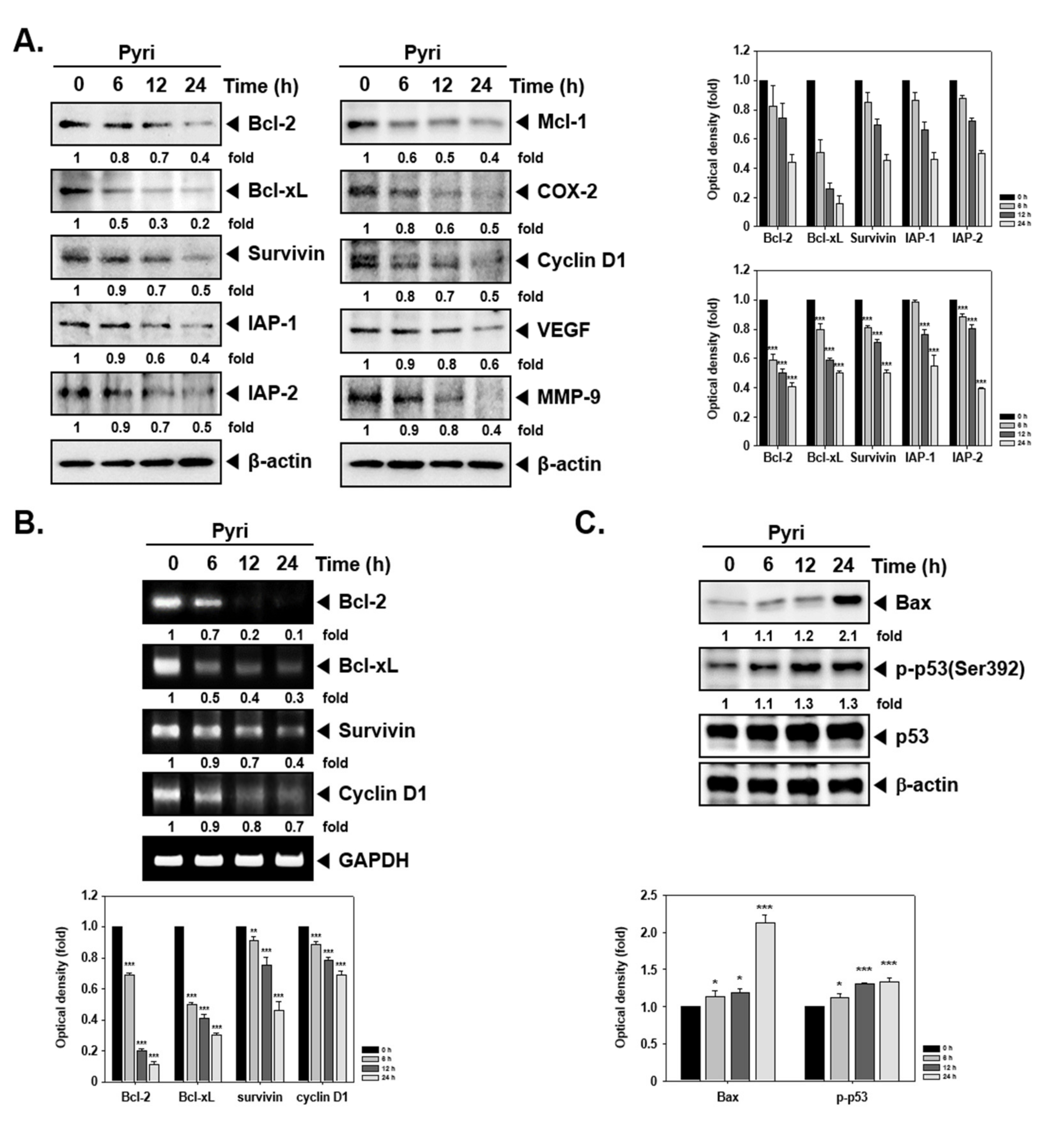
2.3. Pyri Suppressed the Protein and mRNA Expression of Anti-Apoptotic and Oncogenic Markers
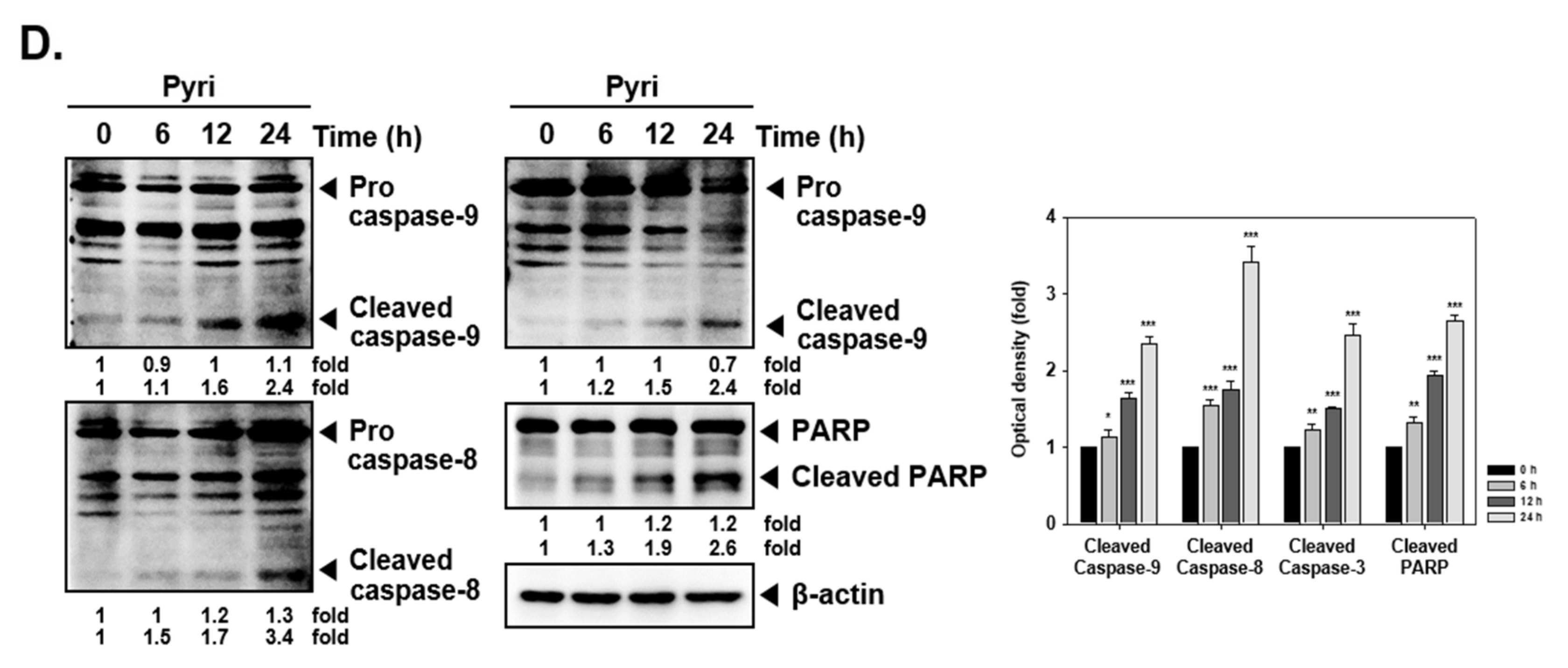
2.4. Pyri Induced Apoptosis through PARP and Caspase Cleavage
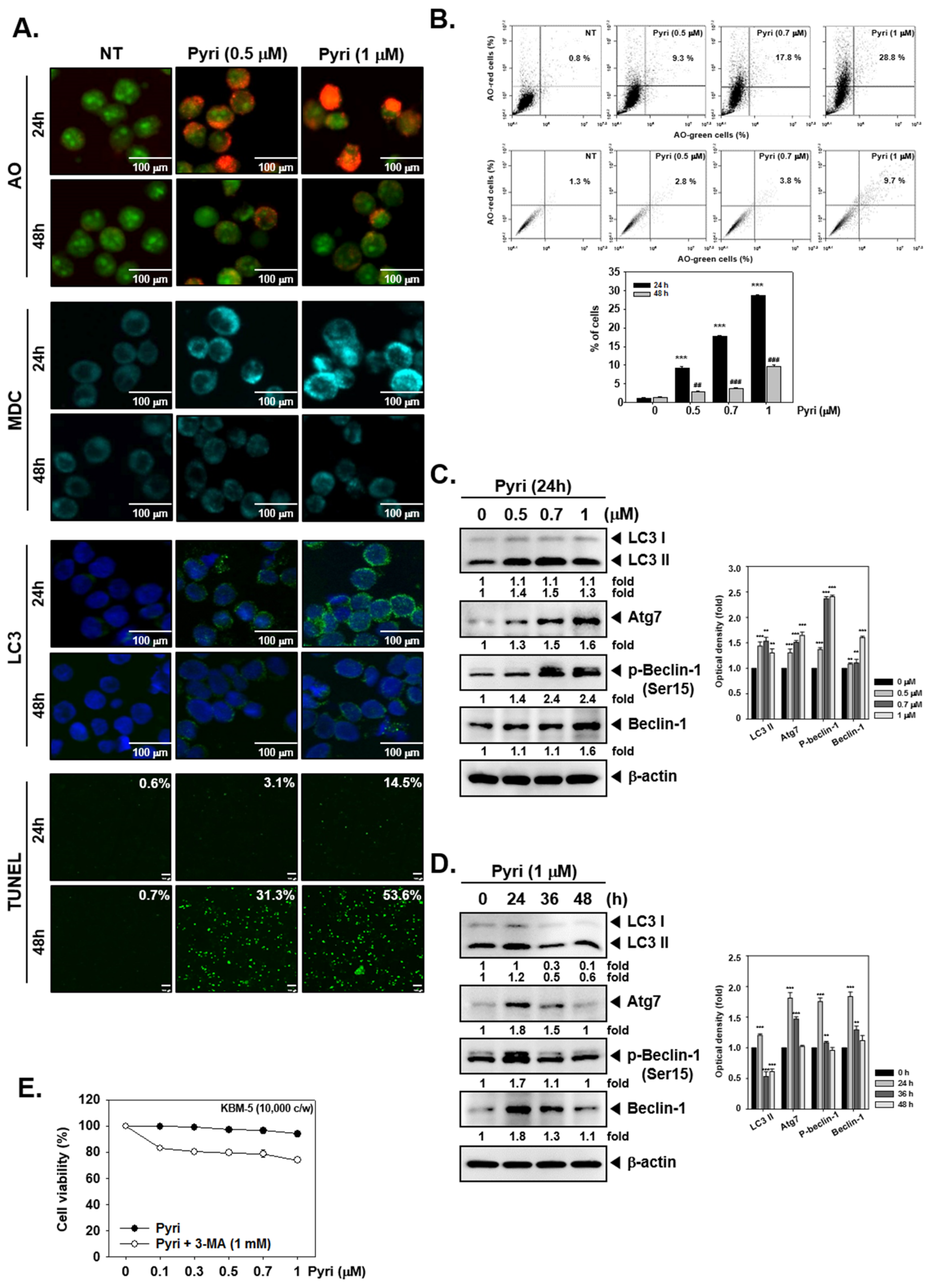
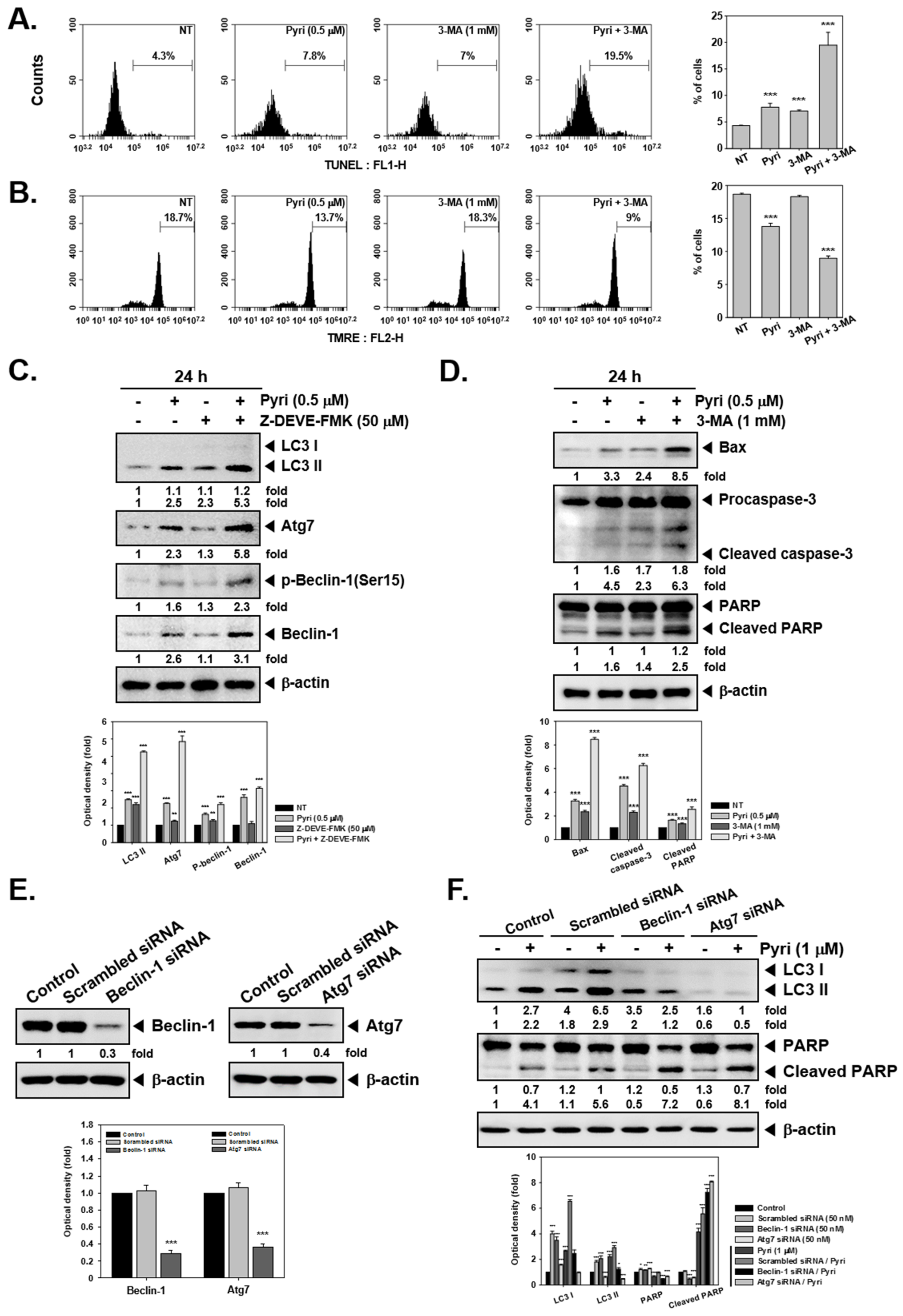
2.5. Pyri Increased the Autophagy-Induced Cell Death in KBM5 Cells
2.6. Pyri Increased the Expression of Autophagy Markers Including LC3II in KBM5 Cells
2.7. Pyri Induced Cell Death by Alternative Pathways upon Autophagy Inhibition
2.8. Pyri Induced LC3II Expression through Causing Beclin-1 and Atg7 Activatio
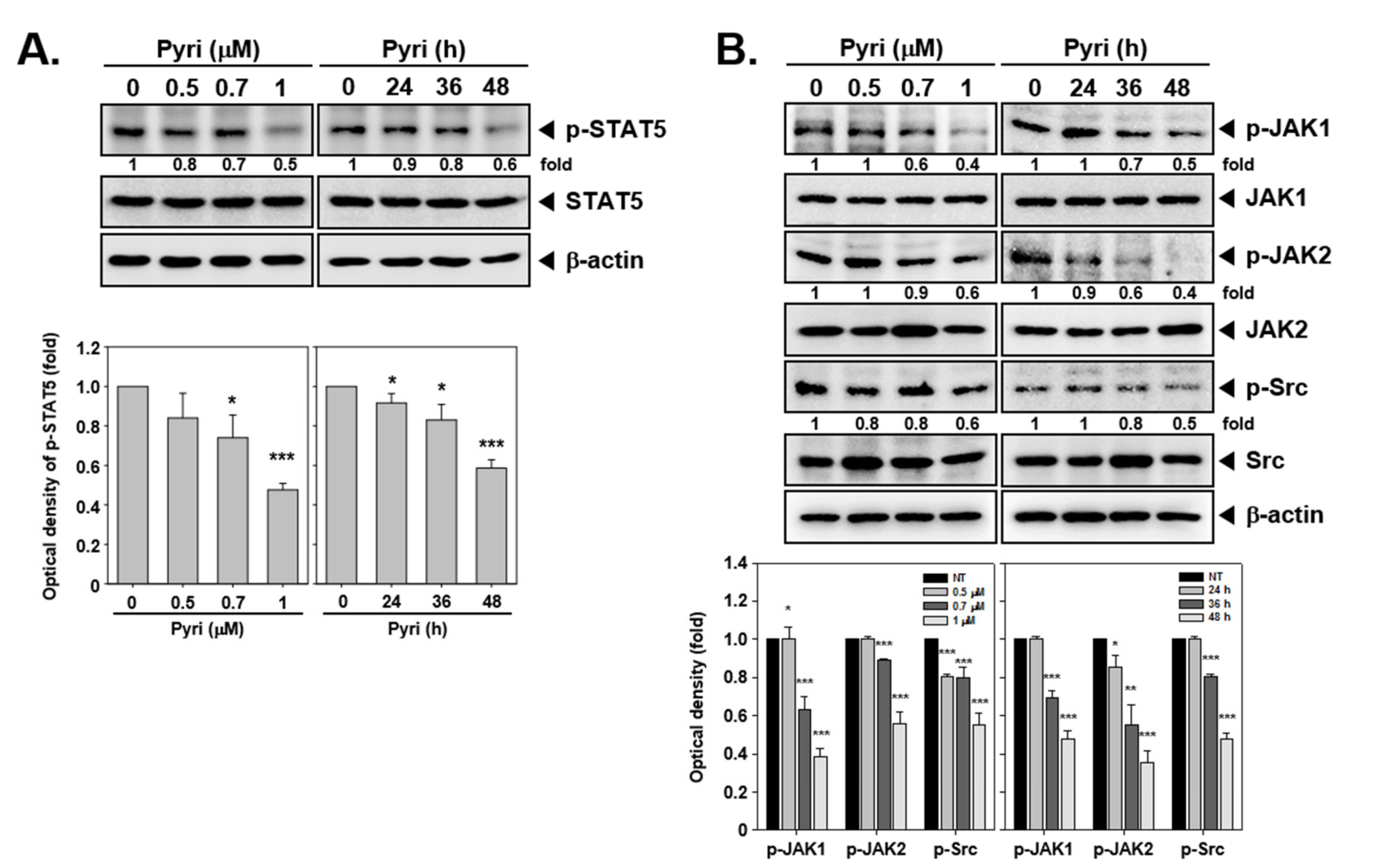
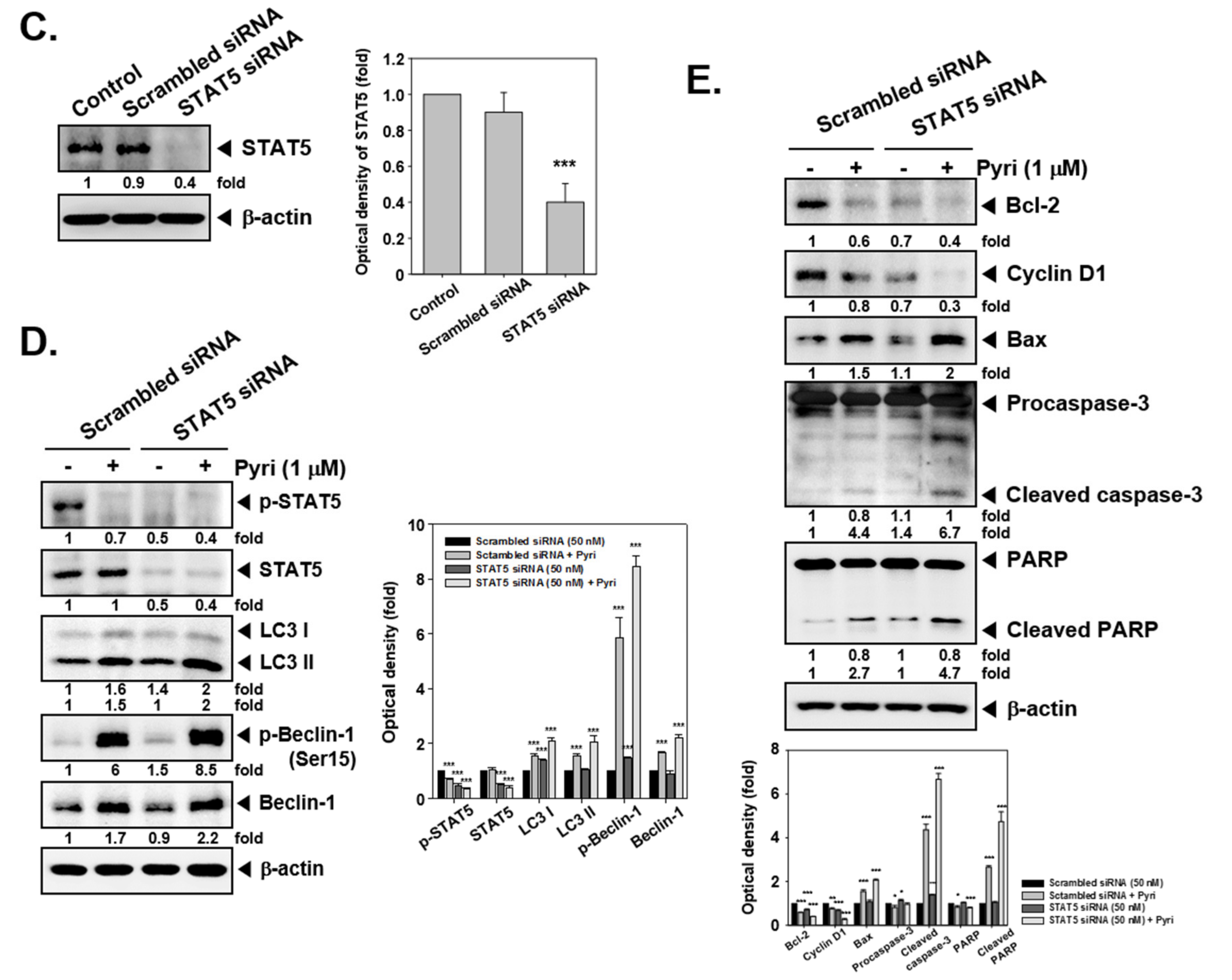
2.9. Pyri Suppressed STAT5 Phosphorylation in KBM5 Cells
2.10. Pyri Down-Regulated the STAT5 Upstream Signals JAK1/2, and Src Activation
2.11. Pyri Induced Apoptosis and Autophagy in STAT5 Deleted Cells
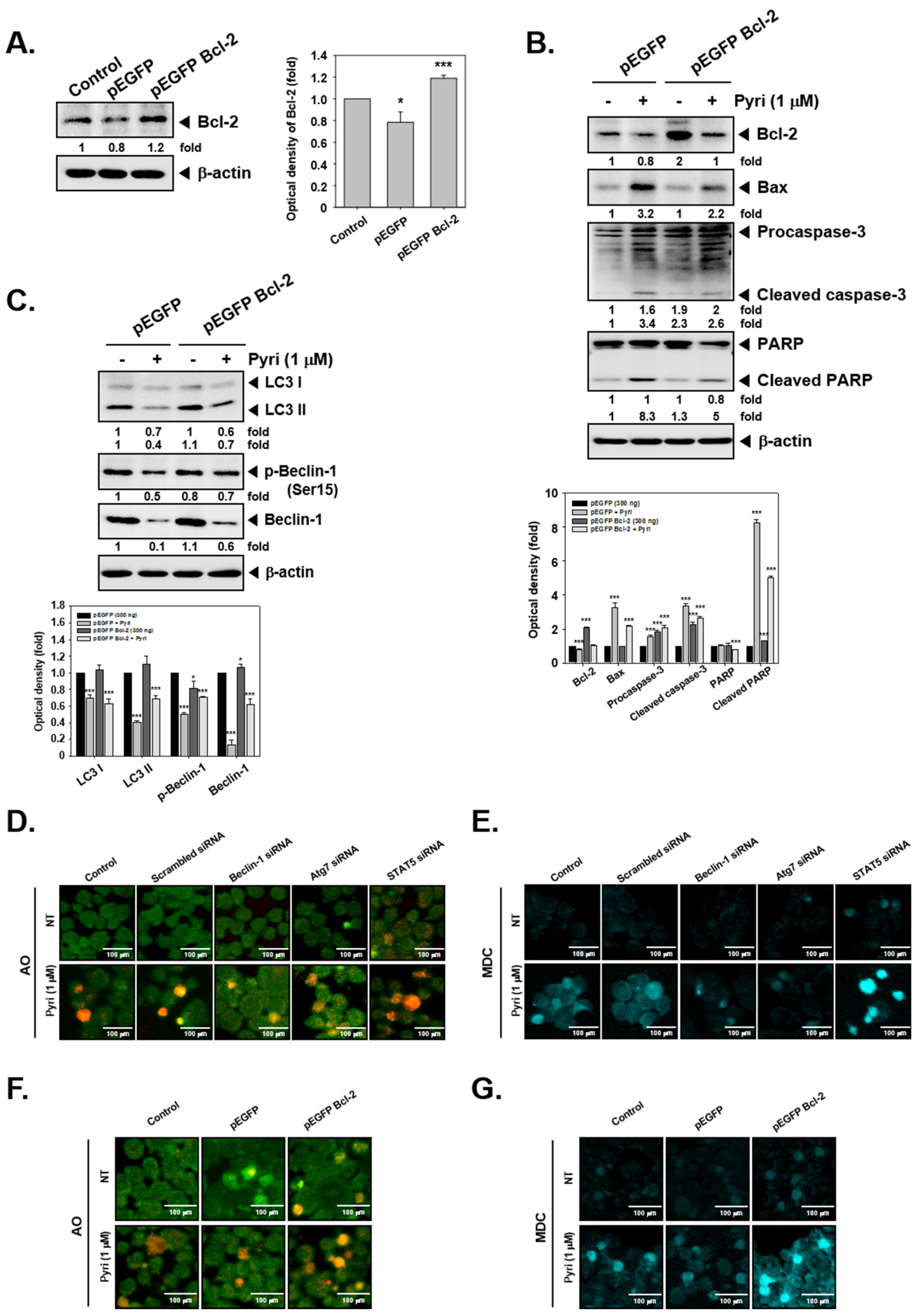
2.12. Bcl-2 Overexpression Induced the Autophagy Activation in KBM5 Cells
2.13. Effect on Induction of Autophagy by Pyri after Beclin-1, Atg7, STAT5, and Bcl-2 Gene Transfection
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Culture Conditions
4.3. MTT Assay
4.4. Western Blot Analysis
4.5. RT-PCR
4.6. Live and Dead Assay
4.7. Acridine Orange Assay
4.8. MDC Staining
4.9. Cell Cycle Analysis
4.10. Annexin V Assay
4.11. TUNEL Assay
4.12. Mitochondrial Membrane Potential (MMP) Assay
4.13. Immunocytochemistry for LC3 Expression
4.14. Knockdown of Beclin-1 and Atg7 by siRNA Transfection
4.15. Blockage of STAT5 Expression by siRNA
4.16. Bcl-2 Overexpression Studies
4.17. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- USPHS/IDSA Prevention of Opportunistic Infections Working Group. 1999 USPHS/IDSA guidelines for the prevention of opportunistic infections in persons infected with human immunodeficiency virus. Clin. Infect. Dis. 2000, 30, S29–S65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bygbjerg, I.C. Pyrimethamine-induced alterations in human lymphocytes in vitro. Mechanisms and reversal of the effect. Acta Pathol. Microbiol. Immunol. Scand. C 1985, 93, 183–188. [Google Scholar] [CrossRef]
- Bygbjerg, I.C.; Odum, N.; Theander, T.G. Effect of pyrimethamine and sulphadoxine on human lymphocyte proliferation. Trans. R Soc. Trop. Med. Hyg. 1986, 80, 295–300. [Google Scholar] [CrossRef]
