4.2. Synthetic Procedures and Analytical Data
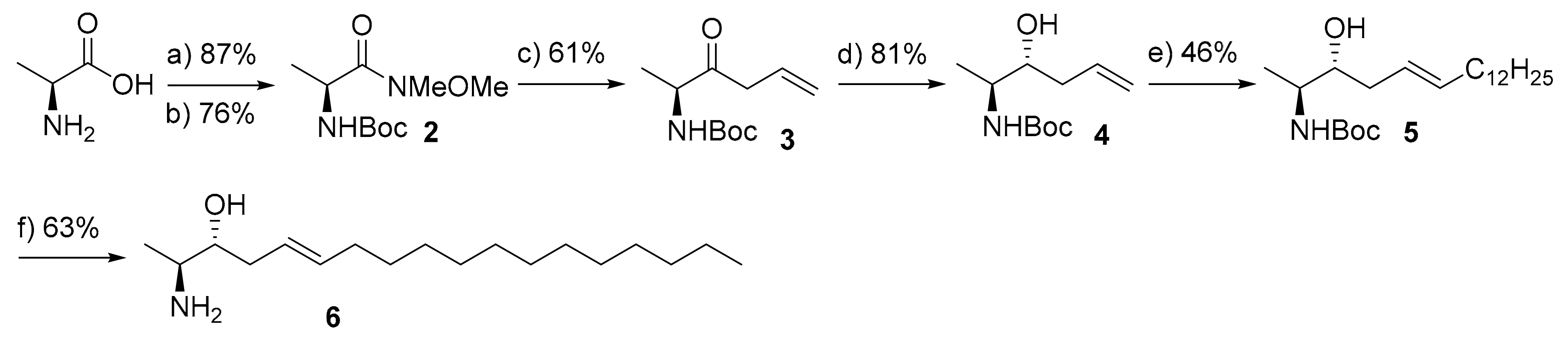
4.2.1. (2S,3R,E)-tert-Butyl-(3-hydroxyoctadec-5-en-2-yl)carbamate (5)
To a stirred solution of compound 4 (210 mg, 1 mmol) and 1-tetradecene (0.8 g, 4 mmol) in dry d-chloroform (5 mL) under argon atmosphere at ambient temperature was added p-benzoquinone (10 mol%) and CuI (2 mol%) followed by a catalytic amount of Grubbs 2nd generation catalyst (10 mol%). The resulting reaction mixture was allowed to stir at ambient temperature for 12 h (as monitored by TLC analysis, no change in the composition of reaction mixture, Pet. ether/ EtOAc 4:1; Rf (adduct) = 0.42; Rf (product) =0.56; visualized with 1.3% ninhydrine). The mixture was concentrated under reduced pressure and the resultant residue was purified by flash column chromatography over silica gel using petroleum ether and ethylacetate as eluents (0–10 % ethylacetate in petroleum ether) to provide the desired product 5 as colorless oil.
Yield: 184 mg (46 %). Rf: 0.56 (Pet. ether /EtOAc 4:1, visualized with 1.3% ninhydrin solution). 1H NMR (CDCl3, 500 MHz, ppm) δ 5.55 (dt, J = 13.6, 6.7 Hz, 1H), 5.44–5.37 (m, 1H), 4.78 (br.s, 1H), 3.74–3.65 (m, 1H), 3.65–3.60 (m, 1H), 2.22–2.15 (m, 1H), 2.09 (dd, J = 15.0, 7.2 Hz, 1H), 2.00 (dd, J = 14.4, 7.2 Hz, 2H), 1.44 (s, 9H), 1.37–1.24 (m, 20H), 1.10 (d, J = 6.8 Hz, 3H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (CDCl3, 126 MHz, ppm) δ 155.71, 134.8, 125.5, 79.4, 73.48, 50.1, 37.2, 32.7, 31.9, 29.7, 29.6, 29.6, 29.5, 29.4, 29.4, 29.2, 28.4, 22.7, 14.1. ESI-MS m/z calcd for C23H45NO3Na [M + Na]+ 406.33; observed: 406.3.
4.2.2. (2S,3R,E)-2-Aminooctadec-5-en-3-ol (6)
To a stirred solution of compound 5 (135 mg, 0.36 mmol) in dry methanol (4 mL) at 0 °C was added dropwise acetyl chloride (260 µL, 3.6 mmol) over a period of 10 min. After being stirred at the same conditions for 30 min and for additional 2 h at ambient temperature (as indicated by TLC analysis for complete deprotection), the mixture was concentrated in vacuo. The residue was diluted with diethylether and washed with sat. NaHCO3 solution. The aqueous layer was extracted again with diethylether and the combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The obtained residue was purified by flash column chromatography over silica gel using isopropanol and ethylacetate as eluents (0–10% isopropanol in ethylacetate) to afford the pure product 6 as a white waxy-white solid.
Yield: 64 mg (63%). R
f: 0.45 (EtOAc/ iso-propanol 4:1, visualized with 1.3% ninhydrin).
= +3.2 (c = 0.65, MeOH).
1H NMR (MeOD, 500 MHz, ppm) δ 5.58 (dd,
J = 14.3, 7.5 Hz, 1H), 5.44 (dd,
J = 14.6, 7.2 Hz, 1H), 3.78–3.72 (m, 1H), 3.26 (dd,
J = 6.7, 2.8 Hz, 1H), 2.29–2.13 (m, 2H), 2.03 (dd,
J = 13.3, 6.6 Hz, 2H), 1.42–1.27 (m, 20H), 1.23 (d,
J = 6.8 Hz, 3H), 0.90 (t,
J = 6.7 Hz, 3H).
13C NMR (MeOD, 126 MHz, ppm) δ 135.4, 126.2, 71.7, 51.9, 37.7, 33.67, 33.0, 30.8, 30.7, 30.6, 30.4, 30.4, 23.8, 14.5, 11.6. ESI-MS:
m/
z calcd for C
18H
38NO [M + H]
+ 284.29; observed 284.3 [
32].
4.2.3. (S,E)-tert-Butyl-(3-oxooctadec-6-en-2-yl)carbamate (8)
A stirred solution of compound 7 (110 mg, 0.47 mmol) and 1-tridecene (0.34 g, 1.9 mmol) in dry d-chloroform (4 mL) under argon atmosphere at ambient temperature was treated a catalytic amount of p-benzoquinone (10 mol%) followed by CuI (2 mol%) and Grubbs catalyst 2nd generation (10 mol%). After the resulting reaction mixture was stirred under reflux for 6 h (as monitored by TLC analysis; no further change in the composition of the reaction mixture; Pet. ether/ EtOAc 4:1; Rf (adduct) = 0.55; Rf (product) =0.68; visualized with 1.3% ninhydrin), the solvent was removed under reduced pressure. The obtained residue was subjected to flash column chromatography over silica gel using petroleum ether and ethylacetate as eluents (0–10% ethylacetate in petroleum ether) to afford the desired product 8 as a waxy-white solid.
Yield: 95 mg (53 %). Rf: 0.47 (Pet. ether /EtOAc 9:1, visualized with 1.3% ninhydrine solution). 1H NMR (CDCl3, 500 MHz, ppm) δ 5.52–5.34 (m, 1H), 5.33–5.25 (m, 1H), 5.20 (br.s, 1H), 4.35–4.20 (m, 1H), 2.59–2.41 (m, 1H), 2.28–2.18 (m, 1H), 1.99–1.84 (m, 4H), 1.37 (s, 9H), 1.30–1.17 (m, 21H), 0.81 (t, J = 7.0 Hz, 3H). 13C NMR (CDCl3, 126 MHz, ppm) δ 209.1, 155.2, 132.0, 127.85, 8.7, 55.1, 39.17, 32.51, 31.9, 29.6, 29.52, 29.4, 29.3, 29.2, 28.3, 26.6, 22.7, 17.9, 14.1. ESI-MS m/z calcd for C23H43NO3Na [M + Na]+ 404.31; observed: 404.3.
4.2.4. (S,E)-2-Aminooctadec-6-en-3-one (9)
To a stirred solution of compound 8 (77 mg, 0.2 mmol) in absolute methanol (2 mL) at 0 °C was dropwisely added acetylchloride (143 µL, 2 mmol) over a period of 10 min. The resulting reaction mixture was allowed to stir at the same conditions for 30 min, gradually warm to ambient temperature, and stirred for additional 2 h; during which a white solid formed. The mixture was filtered, and the residue was washed several times with ice-cooled diethylether to afford a white waxy-solid of crude product. The obtained crude product was further purified by flash column chromatography over silica gel using ethylacetate and isopropanol as eluents (0–10% isopropanol in ethylacetate) to provide the final pure product 9 as a white solid.