- Viora, M.; De Luca, A.; D’Ambrosio, A.; Antinori, A.; Ortona, E. In vitro and in vivo immunomodulatory effects of anti-Pneumocystis carinii drugs. Antimicrob. Agents Chemother. 1996, 40, 1294–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Werff Ten Bosch, J.; Schotte, P.; Ferster, A.; Azzi, N.; Boehler, T.; Laurey, G.; Arola, M.; Demanet, C.; Beyaert, R.; Thielemans, K.; et al. Reversion of autoimmune lymphoproliferative syndrome with an antimalarial drug: Preliminary results of a clinical cohort study and molecular observations. Br. J. Haematol. 2002, 117, 176–188. [Google Scholar] [CrossRef]
- Pierdominici, M.; Giammarioli, A.M.; Gambardella, L.; De Felice, M.; Quinti, I.; Iacobini, M.; Carbonari, M.; Malorni, W.; Giovannetti, A. Pyrimethamine (2,4-diamino-5-p-chlorophenyl-6-ethylpyrimidine) induces apoptosis of freshly isolated human T lymphocytes, bypassing CD95/Fas molecule but involving its intrinsic pathway. J. Pharmacol. Exp. Ther. 2005, 315, 1046–1057. [Google Scholar] [CrossRef] [Green Version]
- Calabretta, B.; Perrotti, D. The biology of CML blast crisis. Blood 2004, 103, 4010–4022. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.Y.; Shanmugam, M.K.; Chinnathambi, A.; Alharbi, S.A.; Shair, O.H.M.; Um, J.Y.; Sethi, G.; Ahn, K.S. Fangchinoline, a Bisbenzylisoquinoline Alkaloid can Modulate Cytokine-Impelled Apoptosis via the Dual Regulation of NF-kappaB and AP-1 Pathways. Molecules 2019, 24, 3127. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Lee, J.H.; Kim, S.H.; Sethi, G.; Ahn, K.S. Artesunate suppresses tumor growth and induces apoptosis through the modulation of multiple oncogenic cascades in a chronic myeloid leukemia xenograft mouse model. Oncotarget 2015, 6, 4020–4035. [Google Scholar] [CrossRef]
- Ahn, K.S.; Sethi, G.; Aggarwal, B.B. Reversal of chemoresistance and enhancement of apoptosis by statins through down-regulation of the NF-kappaB pathway. Biochem. Pharmacol. 2008, 75, 907–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shishodia, S.; Sethi, G.; Ahn, K.S.; Aggarwal, B.B. Guggulsterone inhibits tumor cell proliferation, induces S-phase arrest, and promotes apoptosis through activation of c-Jun N-terminal kinase, suppression of Akt pathway, and downregulation of antiapoptotic gene products. Biochem. Pharmacol. 2007, 74, 118–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danial, N.N.; Rothman, P. JAK-STAT signaling activated by Abl oncogenes. Oncogene 2000, 19, 2523–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.K.; Langdon, W.Y.; Varticovski, L. Tyrosine phosphorylation of p120cbl in BCR/abl transformed hematopoietic cells mediates enhanced association with phosphatidylinositol 3-kinase. Oncogene 1997, 14, 2217–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuchi, D.; Kurosu, T.; Kida, A.; Jin, Z.H.; Jin, A.; Arai, A.; Miura, O. BCR/ABL activates Rap1 and B-Raf to stimulate the MEK/Erk signaling pathway in hematopoietic cells. Biochem. Biophys Res. Commun. 2005, 326, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Arora, L.; Kumar, A.P.; Arfuso, F.; Chng, W.J.; Sethi, G. The Role of Signal Transducer and Activator of Transcription 3 (STAT3) and Its Targeted Inhibition in Hematological Malignancies. Cancers 2018, 10, 327. [Google Scholar] [CrossRef] [Green Version]
- Ong, P.S.; Wang, L.Z.; Dai, X.; Tseng, S.H.; Loo, S.J.; Sethi, G. Judicious Toggling of mTOR Activity to Combat Insulin Resistance and Cancer: Current Evidence and Perspectives. Front. Pharmacol. 2016, 7, 395. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Talpaz, M.; Giles, F.; O’Brien, S.; Cortes, J. New insights into the pathophysiology of chronic myeloid leukemia and imatinib resistance. Ann. Intern. Med. 2006, 145, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.S.; Yang, S.F.; Sethi, G.; Hu, D.N. Natural bioactives in cancer treatment and prevention. Biomed. Res. Int. 2015, 2015, 182835. [Google Scholar] [CrossRef] [Green Version]
- Shanmugam, M.K.; Warrier, S.; Kumar, A.P.; Sethi, G.; Arfuso, F. Potential Role of Natural Compounds as Anti-Angiogenic Agents in Cancer. Curr. Vasc. Pharmacol. 2017, 15, 503–519. [Google Scholar] [CrossRef]
- Yang, S.F.; Weng, C.J.; Sethi, G.; Hu, D.N. Natural bioactives and phytochemicals serve in cancer treatment and prevention. Evid. Based Complement. Alternat. Med. 2013, 2013, 698190. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Targeting TNF-related apoptosis-inducing ligand (TRAIL) receptor by natural products as a potential therapeutic approach for cancer therapy. Exp. Biol. Med. 2015, 240, 760–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, M.K.; Ong, T.H.; Kumar, A.P.; Lun, C.K.; Ho, P.C.; Wong, P.T.; Hui, K.M.; Sethi, G. Ursolic acid inhibits the initiation, progression of prostate cancer and prolongs the survival of TRAMP mice by modulating pro-inflammatory pathways. PLoS ONE 2012, 7, e32476. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Lee, J.H.; Ko, J.H.; Chinnathambi, A.; Alharbi, S.A.; Shair, O.H.M.; Sethi, G.; Ahn, K.S. Formononetin Regulates Multiple Oncogenic Signaling Cascades and Enhances Sensitivity to Bortezomib in a Multiple Myeloma Mouse Model. Biomolecules 2019, 9, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.T.; Kim, C.; Lee, J.H.; Chinnathambi, A.; Alharbi, S.A.; Shair, O.H.M.; Sethi, G.; Ahn, K.S. Cycloastragenol can negate constitutive STAT3 activation and promote paclitaxel-induced apoptosis in human gastric cancer cells. Phytomedicine 2019, 59, 152907. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Wyllie, A.H.; Kerr, J.F.; Currie, A.R. Cell death: The significance of apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar]