Yield: 38 mg (67%). R
f: 0.51 (EtOAc/iso-propanol 4:1, visualized with KMnO
4 solution).
= +13.1 (c = 1.1, MeOH).
1H NMR (MeOD, 500 MHz, ppm) δ 5.55–5.47 (m, 1H), 5.45–5.39 (m, 1H), 4.14 (q,
J = 7.3 Hz, 1H), 2.76–2.69 (m, 1H), 2.66–2.58 (m, 1H), 2.35–2.27 (m, 2H), 1.98 (q,
J = 6.6 Hz, 2H), 1.53–1.49 (m, 4H), 1.37–1.27 (m, 17H), 0.90 (t,
J = 7.0 Hz, 3H).
13C NMR (MeOD, 126 MHz, ppm) δ 206.8, 133.1, 129.1, 55.9, 39.3, 33.6, 33.1, 30.8, 30.7, 30.6, 30.5, 30.3, 27.2, 23.7, 15.7, 14.4. ESI-MS:
m/
z calcd for C
11H
36NO [M + H]
+ 282.28; observed 282.3 [
32].
4.2.5. (S)-tert-Butyl-(3-oxooctadecan-2-yl)carbamate (10)
The 1-pentadecylmagnesium bromide solution was synthesized as follows; A mixture of magnesium turnings (0.51 g, 21 mmol) in anhydrous THF (4 mL) under an argon atmosphere at ambient temperature was treated with drops of 1,2-dibromoethane. The resulting mixture was allowed to stir at the same conditions for 20 min, before it was treated dropwisely with a solution of 1-bromopentadecane (1.5 g, 5.2 mmol, in 2 mL of anhydrous THF) over a period of 20 min. The resulting reaction mixture was allowed to stir for additional 3 h at 35 °C to afford a transparent solution of 1-pentadecylmagnesium bromide which was immediately used in the next step.
A stirred solution of Weinreb amide derivative 2 (0.35 g, 1.5 mmol) in anhydrous THF (15 mL) under an argon atmosphere at 0 °C was treated dropwisely with a freshly prepared 1-pentadecylmagnesium bromide solution (6 mL, 5.1 mmol, 1 M solution in Et2O) over a period of 10 min. After being stirred at ambient temperature for 2 h (as monitored by TLC analysis; Pet. ether / EtOAc 4:1; Rf (adduct) = 0.2; Rf (product) =0.69; visualized with 1.3% ninhydrin solution), the reaction mixture was cooled again to 0 °C and subsequently quenched with an ice-cold 1 M HCl solution (40 mL). The resulting mixture was diluted with diethylether (50 mL), and the layer were separated. The aqueous layer was extracted with diethylether (3 × 50 mL), and the combined organic phases were washed with saturated NaHCO3 solution (80 mL) and brine (80 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resultant crude product was purified by flash column chromatography over silica gel using petroleum ether and ethylacetate as eluents (from 5–15% ethylacetate in petroleum ether) to yield compound 10 as white solid.
Yield: 410 mg (71 %). Rf: 0.48 (Pet. ether /EtOAc 9:1, visualized with 1.3% ninhydrine solution). 1H NMR (CDCl3, 500 MHz, ppm) δ 5.28 (d, J = 6.0 Hz, 1H), 4.30 (p, J = 7.0 Hz, 1H), 2.55–2.40 (m, 2H), 1.62–1.54 (m, 2H), 1.43 (s, 9H), 1.31 (d, J = 7.2 Hz, 3H), 1.29–1.21 (m, 24H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR (CDCl3, 126 MHz, ppm) δ 209.9, 155.2, 79.6, 55.0, 39.2, 31.9, 29.7, 29.7, 29.6, 29.4, 29.4, 29.2, 28.3, 23.6, 22.7, 18.0, 14.1. ESI-MS m/z calcd for C23H46NO3 [M + H]+ 384.34; observed: 384.3.
4.2.6. (2S,3R)-tert-Butyl-(3-hydroxyoctadecan-2-yl)carbamate (11)
To a stirred solution of compound 10 (0.34 g, 0.89 mmol) in dry ethanol (2 mL) under argon atmosphere at −78 °C was added portionwise lithium tri-(tert-butoxy)-aluminum hydride (0.57 g, 2.2 mmol) over a period of 20 min. After the resulting reaction mixture was allowed to stir at the same conditions for 2 h (as judged by TLC analysis: Pet. ether / EtOAc 4:1; Rf (adduct) = 0.69; Rf (product) =0.57; visualized with 1.3% ninhydrine solution), an ice-cold 1 M HCl solution (100 mL) was added dropwise to quench the reaction. The resulting mixture was allowed to warm gradually to ambient temperature and subsequently diluted with ethylacetate (100 mL). The layers were separated and the aqueous layer was extracted again with ethylacetate (2 × 100 mL). The combined organic extracts were washed with sat. NaHCO3 solution and brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Flash column chromatography for the obtained residue over silica gel using petroleum ether and ethylacetate as eluents (15–23% ethylacetate in petroleum ether) afforded the desired product 11 as a white solid.
Yield: 285 mg (83 %). Rf: 0.57 (Pet. ether /EtOAc 4:1, visualized with 1.3% ninhydrin solution). 1H NMR (CDCl3, 500 MHz, ppm) δ 4.63 (br. s, 1H), 3.77–3.53 (m, 2 H), 1.46 (s, 9H), 1.45–1.22 (m, 28H), 1.07 (d, J = 6.8 Hz, 3H), 0.88 (t, J = 6.4 Hz, 3H). 13C NMR (CDCl3, 126 MHz, ppm) δ 155.8, 79.6, 74.6, 50.7, 33.6, 32.1, 29.8, 29.7, 29.6, 29.4, 28.5, 26.2, 22.7, 14.4, 14.1. ESI-MS m/z calcd for C23H48NO3 [M + H]+ 386.36; observed: 386.4.
4.2.7. (2S,3R)-2-Aminooctadecan-3-ol (12)
A solution of compound 11 (0.15 g, 0.38 mmol) in dry methanol (4 mL) at 0 °C was treated with acetyl chloride (272 µL, 3.8 mmol) in dropwise over a period of 10 min. After the resulting reaction mixture was stirred at the same conditions for 30 min and for additional 1 h at ambient temperature (as controlled by TLC analysis; Pet. ether/EtOAc 3:2; Rf (adduct) = 0.51; Rf (product) = 0.0; visualized with 1.3% ninhydrin solution), the solvent was removed under reduced pressure. The obtained residue was portioned between diethylether (100 mL) and sat. NaHCO3 solution (90 mL). The layers were separated and the aqueous layer was extracted again with diethylether (2 × 80 mL). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to afford a white waxy solid. The crude product was further purified by flash column chromatography over silica gel using ethylacetate and isopropanol as eluents (0–10% isopropanol in ethylacetate) to provide the final product 12 as a white solid.
Yield: 84 mg (77%). R
f: 0.47 (EtOAc/ iso-propanol 9:1, visualized with 1.3% ninhydrine).
= +8.4 (c = 1.2, MeOH); [ref +5.2 (c = 0.36, MeOH)] [
53].
1H NMR (MeOD, 500 MHz, ppm) δ 3.72–3.68 (m, 1H), 3.27 (qd,
J = 6.8, 3.0 Hz, 1H), 1.53 (dt,
J = 11.0, 8.2 Hz, 1H), 1.45 (dt,
J = 13.6, 4.6 Hz, 2H), 1.38–1.27 (m, 26H), 1.22 (d,
J = 6.8 Hz, 3H), 0.90 (t,
J = 7.0 Hz, 3H).
13C NMR (MeOD, 126 MHz, ppm) δ 71.6, 52.6, 34.0, 33.1, 30.8, 30.8, 30.7, 30.7, 30.6, 30.5, 27.0, 23.7, 14.4, 12.0. ESI-MS:
m/
z calcd for C
18H
40NO [M + H]
+ 286.32; observed 286.3 [
32].
4.2.8. tert-Butyl-(2-oxoheptadecyl)carbamate (15)
1-Pentadecylmagnesium bromide was synthesized as follows; A mixture of magnesium powder (0.51 g, 21 mmol) in dry THF (2 mL) under argon atmosphere was treated with a catalytic drop of 1,2-dibromoethane followed by a dropwise addition of a solution of 1-bromopentadecane (1.5 g, 5.2 mmol, 1 M in dry TFH). The resulting reaction mixture was stirred under reflux for 2 h to afford a transparent solution of 1-pentadecylmagnesium bromide which was used in the next step.
To a stirred solution of compound 14 (330 mg, 1.5 mmol) in dry THF (15 mL) under argon atmosphere at 0 °C was added dropwise a freshly prepared 1-pentadecylmagnesium bromide solution over a period of 20 min. After being stirred for 2 h at ambient temperature (as monitored by TLC analysis; Pet. ether/ EtOAc 4:1; Rf (adduct) = 0.19; Rf (product) = 0.68; visualized with 1.3% ninhydrine), the reaction mixture was added in dropwise to an ice-cooled 1 M HCl solution (100 mL) to quench. The resulting mixture was diluted with ethylacetate (100 mL) and the layers were separated. The aqueous layer was extracted with ethylacetate (2 × 100 mL), and the combined organic layers were washed with sat. NaHCO3 solution and brine, dried over Na2SO4, filtered, and concentrated. The obtained residue was purified by flash column chromatography over silica gel using petroleum ether and ethylacetate as eluents (0–14% ethylacetate in petroleum ether) to afford compound 15 as a white solid.
Yield: 340 mg (62%). Rf: 0.47 (cyclohexane/EtOAc 9:1, visualized with 1.3% ninhydrin solution). 1H NMR (CDCl3, 500 MHz, ppm) δ 4.11 (br.s, 2H), 2.58–2.42 (m, 2H), 1.66–1.58 (m, 2H), 1.44 (s, 9H), 1.28–1.20 (m, 24H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (CDCl3, 126 MHz, ppm): δ 209.5, 155.3, 79.7, 42.6, 40.1, 33.2, 29.8, 29.6, 29.6, 29.5, 29.4, 29.3, 28.6, 23.6, 22.7, 14.2. ESI-LCMS: m/z calcd for C22H43NO3Na [M + Na]+ 392.31; observed 392.3.
4.2.9. (R)-tert-Butyl-(2-hydroxyheptadecyl)carbamate (16)
Lithium tri-(tert-butoxy)-aluminium hydride (0.49 g, 1.9 mmol) was added in portionwise to a stirred solution of compound 15 (285 mg, 0.76 mmol) in dry ethanol (2 mL) under argon atmosphere at −78 °C. After the resulting reaction mixture was stirred at the same conditions for 3 h (as detected by TLC analysis; Pet. ether/ EtOAc 4:1; Rf (adduct) = 0.69; Rf (product) = 0.55; visualized with 1.3% ninhydrin), an ice-cooled 1 M HCl solution (100 mL) was added in dropwise over a period of 20 min. The resulting mixture was allowed to warm to ambient temperature and was extracted with ethylacetate (3 × 100 mL). The combined organic extracts were washed with sat. NaHCO3 solution and brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification of the resultant residue by flash column chromatography over silica gel using petroleum ether and ethylacetate as eluents (10–20% ethylacetate in petroleum ether) afforded the desired product 16 as a white waxy-solid.
Yield: 210 mg (74%). Rf: 0.55 (cyclohexane/EtOAc 4:1, visualized with 1.3% ninhydrin solution). 1H NMR (CDCl3, 500 MHz, ppm): δ 1H NMR (CDCl3, 500 MHz, ppm) δ 4.48 (br. s, 1H), 3.79–3.72 (m, 1H), 3.17–2.89 (m, 2H), 1.47 (s, 9H), 1.47–1.24 (m, 28H), 0.89 (t, J = 6.5 Hz, 3H). 13C NMR (CDCl3, 126 MHz, ppm): δ 155.6, 79.5, 62.3, 43.7, 34.5, 32.6, 29.8, 29.7, 29.6, 29.4, 28.5, 26.3, 22.7, 14.1. ESI-LCMS: m/z calcd for C22H46NO3 [M + H]+ 372.35; observed 372.3.
4.2.10. (R)-1-Aminoheptadecan-2-ol (17)
Acetyl chloride (305 µL, 4.3 mmol) was added in dropwise to a stirred solution of compound 16 (0.16 g, 0.43 mmol) in dry methanol (4 mL) at 0 °C. The resulting reaction mixture was allowed to stir at the same conditions for 30 min and for additional 2 h at ambient temperature (as monitored by TLC analysis), during while a white solid was formed. The mixture was filtrated, and the residue was washed several times with ice-cooled diethylether to afford a white waxy-solid crude product. Purification of the crude product by flash column chromatography over silica gel using isopropanol and ethylacetate as eluents (0–10% isopropanol in ethyacetate) provided the final product 17 as a white solid.
Yield: 76 mg (65%). R
f: 0.39 (EtOAc/iso-propanol 10:1, visualized with 1.3% ninhydrin).
= +1.9 (c = 1.0, MeOH).
1H NMR (MeOD, 500 MHz, ppm) δ 3.77–3.70 (m, 1H), 3.01 (dd,
J = 12.7, 3.0 Hz, 1H), 2.75 (dd,
J = 12.7, 9.5 Hz, 1H), 1.48 (t,
J = 7.8 Hz, 2H), 1.43–1.23 (m, 26H), 0.90 (t,
J = 6.9 Hz, 3H).
13C NMR (MeOD, 126 MHz, ppm) δ 68.8, 46.1, 36.0, 33.1, 30.8, 30.7, 30.7, 30.6, 30.5, 26.4, 23.7. ESI-MS
m/
z clcd for C
17H
38NO [M + H]
+: 272.29; observed 272.3 [
32].
4.2.11. (2S,3R)-tert-Butyl-(13-bromo-3-hydroxytridecan-2-yl-4,5-d2)carbamate (21)
To a stirred solution of compound 20 (660 mg, 1.7 mmol) in dry d-methanol (20 mL) under argon atmosphere at ambient temperature was added a catalytic amount of 10% Pd/C (10 mg) followed by catalytic drop of d-acetic acid. The reaction vessel was evacuated and backfilled with deuterium gas (this process was repeated 2 times) and the resulting heterogeneous reaction mixture was allowed to stir at the same conditions for 14 h (as detected by HPLC analysis for complete reduction reaction). The reaction mixture was filtered through a short pad of Celite, which was rinsed several times with methanol (3 × 50 mL). The combined filtrates were concentrated under reduced pressure and the obtained residue was purified by flash column chromatography over silica gel using cylcohexane and ethylacetate as eluents (10–20% ethylacetate in cyclohexane) to provide compound 21 as a colorless oil.
Yield: 540 mg (81%). Rf: 0.5 (Pet. ether/ EtOAc 4:1, visualized with 1.3% ninhydrin solution). 1H NMR (CDCl3, 500 MHz, ppm): δ 4.76 (s, 1H), 3.67 (d, J = 15.5 Hz, 1H), 3.63 (t, J = 6.3 Hz, 1H), 3.40 (t, J = 6.9 Hz, 2H), 1.88–1.81 (m, 2H), 1.44 (s, 9H), 1.41–1.22 (m, 14H), 1.07 (d, J = 6.8 Hz, 3H).13C NMR (CDCl3, 126 MHz, ppm): δ 156.0, 79.6, 74.6, 50.7, 34.2, 33.6, 33.0, 29.8, 29.6, 29.6, 29.5, 28.9, 28.5, 28.3, 26.2, 14.5. ESI-LCMS: m/z calcd for C18H35D2NO3Br [M + H]+ 396.21; observed 396.2.
4.2.12. (4S,5R)-tert-Butyl-5-(10-bromodecyl-1,2-d2)-2,2,4-trimethyloxazolidine-3-carboxylate (22)
To a stirred solution of compound 21 (515 mg, 1.3 mmol) in anhydrous toluene (13 mL) under an argon atmosphere was added 2,2-dimethoxypropane (0.8 mL, 6.5 mmol), followed by a catalytic amount of p-toluensulfonic acid monohydrate (10 mol%). After being stirred under reflux in a Dean-Stark apparatus for 2 h (as judged by the TLC analysis; Pet. ether/ EtOAc 4:1; Rf (adduct) = 0.5; Rf (product) = 0.78; visualized with 1.3% ninhydrin solution), the reaction mixture was cooled to ambient temperature, diluted with ethylacetate, and successfully quenched with sat. NaHCO3 solution (80 mL). The mixture was transferred to separating funnel and the layers were separated. The aqueous layer was extracted again with ethylacetate (2 × 80 mL) and the combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. Flash column chromatography of the resultant crude residue over silica gel using cyclohexane and ethylacetate as eluents (0% to 10% ethylacetate in cyclohexane) provided the desired product 22 as a colorless oil.