- Cryns, V.; Yuan, J. Proteases to die for. Genes Dev. 1998, 12, 1551–1570. [Google Scholar] [CrossRef] [Green Version]
- Los, M.; Wesselborg, S.; Schulze-Osthoff, K. The role of caspases in development, immunity, and apoptotic signal transduction: Lessons from knockout mice. Immunity 1999, 10, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lazebnik, Y.A.; Kaufmann, S.H.; Desnoyers, S.; Poirier, G.G.; Earnshaw, W.C. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 1994, 371, 346–347. [Google Scholar] [CrossRef] [PubMed]
- Los, M.; Herr, I.; Friesen, C.; Fulda, S.; Schulze-Osthoff, K.; Debatin, K.M. Cross-resistance of CD95- and drug-induced apoptosis as a consequence of deficient activation of caspases (ICE/Ced-3 proteases). Blood 1997, 90, 3118–3129. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar] [PubMed]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef]
- Sethi, G.; Sung, B.; Kunnumakkara, A.B.; Aggarwal, B.B. Targeting TNF for Treatment of Cancer and Autoimmunity. Adv. Exp. Med. Biol. 2009, 647, 37–51. [Google Scholar] [PubMed]
- Ahn, K.S.; Sethi, G.; Jain, A.K.; Jaiswal, A.K.; Aggarwal, B.B. Genetic deletion of NAD(P)H:quinone oxidoreductase 1 abrogates activation of nuclear factor-kappaB, IkappaBalpha kinase, c-Jun N-terminal kinase, Akt, p38, and p44/42 mitogen-activated protein kinases and potentiates apoptosis. J. Biol. Chem. 2006, 281, 19798–19808. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.Y.; Lee, J.H.; Nam, D.; Narula, A.S.; Namjoshi, O.A.; Blough, B.E.; Um, J.Y.; Sethi, G.; Ahn, K.S. Anti-myeloma Effects of Icariin Are Mediated Through the Attenuation of JAK/STAT3-Dependent Signaling Cascade. Front. Pharmacol. 2018, 9, 531. [Google Scholar] [CrossRef]
- Jung, Y.Y.; Shanmugam, M.K.; Narula, A.S.; Kim, C.; Lee, J.H.; Namjoshi, O.A.; Blough, B.E.; Sethi, G.; Ahn, K.S. Oxymatrine Attenuates Tumor Growth and Deactivates STAT5 Signaling in a Lung Cancer Xenograft Model. Cancers 2019, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Shanmugam, M.K.; Kumar, A.P.; Yap, C.T.; Sethi, G.; Bishayee, A. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer 2019, 125, 1228–1246. [Google Scholar] [CrossRef]
- Praharaj, P.P.; Naik, P.P.; Panigrahi, D.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Sethi, G.; Bhutia, S.K. Intricate role of mitochondrial lipid in mitophagy and mitochondrial apoptosis: Its implication in cancer therapeutics. Cell Mol. Life Sci. 2019, 76, 1641–1652. [Google Scholar] [CrossRef]