Yield: 505 mg (89%). Rf: 0.53 (cyclohexane/EtOAc 9:1, visualized with 1.3% ninhydrin solution). 1H NMR (CDCl3, 500 MHz, ppm): δ 3.99–3.76 (m, 2H), 3.40 (t, J = 6.9 Hz, 2H), 1.88–1.81 (m, 2H), 1.61–1.45 (m, 15H), 1.45–1.23 (m, 14H), 1.07 (dd, J = 15.9, 6.2 Hz, 3H). 13C NMR (CDCl3, 126 MHz, ppm): δ 152.1, 151.8, 92.6, 92.2, 79.9, 79.3, 76.6, 76.3, 55.4, 55.4, 34.2, 33.0, 29.6, 29.5, 28.9, 28.7, 28.6, 28.3, 27.5, 25.1, 24.0, 14.5, 13.8. ESI-LCMS: m/z calcd for C21H38D2NO3BrNa [M+Na]+ 458.22; observed 458.2.
4.2.13. (4S,5R)-tert-Butyl-2,2,4-trimethyl-5-(pentadec-11-yn-1-yl-1,2-d2)oxazolidine-3-carboxylate (23)
The lithated 1-pentyne was synthesized as follows; to a stirred solution of 1-pentyne (330 µL, 3.3 mmol) in anhydrous THF (7 mL) under an argon atmosphere at −78 °C was carefully added a solution of t-BuLi (1.8 mL, 3 mmol, 1.7 M solution in pentane) in dropwise over a period of 20 min; the rate of addition was adjusted to keep the internal temperature below −65 °C. The resulting reaction mixture was stirred at the same conditions for 2 h to afford lithiated 1-pentyne solution which was used directly in the next step.
A stirred solution of freshly prepared lithated 1-pentyne at −78 °C was sequentially treated with hexamethylphosphoramide (870 µL, 5 mmol), followed by a dropwise addition of a solution of compound 22 (470 mg, 1.1 mmol, 0.25 M solution in anhydrous THF) over a period of 20 min. After being stirred at the same conditions for 1 h, and for additional 12 h at ambient temperature (as monitored by TLC analysis; Pet. ether/ EtOAc 9:1; Rf (adduct) = 0.53; Rf (product) = 0.64; visualized with Seebach reagent), the reaction mixture was carefully quenched with an ice-cold sat. NH4Cl solution (80 mL). The resulting mixture was diluted with ethylacetate (80 mL) and the layers were separated. The aqueous phase was extracted again with ethylacetate, and the combined organic phases were subsequently washed with sat. NaHCO3 solution and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The obtained residue was purified by flash column chromatography over silica gel using petroleum ether and ethylacetate as eluents (from 0–15% ethylacetate in petroleum ether) to afford the desired product 23 as a pale yellow oil.
Yield: 265 mg (58%). Rf: 0.48 (Pet. ether/ EtOAc 10:1, visualized with 1.3% ninhydrine solution). 1H NMR (CDCl3, 500 MHz, ppm): δ 3.93–3.74 (m, 2H), 2.66–2.27 (m, 4H), 1.78–1.64 (m, 2H), 1.63–1.46 (m, 17H), 1.44–1.22 (m, 14H), 1.09 (dd, J = 15.9, 6.1 Hz, 3H), 0.89 (t, J= 7.3 Hz, 3H). 13C NMR (CDCl3, 126 MHz, ppm): δ 152.1, 151.8, 92.6, 92.2, 83.7, 79.9, 79.3, 77.8, 76.6, 76.4, 55.4, 55.4, 31.6, 30.3, 29.9, 29.7, 29.7, 29.7, 29.6, 29.4, 28.7, 28.6, 28.4, 27.5, 27.4, 25.1, 24.0, 23.0, 22.7, 22.1, 14.5, 14.0, 13.8. ESI-LCMS: m/z calcd for C26H46D2NO3Br [M + H]+ 424.38; observed 424.4.
4.2.14. (4S,5R)-tert-Butyl-2,2,4-trimethyl-5-((Z)-pentadec-11-en-1-yl-1,2-d2)oxazolidine-3-carboxylate (24)
A stirred solution of compound 23 (220 mg, 0.53 mmol) in dry mixture of ethylacetate:DMF (50 mL, 95:5) under an argon atmosphere at ambient temperature was treated with a catalytic amount of Lindlar catalyst. The reaction vessel was evacuated and then backfilled with hydrogen gas (this process was repeated at least 2 times), and the resulting reaction mixture was allowed to stir at the same conditions for 16 h (as monitored by HPLC analysis for complete reduction). The mixture was subsequently filtered through a pad of Celite, which was rinsed several times with ethylacetate (3 × 80 mL). The combined filtrates were concentrated under in vacuo and the obtained residue was purified by flash column chromatography over silica gel using petroleum ether and ethylacetate as eluents (from 0–10% ethylacetate in petroleum ether) to provide the desired compound 24 as a colorless oil.
Yield: 155 mg (69%). Rf: 0.47 (Pet. ether/ EtOAc 10:1, visualized with 1.3% ninhydrin solution). 1H NMR (CDCl3, 500 MHz, ppm): δ 3 5.40–5.32 (m, 2H), 3.98–3.77 (m, 2H), 2.04–1.98 (m, 4H), 1.60–1.45 (m, 16H), 1.41–1.23 (m, 17H), 1.08 (dd, J = 16.1, 6.2 Hz, 3H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3, 126 MHz, ppm): δ 152.1, 151.8, 130.2, 129.8, 92.6, 92.2, 79.9, 79.3, 76.6, 76.4, 55.5, 55.4, 31.6, 30.3, 29.9, 29.7, 29.7, 29.7, 29.6, 29.4, 28.7, 28.6, 28.4, 27.5, 27.4, 25.1, 24.0, 23.3, 23.0, 14.5, 14.0, 13.8. ESI-LCMS: m/z calcd for C26H46D2NO3Br [M + H]+ 426.39; observed 426.4.
4.2.15. (2S,3R,Z)-2-Aminooctadec-14-en-4,5-d2-3-ol (25)
To a stirred solution of compound 24 (0.12 g, 0.28 mmol) in dry methanol (3 mL) at 0 °C was added dropwise acetyl chloride (200 µL, 2.8 mmol). After being stirred at the same conditions for 30 min, and for additional 90 min at ambient temperature (as monitored by TLC, visualized with KMnO4 solution), the reaction mixture was concentrated under reduced pressure. The resultant residue was taken up in diethylether (50 mL) and sequentially washed with saturated NaHCO3 solution (50 mL) and brine solution (80 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. Flash column chromatography of the obtained crude product over silica gel using ethylacetate and isopropanol as eluents (from 0–10% isopropanol in ethylacetate) provided the desired product 25 as a white solid.
Yield: 62 mg (76%). Rf: 0.46 (EtOAc/ iso-propanol 4:1, visualized with 1.3% ninhydrin solution). = +11.6 (c = 0.73, MeOH). 1H NMR (MeOD, 500 MHz, ppm): δ 5.39–5.31 (m, 2H), 3.72–3.68 (m, 1H), 3.27 (qd, J = 6.8, 3.0 Hz, 1H), 2.06–1.99 (m, 2H), 1.46–1.28 (m, 18H), 1.22 (d, J = 6.8 Hz, 3H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (MeOD, 126 MHz, ppm): δ 131.0, 130.6, 71.6, 52.6, 30.8, 30.7, 30.6, 30.3, 30.3, 28.1, 23.9, 14.1, 12.0. ESI-LCMS: m/z calcd for C18H36D2NO [M + H]+ 286.31; observed 286.3.