- Mizushima, N. The pleiotropic role of autophagy: From protein metabolism to bactericide. Cell Death Differ. 2005, 12, 1535–1541. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Bio. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Kroemer, G.; Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Invest. 2009, 119, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Borghouts, C.; Brendel, C.; Moriggl, R.; Delis, N.; Brill, B.; Vafaizadeh, V.; Groner, B. Stat5 Exerts Distinct, Vital Functions in the Cytoplasm and Nucleus of Bcr-Abl(+) K562 and Jak2(V617F)(+) HEL Leukemia Cells. Cancers 2015, 7, 503–537. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.; Sexl, V.; Valent, P.; Moriggl, R. Inhibition of STAT5: A therapeutic option in BCR-ABL1-driven leukemia. Oncotarget 2014, 5, 9564–9576. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Cho, S.K.; Kapoor, S.; Kumar, A.; Vali, S.; Abbasi, T.; Kim, S.H.; Sethi, G.; Ahn, K.S. beta-Caryophyllene oxide inhibits constitutive and inducible STAT3 signaling pathway through induction of the SHP-1 protein tyrosine phosphatase. Mol. Carcinog. 2014, 53, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, C.; Sethi, G.; Ahn, K.S. Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget 2015, 6, 6386–6405. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Chiang, S.Y.; Nam, D.; Chung, W.S.; Lee, J.; Na, Y.S.; Sethi, G.; Ahn, K.S. Capillarisin inhibits constitutive and inducible STAT3 activation through induction of SHP-1 and SHP-2 tyrosine phosphatases. Cancer Lett. 2014, 345, 140–148. [Google Scholar] [CrossRef]
- Mohan, C.D.; Bharathkumar, H.; Bulusu, K.C.; Pandey, V.; Rangappa, S.; Fuchs, J.E.; Shanmugam, M.K.; Dai, X.; Li, F.; Deivasigamani, A.; et al. Development of a novel azaspirane that targets the Janus kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 34296–34307. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Lee, S.G.; Yang, W.M.; Arfuso, F.; Um, J.Y.; Kumar, A.P.; Bian, J.; Sethi, G.; Ahn, K.S. Formononetin-induced oxidative stress abrogates the activation of STAT3/5 signaling axis and suppresses the tumor growth in multiple myeloma preclinical model. Cancer Lett. 2018, 431, 123–141. [Google Scholar] [CrossRef]
- Bandapalli, O.R.; Schuessele, S.; Kunz, J.B.; Rausch, T.; Stutz, A.M.; Tal, N.; Geron, I.; Gershman, N.; Izraeli, S.; Eilers, J.; et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica 2014, 99, E188–E192. [Google Scholar] [CrossRef]