4.2.16. (S)-tert-Butyl-(1-hydroxy-3-oxopent-4-en-2-yl)carbamate (28)
To a stirred solution of compound 27 (1.45 g, 5.9 mmol) in dry THF (30 mL) at −78 °C under argon atmosphere was added dropwise a solution of n-butyllithium (n-BuLi, 2.1 mL, 5.3 mmol, 2.5 M in hexane). The resulting mixture was stirred at the same conditions for 30 min, before a solution of vinylmagnesium bromide (14.8 mL, 14.8 mmol, 1 M solution in THF) was added dropwise; the addition rate was adjusted so as to keep the internal temperature at −40 °C and it took 25 min to complete. After being stirred at −40 °C for 30 min, gradually warmed to ambient temperature and stirred for additional 4 h (as monitored by TLC analysis), the reaction mixture was added dropwise to an ice-cold 1 M HCl solution (100 mL) to quench; again, so as to keep the internal temperature below −10 °C and it took 30 min to complete. The resultant mixture was subsequently diluted with ethylacetate (100 mL) and the layers were separated. The aqueous layer was extracted several times with ethylacetate (4 × 80 mL). The combined organic extracts were washed with saturated NaHCO3 solution (150 mL) and brine (150 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. Flash column chromatography of the resultant crude mixture over silica gel using cyclohexane and ethylacetate as eluents (from 25–40% ethylacetate in cyclohexane) afforded compound 28 as a colorless oil.
Yield: 920 mg, (73%). Rf: 0.45 (Cyclohexane/ EtOAc 1:1, visualized with 1.3% ninhydrin). 1H NMR (CDCl3, 500 MHz, ppm): δ 6.57 (dd, J = 17.4, 10.5 Hz, 1H), 6.44 (dd, J = 17.5, 1.0 Hz, 1H), 5.93 (d, J = 10.5 Hz, 1H), 5.69 (br.s, 1H), 4.66 (m, 1H), 3.95 (dd, J = 11.6, 3.6 Hz, 1H), 3.90 (dd, J = 11.6, 4.2 Hz, 1H), 1.46 (s, 9H). 13C NMR (CDCl3, 125 MHz, ppm): δ 196.4, 156.1, 132.7, 130.7, 80.4, 63.6, 59.9, 28.3. ESI-LCMS: m/z calcd for C10H18NO4 [M + H]+ 216.12; observed 216.1.
4.2.17. (2S,4E)-tert-Butyl-(1-hydroxy-3-oxooctadec-4-en-2-yl)carbamate (29)
To a stirred solution of compound 28 (155 mg, 0.72 mmol) in dry DCM (4 mL) under argon atmosphere was added 1-pentadecene (782 µL, 2.9 mmol), followed by a catalytic amount of Grubbs Catalyst 2nd Generation (as a solid in one portion, 7 mol%). The resulting reaction mixture was allowed to stir under reflux and was monitored by TLC analysis until no further change in the composition of the reaction mixture (6 h, visualized with KMNO4). The solvent was subsequently removed under reduced pressure and the resultant residue was directly purified by flash column chromatography over silica gel using cyclohexane and ethylacetate as eluents (from 20–27% ethylacetate in cyclohexane) to afford the desired product 29 as a yellow oil.
Yield: 190 mg (67%). Rf: 0.62 (cyclohexane/ethylacetate 3:2, visualized with 1.3% ninhydrin solution). 1H NMR (CDCl3, 500 MHz, ppm): δ 7.06 (dt, J = 15.7, 6.9 Hz, 1H), 6.27 (d, J = 15.7 Hz, 1H), 5.73 (br.s, 1H), 4.62 (s, 1H), 3.93 (dd, J = 11.5, 3.4 Hz, 1H), 3.85 (dd, J = 11.5, 4.5 Hz, 1H), 2.27–2.22 (m, 2H), 1.45 (s, 9H), 1.33–1.24 (m, 22H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR (CDCl3, 125 MHz, ppm): δ 195.9, 156.4, 151.2, 126.5, 80.5, 64.5, 60.1, 32.9, 32.1, 22.8, 14.3. ESI-LCMS: m/z calcd for C23H44NO4 [M + H]+ 398.33; observed 398.3.
4.2.18. (2S)-2-Amino-1-hydroxyoctadecan-3-one-4,5-d2 (30)
A stirred solution of compound 29 (100 mg, 0.25 mmol) in anhydrous d-methanol (5 mL) under an argon atmosphere at ambient temperature was treated with a catalytic amount of 10% Pd/C (6 mg) followed by 2-drops of d-acetic acid. After being evacuated, the reaction vessel was backfilled with deuterium gas and the resulting heterogeneous reaction mixture was allowed to stir for 16 h at the same conditions. The suspension was subsequently filtered through a short pad of Celite, which was rinsed several times with chloroform (3 × 50 mL). The filtrates were combined and concentrated under reduced pressure to afford N-Boc-3-ketosphinganine-d2 as an oily residue. The obtained residue was dissolved in methanol (15 mL) and subsequently treated with 4N HCl-THF solution (3 mL). After the resulting reaction mixture was allowed to stir under reflux for 2 h (as monitored by TLC analysis; cyclohexane / EtOAc 3:2; Rf (adduct) = 0.62; Rf (product) =0.01; visualized with 1.3% ninhydrin solution), the solvent was removed under reduced pressure. The obtained residue was taken up in n-hexane (5 mL) and the mixture was vigorously stirred for 30 min, during which a white precipitate was formed. The obtained white precipitate was filtered off and subsequently washed with ice-cold diethylether to provide the desired product 30 as a white solid.
Yield: 46 mg (63%, over 2 steps). Rf: 0.32 (EtOAc/ iso-propanol 10:1, visualized with 1.3% ninhydrin solution). = +11.7 (c = 0.78, MeOH). 1H NMR (MeOD, 500 MHz, ppm): δ 4.18 (t, J = 3.8 Hz, 1H), 4.10 (dd, J = 12.1, 4.2 Hz, 1H), 3.98 (dd, J = 12.1, 3.5 Hz, 1H), 2.62 (dd, J = 14.1, 7.1 Hz, 1H), 1.60 (m, 1H), 1.30 (m, 24H), 0.90 (t, J = 7.0 Hz, 3H). 13C NMR (MeOD, 126 MHz, ppm): δ 205.3, 62.2, 60.2, 33.1, 30.8, 30.7, 30.6, 30.5, 30.4, 29.9, 23.7, 14.4. ESI-LCMS: m/z calcd for C18H36D2NO [M + H]+ 302.3; observed 302.30.
4.2.19. 2-(Undec-10-en-1-yloxy)-tetrahydro-2H-pyran (33)
To a stirred solution of compound 32 (9.4 g, 28.3 mmol) in anhydrous THF (280 mL) at ambient temperature under an argon atmosphere was added tert-BuOK (3.5 g, 31.13 mmol) in portionwise over a period of 30 min. After being stirred under reflux for 12 h (as monitored by TLC analysis, cyclohexane/EtOAc 95:5; Rf (adduct) = 0.4; Rf (product) = 0.58; visualized with ceric ammonium molybdate solution), the reaction was cooled to room temperature and subsequently quenched with water (200 mL). The layers were separated and the aqueous layer was extracted with petroleum ether (3 × 100 mL). The combined organic extracts were washed with brine (200 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. Purification of the resultant oil by flash column chromatography over silica gel using petroleum ether and ethylacetate as eluents (from 0–9% ethylacetate in petroleum ether) provided compound 33 as colorless oil.
Yield: 4.6 g (64 %). Rf: 0.67 (Pet. ether/ EtOAc 9:1, visualized with ceric ammonium molybdate solution). 1H-NMR (126 MHz, CDCl3, ppm): 5.81 (ddt, J = 16.9, 6.7, 1.3 Hz, 1H), 5.12–4.93 (m, 2H), 4.58–4.54 (m, 1H), 3.89–3.69 (m, 2 H), 3.64–3.31 (m, 2H), 2.11–1.98 (m, 2H), 1.89–1.65 (m, 2H), 1.64–1.46 (m, 6H), 1.44–1.27 (m, 12H). 13C-NMR (126 MHz, CDCl3, ppm): 139.17, 113.79, 98.84, 67.65, 62.24, 33.87, 30.76, 29.69, 29.56, 29.37, 29.14, 28.75, 26.24, 25.53, 19.56. ESI-MS m/z calcd for C16H31O2 [M + H]+: 255.23; observed 255.2.
4.2.20. Undec-10-en-1-ol (34)
A stirred solution of compound 33 (4.4 g, 17.1 mmol) in ethanol (85 mL) at ambient temperature was treated with a catalytic amount of pyridinium p-toluenesulfonate (PPTS) (0.43 g, 1.71 mmol, 10 mol%). After the resulting reaction mixture was allowed to stir under reflux for 2 h (as judged by TLC analysis for almost a complete deprotection; Pet. ether/ EtOAc 9:1; Rf (adduct) = 0.67; Rf (product) = 0.39; visualized with ceric ammonium molybdate solution), the solvent was removed under reduced pressure. The resultant oily residue was diluted with diethylether (100 mL) and subsequently washed with saturated NaHCO3 solution (100 mL). The aqueous layer was extracted again with diethylether (3 × 80 mL), and the combined organic extracts were washed with brine (150 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The obtained crude product was purified by flash column chromatography over silica gel using cyclohexane and ethylacetate as eluents (from 15–20% ethylacetate in cyclohexane) to furnish compound 34 as colorless oil.
Yield: 2.3 g (79 %). Rf: 0.48 (Pet. ether/ EtOAc 4:1, visualized with ceric ammonium molybdate solution). 1H NMR (500 MHz, CDCl3, ppm) δ 5.81 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 4.99 (ddd, J = 17.1, 3.7, 1.6 Hz, 1H), 4.92 (ddt, J = 10.2, 2.3, 1.2 Hz, 1H), 3.63 (t, J = 6.7 Hz, 2H), 2.06–2.01 (m, 2H), 1.56 (dq, J = 13.4, 6.7 Hz, 2H), 1.42–1.26 (m, 12H). 13C NMR (126 MHz, CDCl3, ppm) δ 139.4, 114.2, 63.2, 33.9, 32.9, 29.7, 29.5, 29.2, 29.1, 25.9. ESI-MS m/z calcd for C11H23O [M + H]+: 171.17; observed 171.2.
4.2.21. Undec-10-en-1-yl-4-methylbenzenesulfonate (36)
A stirred solution of 10-undecenol 34 (1.1 g, 6.25 mmol) in dry dichloromethane (16 mL) under argon atmosphere at 0 °C was treated with pyridine (2 mL, 25 mmol), followed by a portionwise addition of tosyl chloride (1.5 g, 7.8 mmol). After being stirred at the same condition for 1 h and for additional 16 h at ambient temperature (as monitored by TLC analysis; Pet. ether/ EtOAc 4:1; Rf (adduct) = 0.48; Rf (product) = 0.77; visualized with KMnO4 solution), the reaction mixture was diluted with CH2Cl2 (100 mL) and carefully quenched with saturated NaHCO3 solution (100 mL). The layers were separated, and the aqueous layer was extracted several times with CH2Cl2 (3 × 80 mL). The combined organic layers were successively washed with saturated CuSO4 solution (150 mL) and brine (150 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. Flash column chromatography of the resultant crude product over silica gel using petroleum ether and ethylacetate as eluents (from 0–10% ethylacetate in petroleum ether) afforded the desired product 36 as white waxy solid.
Yield: 1.7 g (84 %). Rf: 0.57 (petroleum ether /EtOAc 9:1, visualized with KMnO4 solution). 1H NMR (500 MHz, CDCl3, ppm) δ 4.58 (dd, J = 4.4, 2.8 Hz, 1H), 3.88 (ddd, J = 11.1, 7.0, 3.9 Hz, 1H), 3.74 (dt, J = 9.6, 6.9 Hz, 1H), 3.50 (ddd, J = 8.1, 6.5, 4.6 Hz, 1H), 3.46–3.34 (m, 3H), 1.92–1.78 (m, 3H), 1.79–1.66 (m, 1H), 1.63–1.49 (m, 6H), 1.46–1.25 (m, 12H). 13C NMR (126 MHz, CDCl3, ppm) δ 99.01, 67.82, 62.52, 34.22, 32.97, 30.93, 29.88, 29.60, 29.57, 29.51, 28.88, 28.30, 26.36, 25.65, 19.86. ESI-MS m/z calcd for C18H28NaO3S [M + Na]+: 347.17; observed 347.2.
4.2.22. (S)-13-Methylpentadec-1-ene (39)
The Grignard reagent of (S)-1-bromo-2-methylbutane (38) was synthesized as follows; A mixture of magnesium turnings (0.3 g, 12.5 mmol) in anhydrous THF (3 mL) under an argon atmosphere was treated with 2 drops of 1,2-dibromoethane (in order to activate the magnesium), followed by a solution of (S)-1-bromo-2-methylbutane (1.2 g, 8.3 mmol, in 5 mL of anhydrous THF). After the reaction had begun, the reaction mixture was allowed to stir for 90 min at 30 °C to yield a transparent Grignard solution which was immediately used in the next step.
Coupling reaction: According to Effenberger and Heid [
69], and Tamura and Kochi [
70] with some modifications. To a stirred solution of tosylate
36 (0.9 g, 2.8 mmol) in anhydrous THF (25 mL) under argon atmosphere at −78 °C was added dropwise a catalytic amount of dilithium tetrachlorocuprate (Li
2CuCl
4) solution (1.4 mL, 0.1 M in THF). The resulting mixture was stirred for 10 min at the same conditions, before a freshly prepared Grignard solution
38 was added dropwise over a period of 20 min. After the resulting reaction mixture was allowed to stir at ambient temperature for 16 h, during which a dark solution formed, (as monitored by TLC analysis; Pet. ether/ EtOAc 9:1; R
f (adduct) = 0.57; R
f (product) = 1; visualized with KMnO
4 solution), the reaction mixture was cooled to 0 °C and successfully quenched with an ice-cold saturated NH
4Cl solution (100 mL). The resulting mixture was diluted with n-pentane (100 mL), and the layers were separated. The aqueous layer was extracted with n-pentane (3 × 50 mL), and the combined organic layers were washed with brine, dried over anhydrous Na
2SO
4, filtered, and concentrated in vacuo (
Caution: temperature of rotatory evaporator shouldn’t exceed 25 °C). Purification of the obtained crude mixture by flash column chromatography over silica gel using n-pentane as eluent afforded the desired product
39 as colorless oil.
Yield: 0.42 g (49 %). Rf: 0.59 (Pet. ether, visualized with KMnO4 solution). 1H NMR (500 MHz, CDCl3, ppm) δ 5.82 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 4.99 (ddd, J = 17.1, 3.7, 1.6 Hz, 1H), 4.93 (ddt, J = 10.2, 2.3, 1.2 Hz, 1H), 2.07–2.02 (m, 2H), 1.40–1.25 (m, 26H), 1.17–1.07 (m, 6H), 0.88–0.83 (m, 19H). 13C NMR (126 MHz, CDCl3, ppm) δ 139.4, 114.2, 36.8, 34.9, 34.6, 34.1, 34.0, 30.2, 29.9, 29.8, 29.8, 29.7, 29.7, 29.3, 29.1, 27.3, 19.4, 11.6, 11.6.
4.2.23. (4S,5R)-tert-Butyl-4-(hydroxymethyl)-2,2-dimethyl-5-((S,E)-13-methylpentadec-1-en-1-yl)oxazolidine-3-carboxylate (41)
To a stirred solution of compound 40 (82 mg, 0.32 mmol.) in dry dichloromethane (3 mL), under argon atmosphere, was added the cross partner 39 (0.36 g, 1.6 mmol), followed by a catalytic amount of (1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene)-dichloro-(phenylmethylene) (tricyclohexylphosphine)-ruthenium, Grubbs catalyst 2nd generation, 8 mol%). After the resulting reaction mixture was allowed to stir under reflux for 12 h (until the TLC analysis showed no further change in the composition of the reaction mixture; Pet. ether/ EtOAc 7:3; Rf (adduct) = 0.45; Rf (product) = 0.6; visualized with 1.3% ninhydrin), the solvent was removed under reduced pressure. The resultant crude residue was subsequently purified by flash column chromatography over silica gel using cyclohexane and ethylacetate as eluents (from 20–25% ethylacetate in cyclohexane) to afford the desired product 41 as pale-yellow oil.
Yield: 88 mg (61.4%). Rf: 0.48 (cyclohexane/ethylacetate 8:2, visualized with 1.3% ninhydrin). 1H NMR (CDCl3, 500 MHz, ppm): δ 5.92–5.84 (m, 1H), 5.63–5.41 (m, 1H), 4.57 (t, J = 6.5 Hz, 1H), 4.11–4.04 (m, 1H), 3.89–3.43 (m, 3H), 2.06 (dd, J = 10.3, 6.4 Hz, 2H), 1.67–1.46 (m, 15H), 1.42–1.25 (m, 21H), 1.16–1.07 (m, 2H), 0.87–0.82 (m, 6H). 13C NMR (CDCl3, 125 MHz, ppm): δ 154.6, 137.6, 123.4, 93.1, 81.4, 80.4, 63.9, 62.2, 36.8, 34.5, 32.5, 30.2, 29.8, 29.8, 29.7, 29.6, 29.6, 29.4, 28.5, 27.3, 19.4, 11.6. ESI-MS m/z calcd for C27H52NO4 [M + H]+: 454.39; observed 454.4.