- Vafaizadeh, V.; Klemmt, P.; Brendel, C.; Weber, K.; Doebele, C.; Britt, K.; Grez, M.; Fehse, B.; Desrivieres, S.; Groner, B. Mammary Epithelial Reconstitution with Gene-Modified Stem Cells Assigns Roles to Stat5 in Luminal Alveolar Cell Fate Decisions, Differentiation, Involution, and Mammary Tumor Formation. Stem Cells 2010, 28, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Gouilleux, F.; Wakao, H.; Mundt, M.; Groner, B. Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO J. 1994, 13, 4361–4369. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Yang, S.; Zhu, X.; Sun, D.; Sun, D.; Jiang, X.; Zhang, C.; Wang, L. The RAGE/STAT5/autophagy axis regulates senescence in mesangial cells. Cell Signal. 2019, 62, 109334. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Joza, N.; Tasdemir, E.; Maiuri, M.C.; Hengartner, M.; Abrams, J.M.; Tavernarakis, N.; Penninger, J.; Madeo, F.; Kroemer, G. No death without life: Vital functions of apoptotic effectors. Cell Death Differ. 2008, 15, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Oberst, A.; Bender, C.; Green, D.R. Living with death: The evolution of the mitochondrial pathway of apoptosis in animals. Cell Death Differ. 2008, 15, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Manu, K.A.; Shanmugam, M.K.; Li, F.; Chen, L.; Siveen, K.S.; Ahn, K.S.; Kumar, A.P.; Sethi, G. Simvastatin sensitizes human gastric cancer xenograft in nude mice to capecitabine by suppressing nuclear factor-kappa B-regulated gene products. J. Mol. Med. 2014, 92, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Sethi, G.; Shanmugam, M.K.; Warrier, S.; Merarchi, M.; Arfuso, F.; Kumar, A.P.; Bishayee, A. Pro-Apoptotic and Anti-Cancer Properties of Diosgenin: A Comprehensive and Critical Review. Nutrients 2018, 10, 645. [Google Scholar] [CrossRef] [Green Version]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.H.; Ko, J.H.; Lee, H.; Jung, J.; Kong, M.; Lee, J.W.; Lee, J.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; et al. Resveratrol inhibits STAT3 signaling pathway through the induction of SOCS-1: Role in apoptosis induction and radiosensitization in head and neck tumor cells. Phytomedicine 2016, 23, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, C.; Kim, S.H.; Sethi, G.; Ahn, K.S. Farnesol inhibits tumor growth and enhances the anticancer effects of bortezomib in multiple myeloma xenograft mouse model through the modulation of STAT3 signaling pathway. Cancer Lett. 2015, 360, 280–293. [Google Scholar] [CrossRef]