4.2.24. (2S,3R,16S,E)-2-Amino-16-methyloctadec-4-ene-1,3-diol (42)
A stirred solution of compound 41 (65 mg, 0.14 mmol) in absolute methanol (1.5 mL) at 0 °C was carefully treated with acetyl chloride in dropwise (AcCl, 150 µL, 2.1 mmol). After the resulting reaction mixture was allowed to stir at the same conditions for 30 min and for additional 90 min at ambient temperature (as detected by TLC analysis for complete deprotection), the solvent was removed under reduced pressure. The resultant waxy residue was washed with ice-cooled diethylether and subsequently filtered off to provide the desired crude product as a colorless waxy solid. Further purification of the obtained crude product was performed by flash column chromatography over silica gel using ethylacetate and isopropanol as eluents (from 0–5% isopropanol in ethylacetate) to afford compound 42 as a white waxy solid.
Yield: 36 mg (82 %). Rf: 0.31 (EtOAc/ iso-propanol 9:1, visualized with KMnO4 solution). = +6.1 (c = 0.58, MeOH). 1H NMR (MeOD, 500 MHz, ppm) δ 5.88 (ddd, J = 14.9, 7.2, 3.8 Hz, 1H), 5.53–5.47 (m, 1H), 4.34–4.30 (m, 1H), 3.82 (dd, J = 11.6, 4.0 Hz, 1H), 3.69 (dd, J = 11.6, 8.3 Hz, 1H), 3.23 (dt, J = 8.5, 4.3 Hz, 1H), 2.13 (q, J = 7.0 Hz, 2H), 1.48–1.43 (m, 2H), 1.40–1.30 (m, 16H), 1.20–1.11 (m, 2H), 0.92–0.87 (m, 6H). 13C NMR (MeOD, 125 MHz, ppm) δ 136.5, 128.5, 70.9, 59.4, 58.5, 37.8, 35.7, 33.3, 31.1, 30.8, 30.8, 30.7, 30.6, 30.6, 30.4, 30.2, 28.2, 19.6, 11.7. ESI-MS m/z calcd for C19H40NO2 [M + H]+: 314.31; observed 314.3.
4.2.25. ((2S,3R,E)-tert-Butyl-13-bromo-1-((tert-butyldiphenylsilyl)oxy)-3-hydroxytridec-4-en-2-yl)carbamate (44)
A stirred solution of compound 43 (1.1 g, 2.5 mmol), obtained from L-serine in 5-steps, and 10-bromo-decene (2.2 g, 10 mmol) in dry d-chloroform (25 mL) under argon atmosphere was treated with p-benzoquinone (10 mol%) and CuI (3 mol%), followed by Grubbs 2nd generation catalyst (10 mol%). The resulting reaction mixture was allowed to stir at 35 °C for 14 h (as judged by TLC analysis, no change in the mixture), and subsequently the solvent was removed under reduced pressure. The obtained crude mixture was subjected to flash column chromatography over silica gel using ethylacetate and petroleum ethers as mobile phase (10–15% ethylacetate in petroleum ether) to furnish the desired E-isomer 44 as pale-yellow oil.
Yield: 810 mg (51 %). Rf: 0.32 (PE/ EA 9:1, visualized with KMnO4 solution). 1H NMR (CDCl3, 500 MHz, ppm) δ 7.64 (td, J = 8.0, 1.4 Hz, 4H), 7.47–7.42 (m, 2H), 7.41–7.37 (m, 4H), 5.81–5.74 (m, 1H), 5.48 (ddt, J = 15.4, 6.0, 1.3 Hz, 1H), 5.19 (br.s, 1H), 4.24 (t, J = 5.0 Hz, 1H), 3.90 (dd, J = 10.5, 3.7 Hz, 1H), 3.75 (d, J = 10.2 Hz, 1H), 3.65 (br.s, 1H), 3.40 (t, J = 6.9 Hz, 2H), 2.07–2.00 (m, 2H), 1.88–1.81 (m, 2H), 1.45 (s, 9H), 1.41–1.27 (m, 9H), 1.07 (s, 9H). 13C NMR (CDCl3, 126 MHz, ppm) δ 156.0, 135.7, 133.4, 132.8, 132.7, 130.1, 130.1, 129.4, 128.0, 79.6, 74.4, 64.3, 55.2, 34.1, 33.0, 32.4, 29.4, 29.3, 29.2, 28.8, 28.5, 28.3, 27.0, 19.3. ESI-MS m/z calcd for C34H53NO4BrSi [M + H]+: 646.29; observed 646.3.
4.2.26. ((2S,3R)-tert-Butyl-13-bromo-1-((tert-butyldiphenylsilyl)oxy)-3-hydroxytridecan-2-yl)-carbamate (45)
A stirred solution of compound 44 (790 mg, 1.25 mmol) in dry THF (12 mL) at ambient temperature under argon atmosphere was treated with 10% Pd/C (12 mg) followed by a drop of acetic acid. After the argon was evacuated and replaced by hydrogen gas, the obtained reaction mixture was stirred at ambient temperature for 16 h (as judged by UPLC analysis for a complete reduction of starting material), the mixture was allowed to filter through Celite. The collected rinsed filtrates were concentrated under reduced pressure. Flash column chromatography of crude product was performed over silica gel using petroleum ether and ethylacetate as eluents (10–15% ethylacetate in petroleum ether) to provide compound 45 as white waxy-solid.
Yield: 680 mg (86 %). Rf: 0.44 (PE/ EA 4:1, visualized with 1.3% ninhydrin solution). 1H NMR (CDCl3, 500 MHz, ppm) δ 7.68–7.47 (m, 4H), 7.47–7.42 (m, 6H), 5.29 (d, J = 8.1 Hz, 1H), 3.92 (dd, J = 10.8, 3.6 Hz, 1H), 3.82 (d, J = 8.5 Hz, 1H), 3.69–3.65 (m, 1H), 3.56 (d, J = 6.7 Hz, 1H), 3.41 (t, J = 6.9 Hz, 2H), 1.89–1.82 (m, 2H), 1.53–1.40 (m, 12H), 1.34–1.25 (m, 12H), 1.07 (s, 9H). 13C NMR (CDCl3, 126 MHz, ppm) δ 155.9, 135.7, 132.7, 130.2, 130.1, 128.1, 128.0, 79.6, 74.0, 64.3, 54.7, 34.6, 34.2, 33.0, 29.7, 29.6, 29.6, 28.9, 28.6, 28.3, 27.0, 26.0, 19.3. ESI-MS m/z calcd for C34H55NO4BrSi [M + H]+: 648.31; observed 648.3.
4.2.27. (4S,5R)-tert-Butyl-5-(10-bromodecyl)-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyloxazolidine-3-carboxylate (46)
To a stirred solution of compound 45 (655 mg, 1 mmol) in dry toluene (10 mL) under argon atmosphere was added 2,2-dimethoxy-propane (mL, mmol), followed by a catalytic amount of p-toluenesulfonic acid monohydrate (10 mol%). After the obtained mixture was stirred at 80 °C for 3 h (as monitored by TLC analysis for a complete reaction), the mixture was cooled to ambient temperature and subsequently quenched by saturated NaHCO3 solution. The resultant mixture was diluted with ethylacetate and the layers were separated. The aqueous layer was extracted with ethylacetate (2 × 70 mL), and the collected organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. Purification of resultant crude product by flash column chromatography using petroleum ether and ethylacetate as mobile phases (0–5% ethylacetate in petroleum ether) afforded compound 46 as a pale-yellow oil.