- Carrington, E.M.; Zhan, Y.F.; Brady, J.L.; Zhang, J.G.; Sutherland, R.M.; Anstee, N.S.; Schenk, R.L.; Vikstrom, I.B.; Delconte, R.B.; Segal, D.; et al. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ. 2017, 24, 878–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, M.T. Anti-apoptosis and cell survival: A review. Bba-Mol. Cell Res. 2011, 1813, 238–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A molecular biomarker in cancer. Indian J. Med. Res. 2015, 141, 389–397. [Google Scholar]
- Goradel, N.H.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology-Basel 2005, 69, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef] [Green Version]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Kirtonia, A.; Sethi, G.; Garg, M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Manu, K.A.; Shanmugam, M.K.; Ramachandran, L.; Li, F.; Siveen, K.S.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Arfuso, F.; Kumar, A.P.; et al. Isorhamnetin augments the anti-tumor effect of capecitabine through the negative regulation of NF-kappaB signaling cascade in gastric cancer. Cancer Lett. 2015, 363, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Canc. Res. 2011, 30. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar]
- Kraft, C.; Martens, S. Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 2012, 24, 496–501. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carames, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum Dis. 2012, 71, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Bouderlique, T.; Vuppalapati, K.K.; Newton, P.T.; Li, L.; Barenius, B.; Chagin, A.S. Targeted deletion of Atg5 in chondrocytes promotes age-related osteoarthritis. Ann. Rheum Dis. 2016, 75, 627–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carames, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy Is a Protective Mechanism in Normal Cartilage, and Its Aging-Related Loss Is Linked With Cell Death and Osteoarthritis. Arthritis Rheum-Us 2010, 62, 791–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, A.; Borghouts, C.; Brendel, C.; Moriggl, R.; Delis, N.; Brill, B.; Vafaizadeh, V.; Groner, B. The inhibition of stat5 by a Peptide aptamer ligand specific for the DNA binding domain prevents target gene transactivation and the growth of breast and prostate tumor cells. Pharmaceuticals 2013, 6, 960–987. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, C.; Um, J.Y.; Sethi, G.; Ahn, K.S. Casticin-Induced Inhibition of Cell Growth and Survival Are Mediated through the Dual Modulation of Akt/mTOR Signaling Cascade. Cancers 2019, 11, 254. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Shanmugam, M.K.; Chen, L.; Chatterjee, S.; Basha, J.; Kumar, A.P.; Kundu, T.K.; Sethi, G. Garcinol, a polyisoprenylated benzophenone modulates multiple proinflammatory signaling cascades leading to the suppression of growth and survival of head and neck carcinoma. Cancer Prev. Res. 2013, 6, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.H.; Jung, S.H.; Chinnathambi, A.; Alahmadi, T.A.; Alharbi, S.A.; Sethi, G.; Ahn, K.S. Attenuation of STAT3 Signaling Cascade by Daidzin Can Enhance the Apoptotic Potential of Bortezomib against Multiple Myeloma. Biomolecules 2019, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, A.; Loo, S.Y.; Rajendran, P.; Manu, K.A.; Perumal, E.; Li, F.; Shanmugam, M.K.; Siveen, K.S.; Park, J.I.; Ahn, K.S.; et al. An anthraquinone derivative, emodin sensitizes hepatocellular carcinoma cells to TRAIL induced apoptosis through the induction of death receptors and downregulation of cell survival proteins. Apoptosis 2013, 18, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.S.; Sethi, G.; Aggarwal, B.B. Simvastatin potentiates TNF-alpha-induced apoptosis through the down-regulation of NF-kappaB-dependent antiapoptotic gene products: Role of IkappaBalpha kinase and TGF-beta-activated kinase-1. J. Immunol. 2007, 178, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Chinnathambi, A.; Alharbi, S.A.; Shair, O.H.M.; Sethi, G.; Ahn, K.S. Farnesol abrogates epithelial to mesenchymal transition process through regulating Akt/mTOR pathway. Pharmacol. Res. 2019, 150, 104504. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, Y.Y.; Kim, C.; Ha, I.J.; Lee, S.-G.; Lee, J.; Um, J.-Y.; Ahn, K.S. Pyrimethamine Modulates Interplay between Apoptosis and Autophagy in Chronic Myelogenous Leukemia Cells. Int. J. Mol. Sci. 2021, 22, 8147. https://doi.org/10.3390/ijms22158147
Jung YY, Kim C, Ha IJ, Lee S-G, Lee J, Um J-Y, Ahn KS. Pyrimethamine Modulates Interplay between Apoptosis and Autophagy in Chronic Myelogenous Leukemia Cells. International Journal of Molecular Sciences. 2021; 22(15):8147. https://doi.org/10.3390/ijms22158147
Chicago/Turabian StyleJung, Young Yun, Chulwon Kim, In Jin Ha, Seok-Geun Lee, Junhee Lee, Jae-Young Um, and Kwang Seok Ahn. 2021. "Pyrimethamine Modulates Interplay between Apoptosis and Autophagy in Chronic Myelogenous Leukemia Cells" International Journal of Molecular Sciences 22, no. 15: 8147. https://doi.org/10.3390/ijms22158147
APA StyleJung, Y. Y., Kim, C., Ha, I. J., Lee, S. -G., Lee, J., Um, J. -Y., & Ahn, K. S. (2021). Pyrimethamine Modulates Interplay between Apoptosis and Autophagy in Chronic Myelogenous Leukemia Cells. International Journal of Molecular Sciences, 22(15), 8147. https://doi.org/10.3390/ijms22158147