Yield: 500 mg (72 %). Rf: 0.53 (PE/ EA 9:1, visualized with 1.3% ninhydrin solution). 1H NMR (CDCl3,500 MHz, ppm) δ 7.75–7.62 (m, 4H), 7.46–7.35 (m, 6H), 4.10–4.00 (m, 1H), 3.85 (ddd, J = 17.3, 7.4, 4.8 Hz, 1H), 3.64–3.49 (m, 1H), 3.40 (t, J = 6.9 Hz, 3H), 1.89–1.81 (m, 2H), 1.54–1.38 (m, 12H), 1.34–1.25 (m, 16H), 1.06 (s, 9H). 13C NMR (CDCl3, 126 MHz, ppm) δ 152.2, 151.8, 135.9, 135.9, 135.8, 135.7, 133.9, 133.5, 133.3, 130.2, 130.1, 129.9, 129.8, 129.7, 128.0, 128.0, 127.8, 127.8, 92.5, 92.2, 80.0, 79.7, 76.4, 64.4, 61.8, 61.2, 60.8, 34.2, 33.0, 29.9, 29.6, 29.6, 28.9, 28.6, 28.5, 28.3, 27.1, 27.0, 24.8, 23.4, 19.3. ESI-MS m/z calcd for C37H59NO4BrSi [M + H]+: 688.34; observed 688.3.
4.2.28. (4S,5R)-tert-Butyl-4-(((tert-butyldiphenylsilyl)-oxy)-methyl)-2,2-dimethyl-5-(pentadec-11-yn-1-yl)oxazolidine-3-carboxylate (47)
1-Pentyne (270 µL, 2.72 mmol) in dry THF (3 mL) under argon atmosphere at −78 °C was treated in dropwise with tert-BuLi solution (1.5 mL, 2.45 mmol, 1.7 M solution in pentane) over a period of 20 min. After the resulting mixture was allowed to stir at the same conditions for 2 h, HMPA (590 µL, 3.4 mmol) was added followed by a solution of compound 46 (470 mg, 0.68 mmol, 0.25 M in THF). The resulting reaction mixture was stirred at the same conditions for 1 h, worm gradually to ambient temperature and stirred for additional 14 h (as indicated by TLC for almost a complete reaction). The mixture was cooled again to 0 °C and subsequently quenched with 1 M NH4Cl solution. The obtained mixture was extracted with ethylacetat (3 × 80 mL), and the combined organic extracts were washed with saturated NHCO3 solution and brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resultant crude mixture was purified by flash column chromatography using petroleum ether and ethylacetate as eluents (0–5% ethylacetate in petroleum ether) to provide compound 47 as a pale-yellow oil.
Yield: 290 mg (63 %). Rf: 0.51 (PE/ EA 10:1, visualized with 1.3% ninhydrin solution). 1H NMR (CDCl3,500 MHz, ppm) δ 7.74–7.58 (m, 4H), 7.43–7.31 (m, 6H), 4.10–4.07 (m, 1H), 3.82–3.73 (m, 2H), 3.68–3.53 (m, 1H), 2.31–2.16 (m, 2H), 2.15–1.99 (m,2H), 1.76–1.69 (m, 2H), 1.68–1.43 (m, 17H), 1.39–1.28 (m, 14H), 1.05 (s, 9H), 0.91 (t, J = 7.8 Hz, 3H). 13C NMR (CDCl3, 126 MHz, ppm) δ 151.7, 136.0, 135.9, 135.8, 135.7, 133.8, 133.5, 133.4, 130.3, 130.2, 130.1, 129.9, 129.8, 128.1, 127.9, 91.6, 79.9, 79.7, 78.1, 77.4, 76.8, 75.6, 63.3, 62.8, 35.6, 34.3, 33.5, 32.9, 30.0, 30.8, 30.7, 29.9, 29.7, 28.8, 28.6, 28.4, 27.4, 27.1, 24.4, 23.8, 14.1. ESI-MS m/z calcd for C37H59NO4BrSi [M + H]+: 676.48; observed 676.5.
4.2.29. (4S,5R)-tert-Butyl-4-(((tert-butyldiphenylsilyl)oxy)methyl)-2,2-dimethyl-5-((Z)-pentadec-11-en-1-yl)oxazolidine-3-carboxylate (48)
Compound 47 (270 mg, 0.4 mmol) in a mixture of ethylacetate and DMF (40 mL, 5:1) under argon atmosphere at ambient temperature was treated with Lindlar catalyst. After the reaction vessel was evacuated from argon and subsequently connected to a hydrogen balloon, the reaction mixture was stirred at ambient temperature for 12 h (as judged by UPLC analysis for a complete reaction). The mixture was then filtered through Celite, and the collected filtrates were concentrated under reduced pressure. The resultant crude mixture was purified by column chromatography over silica gel using ethylacetate and petroleum ether as eluents (0–5% ethylacetate in petroleum ether) to yield the desired Z-isomer of compound 48 as a colourless oil.
Yield: 200 mg (74 %). Rf: 0.59 (PE/ EA 9:1, visualized with 1.3% ninhydrin solution). 1H NMR (CDCl3, 500 MHz, ppm) δ 7.76–7.61 (m, 4H), 7.43–7.32 (m, 6H), 5.45–5.33 (m, 2H), 4.12–4.03 (m, 1H), 3.79 (dd, J = 7.6 Hz, 1H), 3.67–3.52 (m, 1H), 2.11-1.97 (m, 2H), 1.95-1-89 (m,2H), 1.78–1.64 (m, 2H), 1.62–1.41 (m, 15H), 1.38–1.26 (m, 16H), 1.05 (s, 9H), 0.89 (t, J = 7.9 Hz, 3H). 13C NMR (CDCl3, 126 MHz, ppm) δ 152.6, 135.9, 135.9, 135.8, 135.7, 133.8, 133.5, 133.4, 131.7, 131.2, 130.2, 130.2, 129.9, 129.8, 129.8, 128.1, 128.0, 127.8, 91.9, 80.0, 79.7, 75.8, 63.3, 62.8, 35.6, 34.2, 33.9, 33.0, 30.8, 30.8, 30.6, 29.9, 29.7, 28.6, 28.5, 28.2, 27.7, 27.1, 24.8, 23.9, 14.2. ESI-MS m/z calcd for C42H68NO4Si [M + H]+: 678.49; observed 678.5.
4.2.30. (2S,3R,Z)-2-Aminooctadec-14-ene-1,3-diol (49)
A stirred solution of 48 (175 mg, 0.26 mmol) in THF (5 mL) was treated with tert-butyl ammonium fluoride (TBAF) (330 mg, 1.05 mmol). After the resulting mixture was stirred at 55 °C for 3 h (as judged by TLC for complete deprotection), the reaction was cooled to ambient temperature and quenched with saturated NaHCO3 solution. The obtained mixture was extracted with ethylacetate (3 × 50 mL), and the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude mixture was allowed to filter through a pad of silica gel. After the resultant crude product was dissolved in methanol (5 mL) and cooled to 0 °C, acetyl chloride was added (372 µL, 5.2 mmol). The resulting reaction mixture was allowed to stir at 0 °C for 1 h and for additional 2 h at ambient temperature (as detected by TLC analysis for a complete deprotection), and subsequently quenched with saturated NaHCO3 solution. The mixture was extracted with DCM (3 × 40 mL) and the combined extracted were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. Purification of resultant crude product was performed by column chromatography using ethylacetate and iso-propanol as mobile phases (0–10% iso-propanol in ethylacetate) furnished the desired 14Z-sphingosine 49 as a white waxy-solid.
Yield: 34 mg (43 %). Rf: 0.46 (EA/ iso-Pro 9:2, visualized with KMnO4 solution). = +17.6 (c = 1.15, MeOH). 1H NMR (MeOD, 500 MHz, ppm) δ 5.43–5.32 (m, 2H), 3.84 (dd, J = 11.6, 4.0 Hz, 1H), 3.78 (dt, J = 8.3, 4.2 Hz, 1H), 3.70 (dd, J = 11.5, 8.7 Hz, 1H), 3.20 (dt, J = 8.2, 3.9 Hz, 1H), 2.06–2.00 (m, 2H), 2.00–1.94 (m, 2H), 1.50 (ddd, J = 14.2, 8.6, 2.9 Hz, 2H), 1.41–1.28 (m, 18H), 0.91 (t, J = 8.2 Hz, 3H). 13C NMR (MeOD, 126 MHz, ppm) δ 131.7, 131.0, 70.3, 58.9, 58.5, 35.8, 34.2, 33.6, 30.8, 30.7, 30.7, 30.7, 30.6, 30.6, 30.3, 30.2, 28.1, 27.0, 23.8, 14.1. ESI-MS m/z calcd for C18H38NO2 [M + H]+: 300.29; observed 300.3.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








